
Thiol–maleimide “click” chemistry: evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity†
Abstract
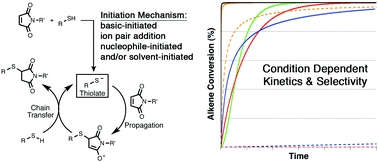
The mechanism and kinetics of thiol–maleimide “click” reactions carried out under a variety of conditions have been investigated computationally and using experimental competition reactions. The influence of three different solvents (chloroform, ethane thiol, and N,N-dimethylformamide), five different initiators (ethylamine, diethylamine, triethylamine, diazabicyclo[2.2.2]octane, and dimethylphenyl-phosphine), and seven different thiols (methyl mercaptan, β-mercaptoethanol, thioacetic acid, methyl thioglycolate, methyl 3-mercaptopropionate, cysteine methyl ester, and thiophenol) on the energetics and kinetics of thiol–maleimide reactions have been examined using density functional methods. Computational and kinetic modeling indicate that the choice of solvent, initiator, and thiol directly influences whether product formation follows a base-, nucleophile-, or ion pair-initiated mechanism (or some combination thereof). The type of mechanism followed determines the overall thiol–maleimide reaction kinetics. Insights from computational studies are then used to understand the selectivity of ternary thiol–maleimide reactions between N-methyl maleimide, thiophenol, and 1-hexanethiol in different combinations of solvents and initiators. The results provide considerable insight into the interplay between reaction conditions, kinetics, and selectivity in thiol–maleimide reactions in particular and thiol-Michael reactions in general, with implications ranging from small molecule synthesis to bioconjugation chemistry and multifunctional materials.
- This article is part of the themed collection: Polymer Chemistry 15th anniversary regional spotlight collection: The Americas
 Please wait while we load your content...
Please wait while we load your content...

