
Structural and relative stabilities, electronic properties and possible reactive routing of osmium and ruthenium borides from first-principles calculations†
Abstract
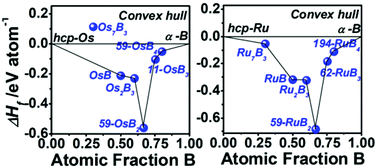
First-principles calculations are employed to provide a fundamental understanding of the structural features and relative stability, mechanical and electronic properties and possible reactive route for osmium and ruthenium borides. The structural searches and calculations of the formation enthalpy identify a low-energy monoclinic phase for OsB3 with P21/m symmetry, an orthorhombic phase for OsB4 with Pmmn symmetry, an orthorhombic phase for RuB3 with Pnma symmetry and a hexagonal phase for RuB4 with P63/mmc symmetry. Also, the structure transition at high pressure is also predicted for MB3 and MB4 (M = Os and Ru). Moreover, among the borides, orthorhombic RuB3 and OsB4 phases are predicted to be potential hard materials with estimated Vickers hardness values of 26.3 and 31.3 GPa, respectively. The analysis on the electronic properties and crystal orbital Hamilton population shows that the directional boron–boron networks, together with the strong metal–boron bonds, are responsible for their excellent mechanical properties. Relative enthalpy calculations with respect to possible constituents are also investigated to assess the prospects for phase formation and an attempt at high-pressure synthesis is suggested to obtain osmium and ruthenium tri- and tetra-borides.
 Please wait while we load your content...
Please wait while we load your content...

