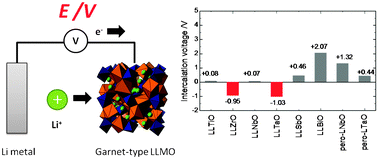
The research and development of rechargeable all-ceramic lithium batteries are vital to realize their considerable advantages over existing commercial lithium ion batteries in terms of size, energy density, and safety. A key part of such effort is the development of solid-state electrolyte materials with high Li+ conductivity and good electrochemical stability; lithium-containing oxides with a garnet-type structure are known to satisfy the requirements to achieve both features. Using first-principles density functional theory (DFT), we investigated the electrochemical stability of garnet-type LixLa3M2O12 (M = Ti, Zr, Nb, Ta, Sb, Bi; x = 5 or 7) materials against Li metal. We found that the electrochemical stability of such materials depends on their composition and structure. The electrochemical stability against Li metal was improved when a cation M was chosen with a low effective nuclear charge, that is, with a high screening constant for an unoccupied orbital. In fact, both our computational and experimental results show that Li7La3Zr2O12 and Li5La3Ta2O12 are inert to Li metal. In addition, the linkage of MO6 octahedra in the crystal structure affects the electrochemical stability. For example, perovskite-type La1/3TaO3 was found, both experimentally and computationally, to react with Li metal owing to the corner-sharing MO6 octahedral network of La1/3TaO3, even though it has the same constituent elements as garnet-type Li5La3Ta2O12 (which is inert to Li metal and features isolated TaO6 octahedra).

 Please wait while we load your content...
Please wait while we load your content...

