
Towards a quantitative understanding of palladium metal scavenger performance: an electronic structure calculation approach†
Abstract
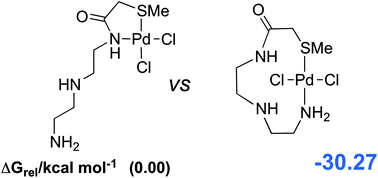
Dispersion corrected density functional theory (DFT-D) has been applied to understand the performance of several palladium metal scavengers. Nine different sulfur-based ligands and three different palladium metal sets have been investigated in detail. Based on a thorough analysis of the thermodynamic binding parameters ΔH, ΔG and ΔS, we have identified the best binding modes for all scavenger ligands. Bis-monodentate coordination is favoured over chelation in ΔH and ΔG values for most of the scavenger ligands. Special attention has been paid to the ligand strain energies, which account for the structural changes of the ligands upon complexation indicating that small (5-membered) chelates are considerably less favourable than expected. Some ligands can use their longest chain (>7-atoms) to yield trans chelates, which ligands with shorter chains (≤6-atoms) are unable to form. A secondary amino nitrogen (RR′NH) is found to be the best donor with highest binding enthalpy for Pd(II) metal systems. In terms of the strength of the initial binding interactions, –SMe > –SH; capping thiols (–SH) as thioethers (–SMe) is therefore suggested to be an effective strategy in scavenger design. These observations mark the beginning of a knowledge base of the full range of possible interactions, leading to the construction of a sulfur ligand database for the design of scavenger systems.
 Please wait while we load your content...
Please wait while we load your content...

