
Simulations of hydrogen sorption in rht-MOF-1: identifying the binding sites through explicit polarization and quantum rotation calculations†
Abstract
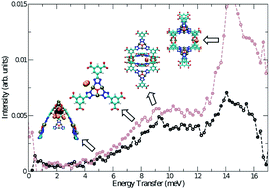
Grand canonical Monte Carlo (GCMC) simulations of hydrogen sorption were performed in rht-MOF-1, a metal–organic framework (MOF) that consists of isophthalate groups joined by copper paddlewheel clusters and Cu3O trimers through tetrazolate moeities. This is a charged rht-MOF that contains extra-framework nitrate counterions within the material. For the simulations performed herein, excellent agreement with experiment was achieved for the simulated hydrogen sorption isotherms and calculated isosteric heat of adsorption, Qst, values only when using a polarizable potential. Thermodynamic agreement is demonstrated via comparing to experimental isotherms and binding sites are revealed by combining simulation and inelastic neutron scattering (INS) data. Simulations involving explicit many-body polarization interactions assisted in the determination of the binding sites in rht-MOF-1 through the distribution of the induced dipoles that led to strong adsorbate interactions. Four distinct hydrogen sorption sites were determined from the polarization distribution: the nitrate ions located in the corners of the truncated tetrahedral cages, the Cu2+ ions of the paddlewheels that project into the truncated tetrahedral and truncated octahedral cages (Cu1 ions), the Cu2+ ions of the Cu3O trimers (Cu3 ions), and the sides of the paddlewheels in the cuboctahedral cage. The simulations revealed that the initial sorption sites for hydrogen in rht-MOF-1 are the nitrate ions; this site corresponds to the high initial Qst value for hydrogen (9.5 kJ mol−1) in the MOF. The radial distribution functions, g(r), about the Cu2+ ions at various loadings revealed that the Cu1 ions are the preferred open-metal sorption sites for hydrogen at low loading, while the Cu3 ions become occupied at higher loadings. The validation of the aforementioned sorption sites in rht-MOF-1 was confirmed by calculating the two-dimensional quantum rotational levels about each site and comparing the levels to the transitions that were observed in the experimental INS spectra for hydrogen in the compound. For each binding site, the rotational transitions from j = 0 to j = 1 were in good agreement to certain transitions that were observed in the INS spectra. From these calculations, the assignment of the peaks in the INS spectra for hydrogen in rht-MOF-1 has been made.
 Please wait while we load your content...
Please wait while we load your content...

