
Optimizing a parametrized Thomas–Fermi–Dirac–Weizsäcker density functional for atoms†

Abstract
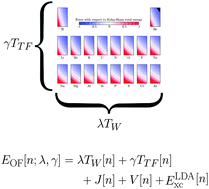
Because of issues with accuracy and transferability of existing orbital-free (OF) density functionals, OF functional development remains an active research area. However, due to numerical difficulties, all-electron self-consistent assessment of OF functionals is limited. Using an all-electron radial OFDFT code, we evaluate the performance of a parametrized OF functional for a wide range in parameter space. Specifically, we combine the parametrized Thomas–Fermi–Weizsäcker kinetic model (λ and γ for the fractions of Weizsäcker and Thomas–Fermi functionals, respectively) with a local density approximation (LDA) for the exchange–correlation functional. In order to obtain the converged results for λ values other than λ = 1, we use the potential scaling introduced in previous work. Because we work within a wide region in parameter space, this strategy provides an effective route towards better understanding of the parameter interplay that allows us to achieve good agreement with the Kohn–Sham (KS) model. Here, our interest lies in total energy, Euler equation eigenvalue, and electronic densities when the parameters are varied between 0.2 and 1.5. We observe that a one-to-one relation between λ and γ defines a region in parameter space that allows the atomic energies to be approximated with a very small average error (less than 3% percent for all the atoms studied) with respect to the KS reference energies. For each atom, the reference KS HOMO eigenvalue can also be reproduced with a similar error, but the one-to-one correspondence between λ and γ belongs to a different region of the same parameter space. Contrary to both properties, the atomic density behaves more smoothly and the error in reproducing the KS reference densities appears more insensitive to variation of the parameters (with mostly an average integrated difference of 0.15–0.20 |e| per electron). These results pave the way towards testing of parameter transferability and further systematic improvement of OF density functionals.
- This article is part of the themed collection: Real-space numerical grid methods in quantum chemistry
 Please wait while we load your content...
Please wait while we load your content...

