
Kinetic regimes in the tandem reactions of H-BEA catalyzed formation of p-xylene from dimethylfuran†
 cd
cd
Abstract
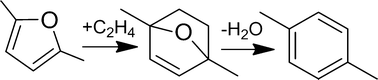
Reaction kinetics and pathways of p-xylene formation from 2,5-dimethylfuran (DMF) and ethylene via cascade reactions of Diels–Alder cycloaddition and subsequent dehydration over H-BEA zeolite (Si/Al = 12.5) were characterized. Two distinct kinetic regimes were discovered corresponding to the rate limiting reaction, namely Diels–Alder cycloaddition and cycloadduct dehydration, as the concentration of Brønsted acid sites decreases. At catalyst loadings with effective acid site concentrations exceeding a critical value (~2.0 mM), the rate of formation of Diels–Alder products becomes constant. Under these conditions, the measured activation energy of 17.7 ± 1.4 kcal mol−1 and reaction orders correspond to the [4 + 2] Diels–Alder cycloaddition reaction of DMF and ethylene. Conversely, at catalyst loadings below the critical value, the formation rate of p-xylene becomes first order in catalyst loading, and the measured activation energy of 11.3 ± 3.5 kcal mol−1 is consistent with dehydration of the Diels–Alder cycloadduct to p-xylene. Experimental comparison between H-BEA and H-Y zeolite catalysts at identical conditions indicates that the micropore structure controls side reactions such as furan dimerization and hydrolysis; the latter is supported via molecular simulation revealing a substantially higher loading of DMF within H-Y than within H-BEA zeolites at reaction conditions.
 Please wait while we load your content...
Please wait while we load your content...

