
An NMR crystallography DFT-D approach to analyse the role of intermolecular hydrogen bonding and π–π interactions in driving cocrystallisation of indomethacin and nicotinamide†
Abstract
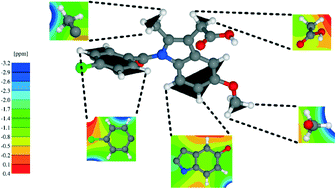
Density functional theory (DFT) calculations using the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional are presented for a 1 : 1 cocrystal formed by indomethacin and nicotinamide (IND-NIC) as well as for crystal structures of the individual components. DFT-D approaches which correct the DFT energy for dispersion effects, specifically the Grimme (G06) and Tkatchenko–Scheffler (TS) schemes, are investigated: for geometry optimisation starting with crystal structures determined experimentally by diffraction and allowing the atomic positions and the unit cell to vary, closest agreement with the experimental unit cell parameters is achieved with the PBE-TS approach (calculated volumes are less than 4% smaller than in experiment). Calculations of solid-state NMR chemical shifts using the GIPAW (gauge including projector augmented wave) approach are presented. Closest agreement between NMR chemical shifts calculated with variable and fixed (experimental) unit cell parameters is also observed for the PBE-TS approach: the root mean squared standard deviation difference is 0.15 ppm (1H) and 0.29 ppm (13C) for PBE-TS, as compared to 0.45 ppm (1H) and 0.68 ppm (13C) with standard PBE. Differences in 1H chemical shifts calculated for the full periodic crystal structure and for isolated molecules extracted from the geometry-optimised crystal structure are presented in conjunction with NICS (nucleus independent chemical shift) maps, so as to separately quantify intermolecular hydrogen bonding and π–π interactions. This analysis is complemented by total energy calculations, including also at the B97D/6-311+G* level of theory with basis set superposition error correction, in order to understand the interactions that drive cocrystallisation.
- This article is part of the themed collection: NMR crystallography
 Please wait while we load your content...
Please wait while we load your content...

