
Experimental and theoretical investigation of the A 3Π–X 3Σ− transition of NH/D–Ne
Abstract
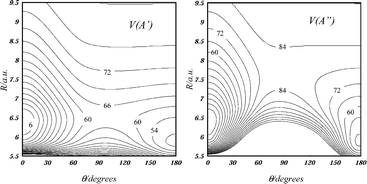
A study of NH/D–Ne was undertaken to investigate the structure of this complex and examine the ability of high-level theoretical methods to predict its properties. The A 3Π–X 3Σ− transition was characterized using laser induced fluorescence measurements. Results from theoretical calculations were used to guide the interpretation of the spectra. Two-dimensional potential energy surfaces were calculated using second-order multireference perturbation theory with large correlation consistent basis sets. The potential energy surfaces were used to predict the ro-vibronic structure of the A–X system. Calculated ro-vibronic energy level patterns could be recognized in the spectra but quantitative discrepancies were found. These discrepancies are attributed to incomplete recovery of the dynamical correlation energy.
 Please wait while we load your content...
Please wait while we load your content...

