
Flux and reflux: metabolite reflux in plant suspension cells and its implications for isotope-assisted metabolic flux analysis†
Abstract
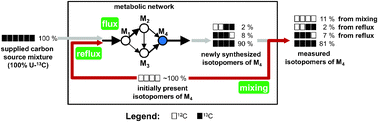
Isotope-assisted metabolic flux analysis (MFA) is a powerful methodology to quantify intracellular fluxes via isotope labeling experiments (ILEs). In batch cultures, which are often convenient, inexpensive or inevitable especially for eukaryotic systems, MFA is complicated by the presence of the initially present biomass. This unlabeled biomass may either mix with the newly synthesized labeled biomass or reflux into the metabolic network, thus masking the true labeling patterns in the newly synthesized biomass. Here, we report a detailed investigation of such metabolite reflux in cell suspensions of the tree poplar. In ILEs supplying 28% or 98% U-13C glucose as the sole organic carbon source, biomass components exhibited lower 13C enrichments than the supplied glucose as well as anomalous isotopomers not explainable by simple mixing of the initial and newly synthesized biomass. These anomalous labeling patterns were most prominent in a 98% U-13C glucose ILE. By comparing the performance of light- and dark-grown cells as well as by analyzing the isotope labeling patterns in aspartic and glutamic acids, we eliminated photosynthetic or anaplerotic fixation of extracellular 12CO2 as explanations for the anomalous labeling patterns. We further investigated four different metabolic models for interpreting the labeling patterns and evaluating fluxes: (i) a carbon source (glucose) dilution model, (ii) an isotopomer correction model with uniform dilution for all amino acids, (iii) an isotopomer correction model with variable dilution for different amino acids, and (iv) a comprehensive metabolite reflux model. Of these, the metabolite reflux model provided a substantially better fit for the observed labeling patterns (sum of squared residues: 538) than the other three models whose sum of squared residues were (i) 4626, (ii) 4983, and (iii) 1748, respectively. We compared fluxes determined using the metabolite reflux model to those determined using an independent methodology involving an excessively long ILE to wash out initial biomass and a minimal reflux model. This comparison showed identical or similar distributions for a majority of fluxes, thus validating our comprehensive reflux model. In summary, we have demonstrated the need for quantifying interactions between initially present biomass and newly synthesized biomass in batch ILEs, especially through the use of ≈100% U-13C carbon sources. Our ILEs reveal a high amount of metabolite reflux in poplar cell suspensions, which is well explained by a comprehensive metabolite reflux model.
 Please wait while we load your content...
Please wait while we load your content...