
A density functional theory study of ethylene hydrogenation on MgO- and γ-Al2O3-supported carbon-containing Ir4 clusters†
Abstract
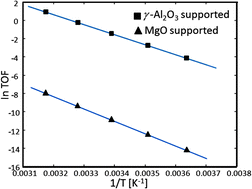
Density functional theory was used to investigate the reaction mechanisms of ethylene hydrogenation on MgO(100)- and γ-Al2O3(110)-supported carbon-containing Ir4 clusters. The cluster supported on γ-Al2O3(110) is more active than that on MgO(100), which is consistent with experimental observations. The present calculations show that the binding energies of reactants on the carbon-containing Ir4 cluster are weaker on the γ-Al2O3 supported catalysts compared to the MgO supported Ir cluster. This relatively weak adsorption energy of ethylene on the γ-Al2O3 surface means that ethylene desorption is easier, hence a higher catalytic activity is achieved. To gain further understanding, the energy decomposition method and micro-kinetic analysis are also introduced.
 Please wait while we load your content...
Please wait while we load your content...

