
Electronic excited states and electronic spectra of biphenyl: a study using many-body wavefunction methods and density functional theories
Abstract
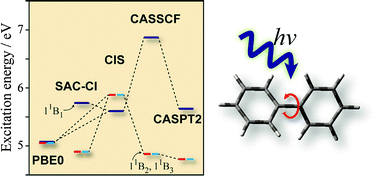
The low-lying electronic excited states of biphenyl were studied using the symmetry-adapted cluster-configuration interaction (SAC-CI), complete active space self-consistent field (CASSCF), complete active space perturbation theory of the second-order (CASPT2), and the time-dependent density functional theory (TDDFT). The molecular geometries in the ground and excited states were optimized using the SAC-CI and TDDFT for singlet and triplet states. The energies of vertical excitations, emissions, and adiabatic transitions were calculated. The TDDFT calculations significantly underestimated the excitation energy of the 11B1 state, while the SAC-CI and CASPT2 provided essentially similar results. The present SAC-CI and CASPT2 calculations concluded that the lowest singlet state of isolated biphenyl is the 11B3 state that takes a planar geometry and the second lowest state is the 11B2 state with a twisted geometry. The present results were consistent with the previous experimental findings. The 11B1 state that has a charge-separated biracial character in the vertical excitation relaxed into a planar quinoid structure in which bond alternations were emphasized. The other states took a benzenoid structure. The ultraviolet (UV) absorption and circular dichroism (CD) spectra below 7 eV were calculated with the SAC-CI method. The valence–Rydberg mixings were found to be significant in the second and higher series of excited states.
 Please wait while we load your content...
Please wait while we load your content...

