
Solvation of copper ions by imidazole: Structures and sequential binding energies of Cu+(imidazole)x, x = 1–4. Competition between ion solvation and hydrogen bonding†‡
Abstract
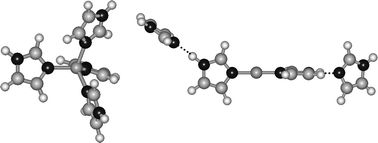
The sequential bond dissociation energies of Cu+(imidazole)x, where x = 1–4 are determined by analysis of the kinetic energy dependence of the collision-induced dissociation with Xe in a guided ion beam tandem mass spectrometer. In all cases, the primary and lowest energy dissociation channel observed is the endothermic loss of an intact imidazole molecule. The primary cross section thresholds are interpreted to yield 0 K and 298 K bond dissociation energies after accounting for the effects of multiple ion–neutral collisions, kinetic and internal energy distributions of the reactants, and dissociation lifetimes. To obtain model structures, vibrational frequencies, rotational constants and energetics for the Cu+(imidazole)x complexes and their dissociation products, density functional theory calculations at the B3LYP/6-31G* level are performed. Theoretical bond dissociation energies are determined from single point energy calculations at the B3LYP/6-311+G(2d,2p) level of theory using the B3LYP/6-31G* optimized geometries. Excellent agreement between theory and experiment is observed for the Cu+(imidazole)x complexes, where x = 1, 2 and 4. In contrast, theory systematically underestimates the strength of the binding in the Cu+(imidazole)3 complex. The ground state structures of the Cu+(imidazole)x complexes and the trends in the sequential bond dissociation energies are explained in terms of stabilization gained from sd hybridization and hydrogen bonding interactions and destabilization arising from ligand–ligand repulsion. The trends in the binding of these complexes are also examined to provide insight into the structural and functional roles that histidine and other ligands play in the behavior of metalloproteins and metalloenzymes.
 Please wait while we load your content...
Please wait while we load your content...

