
Multiscale molecular dynamics simulations of micelles: coarse-grain for self-assembly and atomic resolution for finer details†
Abstract
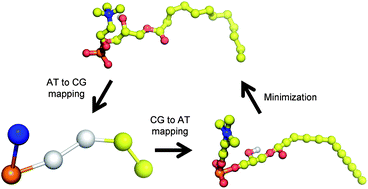
A positional interpolation/extrapolation method for the mapping of coarse-grained (CG) to atomistic (AT) resolution is presented and tested for single-component micelles formed by lysophospholipids of different chain length. The target CG nanoaggregates were self-assembled from random mixtures of surfactants in explicit MARTINI water and equilibrated by molecular dynamics simulations, at the microsecond time scale. The ambiguity inherent in the definition of the CG particles was explored by mapping the same CG structures to AT resolution surfactants of different size. After the conversion, the obtained AT micelles were simulated for 250 ns and the resulting trajectories were analyzed in detail. The mean lifetime of the surfactant–solvent interactions as well as the lateral diffusion coefficients of the surfactant molecules within the micellar aggregates were obtained for the first time. The results suggest that the individual molecules within the micelle behave like lipids in a fluid membrane. The employed mapping back method is efficient and versatile, as it can be applied to diverse combinations of force fields and systems with a minimum of code development. In a general context, this work illustrates the power of multiscale molecular dynamics simulations for the generation and subsequent examination of self-assembled structures, including the fine characterization of structural and dynamic properties of the resulting aggregate.
 Please wait while we load your content...
Please wait while we load your content...

