
Theoretical investigations toward the tandem reactions of N-aziridinyl imine compounds forming triquinanes via trimethylenemethane diyls: mechanisms and stereoselectivity†
Abstract
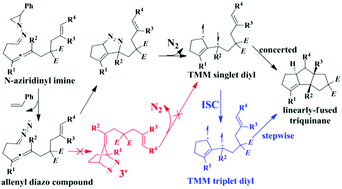
In this paper, we have investigated the tandem reaction mechanism for the N-aziridinyl imine compounds forming triquinanes via trimethylenemethane (TMM) diyls in detail. Based on the calculated results, the reaction is initiated by the cleavage of the N-aziridinyl in the substrate, followed by an intramolecular 1,3-dipolar (3 + 2) cycloaddition preferentially leading to a linearly-fused tetrahydrocyclopentapyrazole intermediate. Next, the intermediate loses N2 to form the singlet TMM diyl M3S, which can then undergo another concerted (3 + 2) cycloaddition to generate the linearly-fused cis–trans or cis–syn triquinane products. In addition, M3S can also undergo intersystem crossing to the triplet TMM diyl M3T, and the six possible reaction pathways associated with M3T have also been identified. The calculated results reveal that the cis–trans fused pathway associated with M3S is energetically preferred with the highest free energy barrier of 25.0 kcal mol−1. In comparison, the cyclization of M3T requires much higher activation free energies (ΔG≠ = 34.4–57.8 kcal mol−1). At the experimental temperature 110 °C, only the linearly-fused cis–trans and cis–syn pathways associated with M3T (ΔG≠ = 34.4 and 35.5 kcal mol−1 respectively) are possible. The calculated results also indicate that for both M3S and M3T, the linearly-fused cis–trans triquinane should be the main product, which is consistent with the experimental observation. At last, conformational and NBO analyses on key transition states identified the cis–trans stereocontrol factors. Further calculations indicate that the methyl substituent on the allene group of the reactant substrate improves the stereoselectivity of the reaction but does not affect the rate-determining step.
 Please wait while we load your content...
Please wait while we load your content...

