
Squeezing water clusters between graphene sheets: energetics, structure, and intermolecular interactions
Abstract
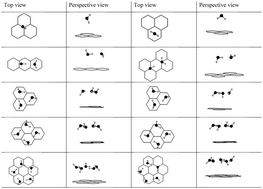
The behavior of water confined at the nanoscale between graphene sheets has attracted much theoretical and experimental attention recently. However, the interactions, structure, and energy of water at the molecular scale underpinning the behavior of confined water have not been characterized by first-principles calculations. In this work we consider small water clusters up to the hexamer adsorbed between graphene sheets using density functional theory calculations with van der Waals corrections. We investigate the effects on structure, energy, and intermolecular interactions due to confinement between graphene sheets. For interlayer distances of about one nanometer or more, the cluster adsorption energy increases approximately linearly with the cluster size by 0.1 eV per molecule in the cluster. As the interlayer distance decreases, the cluster adsorption energy reaches a maximum at 6 to 7 Å with approximately 0.16 eV stabilization energy relative to large interlayer distances. This suggests the possibility of controlling the amount of adsorption in graphene nanomaterials by varying the interlayer distance. We also quantify the intermolecular hydrogen bonding in the clusters by calculating the dissociation energy required to remove one molecule from each cluster. For each cluster size, this is constant for interlayer distances larger than approximately 6 to 8 Å. For smaller distances the intermolecular interaction decreases rapidly thus leading to weaker cohesion between molecules in a squeezed cluster. We expect a mechanism of concerted motion for hydrogen-bonded water molecules confined between graphene sheets, as has been observed for water confined within the carbon nanotubes. Thus, the decrease in the dissociation energy we observed here is consistent with experimental results for water transport through graphene and related membranes that are of interest in nanofiltration. We also calculate the corrugation in the interaction potential between graphene sheets which suggests a switch from very small corrugation to stick–slip behavior at interlayer distances smaller than 6 Å. Our results for gas phase clusters agree reasonably with methods using more demanding quantum chemical methods to treat the van der Waals interactions, thus providing support for the relatively fast density functional theory methods used here for studying water–graphene interactions in nanoscale systems.
 Please wait while we load your content...
Please wait while we load your content...

