
Iron(ii) complexes featuring κ3- and κ2-bound PNP pincer ligands – the significance of sterics†
Abstract
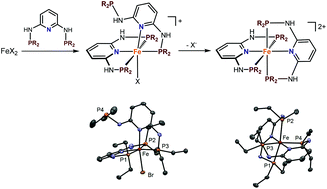
Treatment of anhydrous FeX2 (X = Cl, Br) with 2 equiv. of the sterically little demanding N,N′-bisphosphino-2,6-diaminopyridine based PNP ligands – featuring Ph, biphenol (BIPOL), Me, Et, nPr, and nBu substituents at the phosphorus sites and H, Me, and Ph substituents at the N-linkers – afforded diamagnetic cationic octahedral complexes of the general formula [Fe(κ3-P,N,P-PNP)(κ2-P,N-PNP)X]+ featuring a κ2-P,N bound PNP ligand. With the sterically more encumbered N-methylated ligand PNPMe-Ph the related complex [Fe(κ3-P,N,P-PNPMe-Ph)(κ2-P,N-PNHMe-Ph)Cl]+ rather than [Fe(κ3-P,N,P-PNPMe-Ph)Cl2] was formed. This reaction was accompanied by P–N bond cleavage, thereby forming the κ2-P,N-bound N-diphenylphosphino-N,N′-methyl-2,6-diaminopyridine ligand. In contrast, with the N-phenylated ligands PNPPh-Et and PNPPh-nPr, despite small Et and nPr substituents at the phosphorus sites, complexes [Fe(κ3-P,N,P-PNPPh-Et)Cl2] and [Fe(κ3-P,N,P-PNPPh-nPr)Cl2] were formed, revealing that sterics can be also controlled by substituent variations at the amino N-sites. Depending on the solvent, complexes featuring κ2-P,N-bound ligands undergo facile rearrangement reactions to give dicationic complexes of the type [Fe(κ3-P,N,P-PNP)2]2+ where both PNP ligands are bound in a κ3-P,N,P-fashion. In the presence of either Ag+ or Na+ salts as halide scavengers this reaction takes place within a few minutes. The pendant PR2 arm of the κ3-κ2-complexes is readily oxidized to the corresponding phosphine sulfides upon treatment with elemental sulfur. This was exemplarily shown for [Fe(κ3-P,N,P-PNP-nPr)(κ2-P,N-PNS-nPr)Cl]+. Halide abstraction afforded the dicationic bis-chelated octahedral Fe(II) complex [Fe(κ3-P,N,P-PNP)2]2+ together with the free SNP ligand rather than [Fe(κ3-P,N,P-PNP-nPr)(κ3-S,P,N-PNS-nPr)]2+.
 Please wait while we load your content...
Please wait while we load your content...

