
Density Functional Theory study of the structural and electronic properties of H3PO4/ZSM-5
Abstract
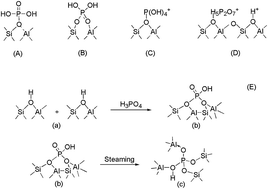
In this work, a Density Functional Theory (DFT) study has been carried out to investigate the structural and electronic properties of H3PO4/ZSM-5 (extra-framework and framework modification). Cluster models at different T sites (T6, T9, and T12) are suggested. The local structure of the extra-framework modified cluster for H3PO4/ZSM-5 shows that there is a hydrogen bond interaction between the hydrogen atoms in H3PO4 and the oxygen atoms in the zeolite framework, involving O4 and Ha. Additionally, the Ozeo always tends to keep in a straight line distribution. Mulliken charge analysis of the cluster models concerned shows that the charge indeed transfers from the oxygen atoms toward P, Al and H atoms. The charge transfer from ZSM-5 to H3PO4 could enhance the interaction between the H3PO4 and the ZSM-5. Based on the DFT study above, the mutual relationship between the acidic sites on ZSM-5 and phosphoric acid might be more clearly confirmed.
 Please wait while we load your content...
Please wait while we load your content...

