
Insights into an intriguing gas sorption mechanism in a polar metal–organic framework with open-metal sites and narrow channels†
Abstract
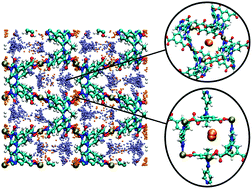
Simulations of H2 and CO2 sorption were performed in the metal–organic framework (MOF), [Cu(Me-4py-trz-ia)]. This MOF was recently shown experimentally to exhibit high uptake for H2 and CO2 sorption and this was reproduced and elucidated through the simulations performed herein. Consistent with experiment, the theoretical isosteric heat of adsorption, Qst, values were nearly constant across all loadings for both sorbates. The simulations revealed that sorption directly onto the open-metal sites was not observed in this MOF, ostensibly a consequence of the low partial positive charges of the Cu2+ ions as determined through electronic structure calculations. Sorption was primarily observed between adjacent carboxylate oxygen atoms (site 1) and between nearby methyl groups (site 2) of the organic linkers. In addition, saturation of the most energetically favorable sites (site 1) is possible only after filling a nearby site (site 2) first due to the MOF topology. This suggests that the lack of dependence on loading for the Qst is due to the concurrent filling of sites 1 and 2, leading to an observed average Qst value.
 Please wait while we load your content...
Please wait while we load your content...

