
The critical role of Ru-like hydrogen adsorption of carbon dots in a carbon-confined Ru system for boosted HER activity†
Zonglin
Liu
 a,
Honglei
Zhang
a,
Dongyue
Liu
a,
Yujie
Feng
a,
Dechang
Jia
a,
Caicai
Li
*b,
Qingfeng
Sun
b,
Yu
Zhou
a,
Zhenhui
Kang
*d and
Baoqiang
Li
*ace
a,
Honglei
Zhang
a,
Dongyue
Liu
a,
Yujie
Feng
a,
Dechang
Jia
a,
Caicai
Li
*b,
Qingfeng
Sun
b,
Yu
Zhou
a,
Zhenhui
Kang
*d and
Baoqiang
Li
*ace
aInstitute for Advanced Ceramics, State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin 150001, China. E-mail: libq@hit.edu.cn
bSchool of Engineering, Zhejiang A & F University, Hangzhou, 311300, China
cLaboratory of Dynamics and Extreme Characteristics of Promising Nanostructured Materials, Saint Petersburg State University, St. Petersburg, 199034, Russia. E-mail: ccli@zafu.edu.cn
dInstitute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Soochow University, Suzhou, 215123, China. E-mail: zhkang@suda.edu.cn
eMIIT Key Laboratory of Advanced Structural-Functional Integration Materials & Green Manufacturing Technology, Harbin Institute of Technology, Harbin 150001, China
First published on 5th January 2024
Abstract
The in-depth understanding of electrocatalysts is the key issue in the fields of clean energy and the chemical industry. Although various carbon-loaded active metal particle catalysts for the electrocatalytic hydrogen evolution reaction (HER) have been developed, the interaction between active metals and the substrate is still unclear. Herein, ruthenium (Ru) nanoparticles loaded on N-doped carbon dots (Ru/NCDs) were constructed to discover the critical roles of the carbon substrate as well as its interaction with Ru. Notably, we show the two critical functions of NCDs in a carbon-confined Ru system. First, the ultra-small Ru nanoparticles increase the hydrogen spillover channels. Second, the carbon substrate shows a Ru-like hydrogen adsorption capacity, supplying sufficient H sources for hydrogen spillover. Thus, the hydrogen spillover is enhanced and the HER process is accelerated. The Ru/NCDs catalyst shows superior HER activity and durability in both acidic and alkaline solutions. This work clarifies the critical role of carbon substrates in carbon-loaded catalysts.
Introduction
The interaction between the active metal component and the substrate has been continuously investigated over the past several decades, and the topic has gained considerable attention in heterogeneous catalysis.1–6 In the field of electrocatalysis, the emphasis is placed on the development and improvement of active centers for supported electrocatalysts, and the interaction relationship between active metals and substrates lacks attention.7,8 For instance, typical commercial Pt/C is currently recognized as the most effective and stable catalyst for the hydrogen evolution reaction (HER), and loading platinum onto carbon with small particles maximizes its activity.9–11 An in-depth understanding of electrocatalysts is the key issue in the fields of clean energy, the chemical industry, and the environment. At present, the interaction between carbon substrates and active components is still far from being clearly elucidated.Ruthenium (Ru) has been considered an alternative successor to platinum (Pt) for the HER because of its promising hydrogen evolution activity and much lower price.12–16 However, the original activity of Ru is unsatisfactory due to its excessively strong hydrogen adsorption.17–23 Loading Ru nanoparticles onto carbon substrates to form a hybrid structure is a valid tactic to enhance the activity and stability of Ru.24–28 Furthermore, with the Ru/C catalyst as a model, it appears that investigating the interaction between ruthenium and carbon is an ideal path to disclose the specific role of carbon in loaded catalysts.
The electrocatalyst of Ru/NCDs was designed and constructed here for trapping Ru nanoparticles on N-doped carbon nanosheets created by stitching N-doped carbon dots. The theoretical prediction was first performed and indicated that hydrogen spillover is closely related to the interfaces between Ru and the substrate. Additionally, the carbon gaps formed during the formation of two-dimensional nanosheets from NCDs show Ru-like adsorption properties. This provides a continuous supply of adsorbed hydrogen for the hydrogen spillover. As expected, the electrochemical tests manifest that Ru/NCDs is an efficient HER electrocatalyst not only in acidic but also in alkaline solutions, which require 15 and 31 mV to achieve 10 mA cm−2, respectively. In addition, Ru/NCDs also exhibited excellent cycling and long-term stability in both media. This work clearly elucidates the critical role of the carbon substrate in Ru/NCDs; it is not only capable of stabilizing nanoparticles, but also shows Ru-like hydrogen adsorption in this carbon-confined Ru system for boosted HER activity.
Results and discussion
Theoretical understanding of hydrogen spillover from Ru to carbon substrates
For visualization, density functional theory (DFT) calculations were employed to explore the hydrogen spillover between Ru and the carbon substrate. HER activity is directly related to the Gibbs free energy of hydrogen adsorption on the catalyst surface (ΔGH*).29 A negative ΔGH* value generally signifies enhanced hydrogen adsorption, whereas a positive ΔGH* value indicates favorable hydrogen desorption, ideally with a ΔGH* value approaching zero for effective HER catalysts.28Fig. 1a depicts the ΔGH* of the top Ru (site-a), Ru–carbon substrate interface (site-b), and carbon substrate (site-c) of Ru/NCDs. The energy change of protons and electrons in the hydrogen evolution process is determined to be zero by fixing the reference potential to the standard hydrogen electrode. The ΔGH* at site-a is −0.29 eV, showing a strongly exothermic process for H* adsorption, which indicates that the adsorption of H* occurs readily at this site but goes against the removal of H*.30 Similarly, ΔGH* values for adjacent sites were estimated, with calculated values for site-b and site-c of −0.05 eV and 0.43 eV, respectively. Notably, site-c exhibits a significantly positive ΔGH* which indicates that the generated H2 is more favorably desorbed at this site. What's more, site-b associated with the Ru–carbon substrate interface has an ideal ΔGH* close to 0 eV, serving as an excellent intermediate state that minimizes the free energy difference between site-a and site-c, thereby facilitating the smooth occurrence of the hydrogen spillover reaction. Specifically, site-a stabilizes the free protons initially, which then progressively travel to site-c through the lower adsorption energy of site-b and ultimately undergo desorption at site-c. | ||
| Fig. 1 DFT calculation of hydrogen spillover channels for Ru/NCDs. (a) Plot of calculated free energy of the proton transfer pathway on Ru/NCDs (inset: optimized H* adsorption structures on different sites). (b) Two-dimensional charge density difference plots and integrated charge curves in the z-axis direction of Ru/NCDs. (c) Schematic diagram of d-band center theory. The calculated –COHP of (d) Ru(I)–H at the top and (e) Ru(II)–H at the interface. | ||
In addition, the hydrogen spillover in Ru/C can be further explained by the electron density difference (EDD) mapping (Fig. S1†). To provide a more intuitive analysis of the electron gain and loss within the model, two-dimensional charge density difference plots and integrated charge curves in the z-axis direction are provided (Fig. 1b). Typically, a significant charge depletion at the interface between Ru and the carbon substrate appears. Meanwhile, the obvious charge loss peak near the interface in the integral charge curve can also be observed. Furthermore, the Bader charge analysis also indicates that the Ru atoms in the near-neighboring carbon layer have a relatively strong electron loss feature (0.24 electrons lost on average), while the next-neighboring layer shows a weak electron gain feature (0.07 electrons gained on average) (Table S1†). All the above results indicate that significant electron depletion occurs at the Ru–carbon substrate interface. In general, unsaturated electrons in the 1s orbitals of active hydrogen species can readily be captured by the electron-rich area.31 Due to the electron loss, the absorption of active hydrogen species at the interface is diminished. Consequently, the interface creates a pathway for the diffusion of H* between Ru and carbon due to the weak adsorption of H*, thereby facilitating the hydrogen spillover process.32
To further elucidate the generation of the hydrogen spillover channel, the density of states (DOS) was determined in order to thoroughly examine the electronic structure alterations of the catalyst in depth. According to the d-band center theory, the adsorption of active hydrogen on Ru is primarily driven by the interaction between the d-band of Ru and the valence band of H, resulting in the formation of bonding and anti-bonding states.33 As revealed in Fig. 1c, compared with the original electronic state, the energy of the bonding state moved down while the energy of the anti-bonding state increased. The bond strength is enhanced when the electrons are filled into the bonding state, while filling electrons in the anti-bonding state leads to a weaker bond. Generally, the closer the d-band center is to the EF, the smaller the proportion of the antibonding state below the EF, resulting in stronger adsorption. Fig. S2† shows the DOS of Ru(I) at the top and Ru(II) at the interface, with the center of the d-band clearly marked. The d-band center of Ru(I) is obviously closer to the EF than that of Ru(II), implying that the former is expected to exhibit stronger hydrogen adsorption. Furthermore, the position of the antibonding orbital relative to the EF can be visually displayed by the crystal orbital Hamilton population (COHP). Fig. 1d depicts the –COHP curve representing the interaction between Ru(I) and adsorbed H*, where the positive part denotes the bonding state and the negative part denotes the anti-bonding state. It can be observed that the anti-bonding component is predominantly situated above the EF. In contrast, a portion of the anti-bonding state formed by Ru(II) and H* is located below the EF (Fig. 1e). As a result of the more filled anti-bonding states, the interaction between Ru(II) and H* is less than that between Ru(I) and H*. Moreover, the bond strength between Ru and H* can be directly reflected by the integrated COHP (ICOHP) curves (Fig. S3†), in which a higher –ICOHP at the EF corresponds to stronger bonding. Because of the smaller –ICOHP at the EF, the bond between Ru(II) and H* is obviously weaker. This situation firmly verifies the prerequisites for hydrogen spillover between Ru and the carbon substrate. Based on the above findings, a mechanism for the hydrogen spillover-assisted HER of Ru/NCDs was proposed, with detailed steps outlined in ESI Note 1.† Specifically, a favorable pathway for hydrogen spillover is generated from Ru to the carbon substrate, with the interface between Ru and the carbon substrate identified as the active site for the hydrogen spillover process.
Confinement effect of NCDs and Ru-like hydrogen adsorption capacity of the carbon substrates
Fig. S4† presents the detailed fabrication process of Ru/NCDs. Specifically, N-doped carbon dots (NCDs) derived from chitosan and melamine will incorporate Ru ions (Ru3+) to form Ru3+–NCDs in the hydrothermal process and eventually complete the zero-dimensional to two-dimensional conversion after pyrolysis. Then, the characteristics of the original NCDs were first investigated. As manifested in Fig. S5a and b,† the transmission electron microscopy (TEM) images as well as the size distribution histogram demonstrate uniformly dispersed NCDs particles with a particle size of about 2.2 nm. In addition, Fig. S5c† shows the high-resolution TEM (HRTEM) images, which reveal a distinct lattice spacing of 0.21 nm for the graphite (002) plane, which aligns with the previously reported characteristics of CDs.34 The characteristics of the NCDs were further characterized via UV-vis absorption spectroscopy and photoluminescence measurements (Fig. S6†). The foregoing results provide evidence of the successful fabrication of NCDs. Furthermore, to disclose the surface composition of NCDs, X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared spectroscopy (FTIR) were also performed. The XPS survey spectrum reveals that NCDs primarily contain C, N, and O elements (Fig. S7a†). The high-resolution XPS spectra illustrate that the surface of NCDs is rich in functional groups, enabling them to effectively bind to metal ions (Fig. S7b–d†).With the above NCDs as sources of carbon substrates, Ru/NCDs were successfully fabricated, incorporating Ru nanoparticles. Fig. 2a displays the X-ray diffraction (XRD) pattern of the Ru/NCDs catalyst. Obviously, the peak at about 26° is attributed to graphene, and several lower diffraction peaks originate from Ru (PDF#89-3942). XPS was also used to determine the chemical states and electronic structures of Ru/NCDs. As illustrated in Fig. 2b, the survey XPS spectrum of Ru/NCDs revealed peaks corresponding to Ru, C, N, and O elements. The Ru/NCDs peaks of Ru0 3p3/2 and Ru0 3p1/2 are presented at binding energies of 484.5 and 462.2 eV, respectively, while the peaks with binding energies of 488.2 and 465.3 eV, respectively, attribute to the 3p3/2 and 3p1/2 of Run+ (Fig. S8†).35 The O 1s, C 1s, and N 1s spectra prove that Ru/NCDs are rich in surface functional groups and that the Ru/NCDs were formed by the combination of Ru nanoparticles and NCDs (Fig. S9–S11†). The FTIR spectrum of Ru/NCDs reveals the inheritance of oxygen-/nitrogen-containing functional groups inherited from NCDs by Ru/NCDs (Fig. 2c). The functional groups facilitate efficient water molecule transportation via robust hydrogen bonding, allowing for rapid proton transport.36
 | ||
| Fig. 2 Ruthenium-like adsorption properties dominated by carbon substrate morphology. (a) XRD pattern of Ru/NCDs. (b) XPS survey scan of Ru/NCDs. (c) FTIR spectrum of Ru/NCDs. (d) TEM image of Ru/NCDs. (e–g) HAADF-STEM images of Ru/NCDs. (h) Calculated free energy diagram for carbon with gaps and Ru/NCDs. | ||
TEM was applied to characterize the microscopic morphology of Ru/NCDs. Among the TEM images, Ru/NCDs have a thin nanosheet morphology that is densely packed with ultra-small nanoparticles (Fig. S12a–c†). Notably, as shown in Fig. 2d, numerous discontinuous or multi-directional interlocking carbon lattices (labeled by arrows and circles) can be observed in the carbon substrate. These discontinuous carbon lattices suggest that there are numerous gaps on the carbon substrate surface. To further validate this result, in-depth characterization was carried out using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). As demonstrated in Fig. 2e and f, although the carbon substrate shows a lamellar structure, there are actually abundant gaps. In addition, the carbon substrate is loaded with numerous ultra-small Ru nanoparticles of approximately 2 nm (Fig. 2g). Furthermore, the uniform dispersion of nanoparticles is observable at various levels of magnification. The high-resolution TEM (HRTEM) result indicates that the Ru nanoparticles on the nanosheets possess an interplanar spacing of 0.158 nm, belonging to the lattice plane (102) of Ru (Fig. S12d†). Additionally, the selected area electron diffraction (SAED) pattern depicted in Fig. S13† illustrates a diffraction ring belonging to the (102) plane of Ru, which echoes the lattice in the HRTEM. The corresponding elemental mapping analysis indicates the coexistence of C, N, O, and Ru elements in Ru/NCDs, in agreement with the XPS results (Fig. S14†). To further verify the role of the gap in the carbon substrate, we examined the hydrogen adsorption energy of the carbon matrix model with a carbon gap using DFT calculations. Hydrogen atoms in the carbon matrix with gaps have an average adsorption energy of −0.3 eV, which is close to that of the Ru site of Ru/NCDs (−0.29 eV) for hydrogen atoms (Fig. 2h and S15†). This indicates that the carbon gap exhibits a hydrogen adsorption capacity comparable to that of Ru, compensating for the diminished hydrogen adsorption capacity at low Ru content and supplying a continuous flow of adsorbed hydrogen, ultimately allowing for smooth hydrogen spillover.
For comparison, Ru/NC catalysts were fabricated by mixing precursors of NCDs with Ru ions (see ESI Materials and methods for details†). The chemical components and surface states of Ru/NC were further examined using XRD and XPS, as detailed in the ESI (Fig. S16–S21†). In contrast to Ru/NCDs, the irregular distribution of Ru nanoparticles in Ru/NC with obvious agglomeration can be observed at different magnifications (Fig. S22a–c†). As shown in Fig. S22d,† the lattice fringe of 0.158 nm corresponds to the (102) plane of Ru. The SAED pattern corresponds well to this result and reveals a clear diffraction ring corresponding to the Ru (102) plane (Fig. S23†). The measurement of Ru nanoparticle size in Ru/NC yielded an average particle size of about 8 nm (Fig. S24†). These results demonstrate that the existence of NCDs effectively limits the growth of Ru nanoparticles, resulting in the formation of ultra-small Ru nanoparticles. The above results indicate that under the same conditions, the existence of NCDs exerts a significant confinement effect on Ru, which can effectively reduce the size of Ru nanoparticles and thus increase the interfacial number between Ru and carbon substrates.
The confinement mechanism of NCDs for Ru/NCD formation
For a comprehensive understanding of the formation process of Ru/NCDs, the chemical structures of NCDs and precursors of Ru/NCDs (i.e. Ru3+–NCDs) were characterized through thermogravimetric analysis (TGA) and derivative thermogravimetric analysis (DTG) (Fig. 3a and b). The degradation processes of NCDs and Ru3+–NCDs exhibit similarities, as both involve multi-step processes (blue line). The distribution of the DTG curve (red line) indicated the weight loss rate, while the peak in the DTG curve served to evaluate the thermal degradation. The initial weight decrease, occurring around 90 °C, is attributed to the loss of bound water. Dehydration and condensation of functional groups on the surface of NCDs and Ru3+–NCDs cause secondary weight loss at around 300 °C.37 Interestingly, more functional groups were found to be present on the surface of NCDs relative to Ru3+–NCDs. This result indicated that the functional groups on the NCDs surface can trap Ru3+. The degradation from 390 °C to 600 °C can be attributed to the decomposition of oxygen-containing functional groups such as hydroxyl groups on NCDs and Ru3+–NCDs surfaces as well as the pyrolyzation of the internal cross-linked carbon network.37,38 The phenomenon can be further supported by the in situ Fourier transform infrared (FTIR) spectrum in Fig. 3c. In situ FTIR spectra can monitor in real-time the change of functional groups on catalyst surfaces with increasing temperature, making it essential for studying the formation mechanism of the catalyst. As can be observed, the functional groups of amino, carboxyl, and hydroxyl groups on the NCDs surface gradually decrease with an increase in temperature,39 in agreement with the results of DTG. Similar findings are evident in the in situ FTIR results of Ru3+–NCDs (Fig. 3d). In conjunction with these results, the formation process of Ru/NCDs is derived from the combination of Ru3+ with functional groups, including amino and carboxyl groups and the dehydration condensation between functional groups during pyrolysis. Thus, a possible confinement mechanism for NCDs in Ru was proposed. Under an inert atmosphere, the formed Ru3+–NCDs gradually became active as the temperature increased, leading to the formation of two-dimensional carbon nanosheets through the dehydration and condensation of surface functional groups. Finally, after pyrolysis, Ru nanoparticles were progressively formed and confined in the cage formed by functional groups, thereby inhibiting Ru nanoparticle development (Fig. 3e). This NCDs cage effect restricts the size of the Ru nanoparticles while also providing some protection for the Ru. Moreover, this confinement effect efficiently increases the interfaces between Ru and carbon substrates. As the active site for hydrogen spillover is the interface between Ru and the carbon substrate, increasing the interfaces can effectively enhance the hydrogen spillover and thereby promote HER activity.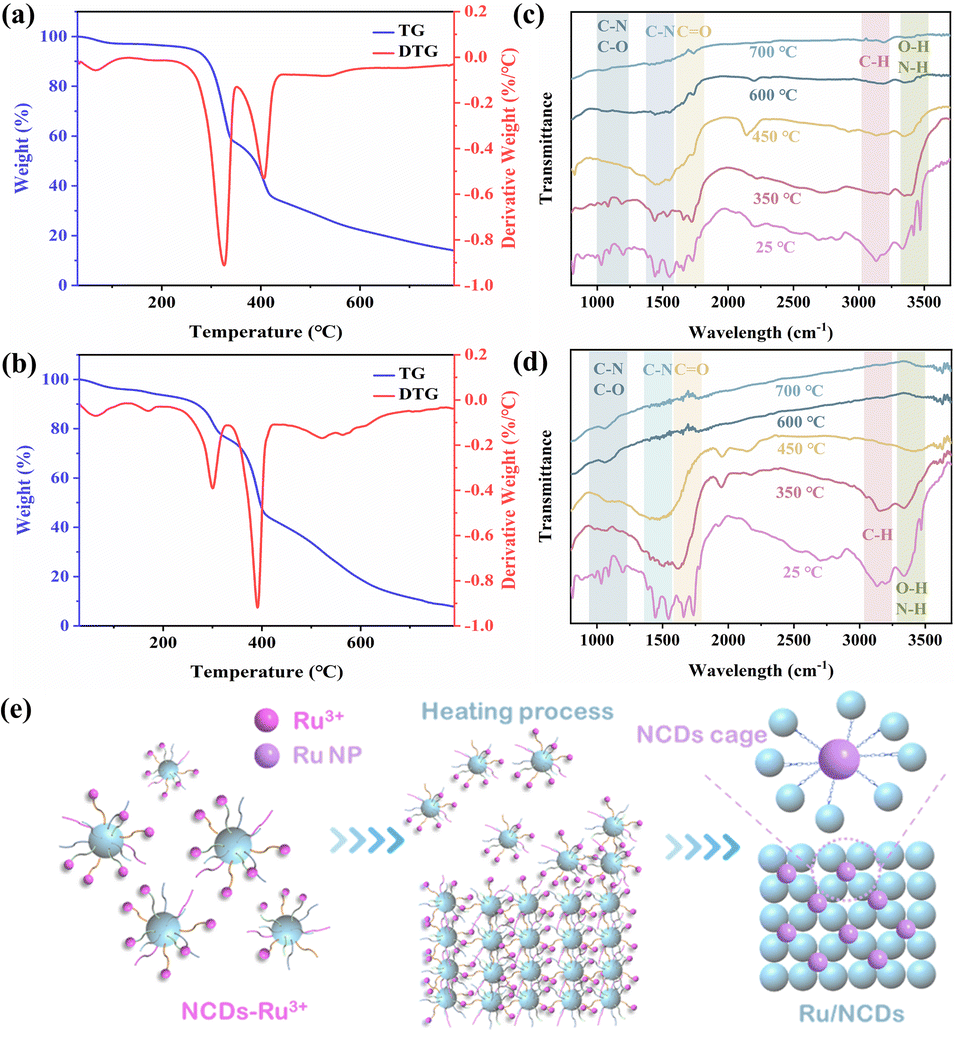 | ||
| Fig. 3 The confinement mechanism of NCDs for Ru/NCDs formation. TGA and DTG thermograms of (a) NCDs and (b) Ru/NCDs. In situ FT-IR spectra of (c) NCDs and (d) Ru/NCDs. (e) Schematic diagram of the “NCDs cages”. | ||
Hydrogen evolution reaction properties
Accordingly, the electrocatalytic activity of Ru/NCDs, as well as their control samples (Ru NPs, Ru/NC, NCDs, NC, and commercial 20% Pt/C) in acidic and alkaline electrolytes was studied in a typical three-electrode system. Therein, the NCDs were heat-treated under the same conditions as those in the preparation of the Ru/NCDs, and later references are to the heat-treated NCDs unless otherwise indicated. The HER properties of Ru/NCDs in 0.5 M H2SO4 were first measured by linear scanning voltammetry (LSV). As illustrated in Fig. 4a, the overpotential of Ru/NCDs required for providing a reference current density of 10 mA cm−2 is only 15 mV, which is lower compared to 21 mV for 20% Pt/C, 29 mV for Ru/NC, 222 mV for Ru NPs, 462 mV for NCDs, and 641 mV for NC, respectively. The Tafel slope was applied to study the HER kinetics of the catalyst. Ru/NCDs display a lower Tafel slope of 25 mV dec−1 than 20% Pt/C (30 mV dec−1) (Fig. 4b), indicating a unique HER mechanism distinct from the traditional Volmer–Tafel process, in which the kinetics of hydrogen adsorption and desorption appear to be no longer constrained.40–44 This mechanism can be attributed to the efficient hydrogen spillover effect at the Ru/NCDs surface. As illustrated in Fig. 4c, the lower Tafel slope and smaller overpotential of Ru/NCDs compared to other contrast catalysts highlight the exceptional activity of Ru/NCDs against HER in an acidic electrolyte. Meanwhile, in contrast to the previously reported catalysts, Ru/NCDs still displayed superior HER activities in an acidic medium (Fig. 4g and Table S2†). The electrochemical impedance spectroscopy (EIS) test was also performed to study the kinetics of hydrogen adsorption on the catalysts. The Nyquist plots (inset of Fig. 4d) are generated using a double parallel equivalent circuit model.41,45 Specifically, the first parallel component (T and R1) represents charge transfer kinetics on the catalyst surface, whereas the second parallel component (Cφ and R2) represents hydrogen adsorption behavior on the catalyst surface.29,45 As displayed in Fig. 4d and Table S3,† Ru/NCDs exhibit the lowest R1 and R2 values, indicating rapid charge and proton transfer rates and increased local H* concentrations on their surfaces. The above results are associated with the enhanced hydrogen spillover and HER activity of Ru/NCDs. Moreover, durability was another key parameter to appraise the electrocatalytic performance. The durability of Ru/NCDs was assessed using cyclic voltammetric (CV) and time-dependent chronopotentiometric tests in 0.5 M H2SO4 solution. As demonstrated in Fig. 4e, the polarization curve of Ru/NCDs after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles is basically the same as the initial test, showing excellent durability. The chronoamperometric test at a constant overpotential further discloses that Ru/NCDs are highly durable, with ignorable current density decay after 20 h (Fig. 4f). All the above results indicate the superior HER activity and durability of Ru/NCDs under acidic conditions.
000 cycles is basically the same as the initial test, showing excellent durability. The chronoamperometric test at a constant overpotential further discloses that Ru/NCDs are highly durable, with ignorable current density decay after 20 h (Fig. 4f). All the above results indicate the superior HER activity and durability of Ru/NCDs under acidic conditions.
 | ||
| Fig. 4 Hydrogen evolution reaction properties in acidic media. (a) HER polarization curves and (b) Tafel slopes of Ru/NCDs, 20% Pt/C, Ru NPs and NCDs in 0.5 M H2SO4. (c) Overpotentials at 10 mA cm−2 (left) and Tafel slopes (right) of Ru/NCDs, 20% Pt/C, Ru NPs and Ru NC in 0.5 M H2SO4. (d) Nyquist plots of Ru/NCDs, Ru NC, Ru NP NCDs and NC in 0.5 M H2SO4 solutions. (e) LSV curves of Ru/NCDs in 0.5 M H2SO4 before and after 10000 cycles. (f) The stability of current–time (I vs. t) for the HER in 0.5 M H2SO4 solution. (g) Comparison of the overpotentials at 10 mA cm−2 and Tafel slopes of the Ru/NCDs with newly reported Ru-based catalysts in acidic electrolytes. | ||
Simultaneously, Ru/NCDs showed superior HER activities in a 1 M KOH solution. Based on the LSV polarization curves, Ru/NCDs only need 31 mV to obtain a current density of 10 mA cm−2, in excess of other electrocatalysts for HER 1 M KOH solution (Fig. 5a). The HER kinetics of Ru/NCDs in alkaline media was also studied using the Tafel slope. As demonstrated in Fig. 5b, the Tafel slope of Ru/NCDs is merely 22 mV dec−1, suggesting a mechanism for hydrogen spillover.42–44Fig. 5c provides a visual representation indicating that Ru/NCDs demonstrate superior alkaline HER activity, as evidenced by their lower overpotential and Tafel slope compared to commercial Pt/C. This suggests that Ru/NCDs exhibit the most favorable performance in alkaline HER, surpassing that of Pt/C. In addition, EIS measurements were conducted to further illustrate the enhanced hydrogen spillover from Ru/NCDs. As presented in Fig. 5d and Table S4,† Ru/NCDs have the minimum R1 value, indicating their best charge-transfer kinetics. Meanwhile, the R2 values of Ru/NCDs are lower than those of Ru/NC and other comparison samples, which indicated that the adsorption capacity of Ru/NCDs for hydrogen was improved, in agreement with the improvement of hydrogen spillover by Ru/NCDs. Furthermore, the electrochemical stability of Ru/NCDs was tested using an accelerated degradation method of CV and a time-dependent chronopotentiometric test to determine its capability in practical applications. The LSV curves of Ru/NCDs indicate that the overpotential remains without a significant change even after 5000 cycles (Fig. 5e). Additionally, the time-dependent chronopotentiometric test of Ru/NCDs reveals that the overpotential remains stable at a continuous current density of 10 mA cm−2 over a 12-h test, which demonstrates excellent durability (Fig. 5f). Fig. 5g and Table S5† conclude the overpotentials and Tafel slopes of the most advanced Ru-based catalysts in alkaline media, indicating that the catalytic activity of Ru/NCDs is also preferable to most of the investigated Ru-based catalysts.
 | ||
| Fig. 5 Hydrogen evolution reaction properties in alkaline media. (a) Polarization curves and (b) the corresponding Tafel plots of Ru/NCDs and the contrast samples in 1.0 M KOH. (c) Overpotentials at 10 mA cm−2 (left) and Tafel slopes (right) of Ru/NCDs, 20% Pt/C, Ru NPs and Ru NC in 1.0 M KOH. (d) Nyquist plots of Ru/NCDs and the contrast samples in 1.0 M KOH. (e) The initial and 5000th polarization curves obtained from Ru/NCDs in 1.0 M KOH. (f) Time-dependent current density curve for Ru/NCDs in 1.0 M KOH. (g) Comparison of the overpotentials at 10 mA cm−2 and Tafel slopes of the Ru/NCDs with those of recently reported Ru-based catalysts in alkaline electrolytes. | ||
Analysis of the activity centers
The superior electrocatalytic performance of Ru/NCDs demonstrated that the confinement strategy using NCDs is an effective method for producing highly active and robust Ru electrocatalysts. To determine the active centers of Ru/NCDs and Ru/NC, thiocyanate ions (SCN−) were added to the electrolyte, which are toxic to the active metal center due to their strong binding capacity to Ru atoms.46 With the addition of SCN−, both Ru/NCDs and Ru/NC exhibit a sharp decrease in current due to the poisoning of Ru, demonstrating that Ru is an actual active center (Fig. S25†). This also implies that the size of the Ru nanoparticles has a strong influence on the enhancement of activity. The ultra-small Ru nanoparticles expose more Ru–C interfaces, effectively enhancing the hydrogen spillover effect and thus improving the HER activity. In addition, reducing the size of nanoparticles can also reveal more active centers, which are linearly related to the electrochemical double-layer capacitance (Cdl) measured by CV. Therefore, the electrochemically active surface areas (ECSA) of Ru/NCDs, Ru/NC, and NCDs were estimated in both alkaline and acidic media (Fig. S26–S28†). As shown in Fig. 6a, the Cdl of Ru/NCDs in alkaline media is 225.7 mF cm−2, a notably higher value compared to that of Ru/NC (106.6 mF cm−2) and NCDs (6.49 mF cm−2). This observation indicates a substantial augmentation in the quantity of active sites when the ultra-small Ru nanoparticles are uniformly dispersed, consequently leading to improved performance. Similarly, as observed in Fig. 6b, Ru/NCDs (210.5 mF cm−2) have the largest Cdl value in acidic media, which is almost 6 times higher than that of Ru/NC (34.9 cm−2), indicating more active sites on Ru/NCDs than Ru/NC. This result confirms that the ultra-small Ru nanoparticles have more active Ru–C interfaces and more reactive sites.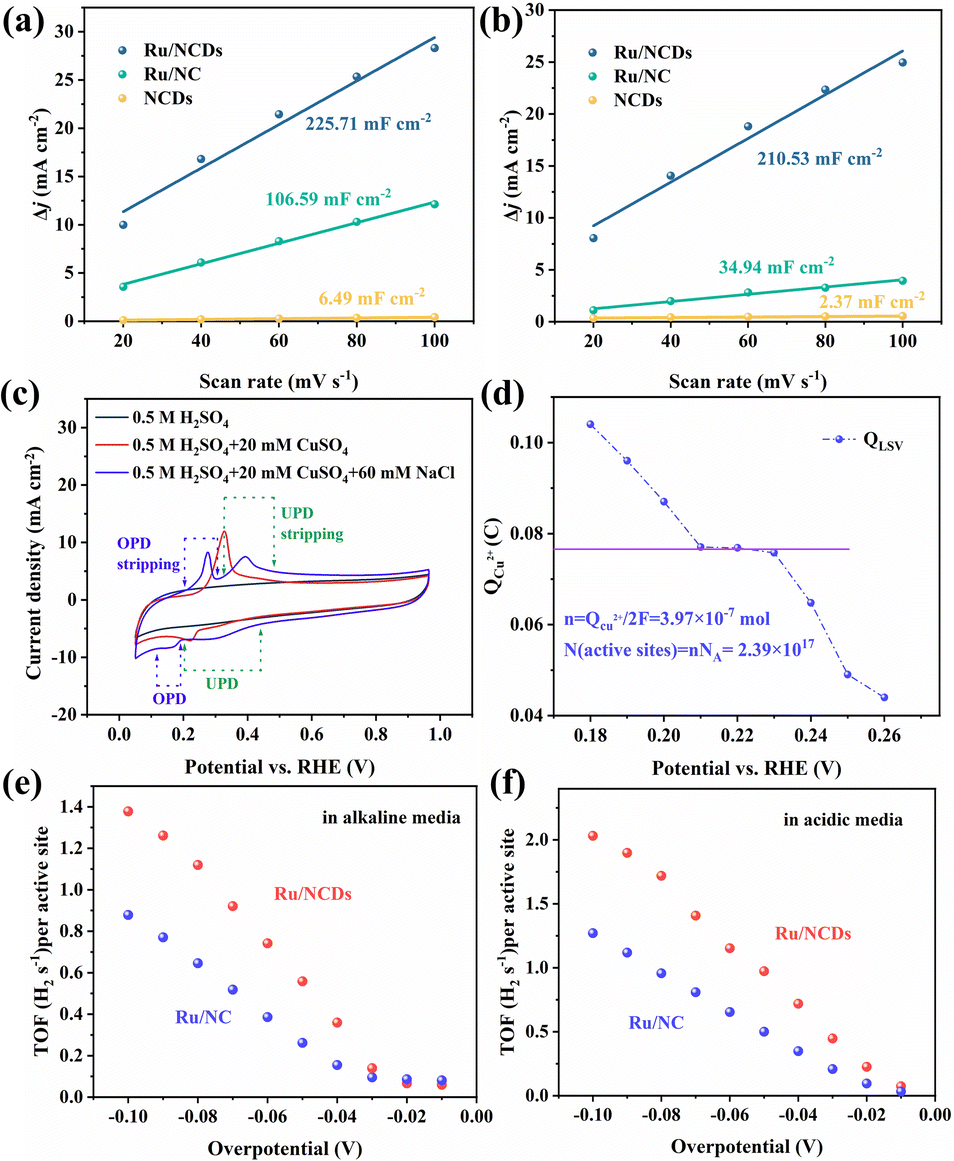 | ||
| Fig. 6 Analysis of the activity centers. Linear fitting curves of the capacitive currents versus different scan rates for Ru/NCDs, Ru/NC, and NCDs in (a) 1.0 M KOH and in (b) 0.5 M H2SO4. (c) Current–voltage scans of Ru/NCDs in different solutions. (d) The charge needed to strip the deposited Cu at various underpotentials in Ru/NCDs. TOF values of Ru/NCDs and Ru/NC at different potentials in (e) 1.0 M KOH and (f) 0.5 M H2SO4. | ||
Furthermore, the number of active centers in Ru/NCDs and Ru/NC was quantitatively investigated using the copper (Cu) underpotential deposition (UPD) method, which works on the mechanism that the active centers in charge of proton reduction also correspond to the reduction of Cu2+ at underpotential.35,47,48 Therefore, the number of active centers is calculated from the charge created during the oxidative stripping of Cu. The CV scans of Ru/NCDs in different electrolytes are presented in Fig. 6c. No oxidation or reduction peaks are observed in the 0.5 M H2SO4 solution, while only a single reduction or oxidation peak can be observed when scanning in a solution of 0.5 M H2SO4 and 20 mM CuSO4 (the typical solution employed for the UPD method). This indicated that the UPD and overpotential deposition (OPD) were not separated. The UPD and OPD peaks are obviously separated in the presence of Cl−, and the peeling region can be observed, which is attributed to the rapid adsorption of Cl− on the Cu UPD layer, thus effectively suppressing the OPD of Cu. The LSV curves of stripped Cu are thus calculated at various overpotentials ranging from 0.18 to 0.3 V (Fig. S29†). The charges for stripping the deposited Cu at various underpotentials are shown in Fig. 6d, in which the platform region is the area where UPD occurs and OPD is inhibited. Therefore, we use the charge obtained in the platform region to estimate the number of active sites, and the final active site density is estimated to be 2.39 × 1017 sites per cm2. As a comparison, the active site density of Ru/NC was also counted using the UPD method. Fig. S30† shows that the active site density of Ru/NC is calculated to be 1.62 × 1017 sites per cm2. To compare the intrinsic catalytic activities of Ru/NCDs and Ru/NC, the turnover frequency (TOF) values of both were calculated based on the above-mentioned number of active sites. The TOF values of Ru/NCDs and Ru/NC in alkaline solutions are shown in Fig. 6e, and the TOF value of Ru/NCDs exceeds that of Ru/NC at different potentials, and the detailed values are shown in Table S6.† A similar effect can be achieved in the acidic solution (Fig. 6f), with the TOF values of Ru/NCDs at various potentials being higher than those of Ru/NC, and the detailed results are shown in Table S7.† It is demonstrated that Ru/NCDs have stronger intrinsic activity than Ru/NC, aligning well with the experimental results, implying that the confinement effect of NCDs can effectively construct more active sites and accomplish the advancement of efficient HER.
Conclusions
In summary, carbon dots were selected as the substrate to fabricate an electrocatalyst of Ru/NCDs with a hydrogen spillover function. Then Ru/NCDs served as a model to reveal the dual roles of the carbon substrate, including the confinement effect of carbon dots and the Ru-like properties of the carbon substrate. Specifically, NCDs not only serve to immobilize the Ru nanoparticles, but also to construct more hydrogen spillover channels and obtain a sufficient supply of adsorbed hydrogen comparable to Ru, which leads to stronger HER activity. Ru/NCDs showed notable activity, with overpotentials of 15 mV and 31 mV at a current density of 10 mA cm−2 under acidic and alkaline conditions, respectively. This research examines the crucial role played by the carbon substrate in carbon-loaded ruthenium-based catalysts, which can offer additional insight into the design and manufacturing of more effective electrocatalysts in the future.Methods
Chemicals
RuCl3·3H2O was purchased from Shanghai Yuanye Bio-Technology Co. Ltd. (Shanghai, China). Chitosan and melamine were purchased from Aladdin Reagents Ltd. (Shanghai, China). Nafion (5 wt%) and 20 wt% Pt/C were purchased from Sigma Aldrich. KOH was purchased from Sinopharm Chemical Reagent Limited Corporation. All chemical reagents were used without further purification. Deionized water (DI) was utilized for all experiments.Characterization
Transmission electron microscope (TEM) images of pristine CDs and Ru/NCDs were generated using an FEI Tecnai G2 F20 transmission electron microscope, in addition with the function of obtaining X-ray energy dispersive spectrometer (EDS) spectra on an EDAX GenesisR TEM. The UV-vis absorption spectrum was captured using a Shimadzu UV-3101 PC spectrophotometer. The fluorescence spectra were acquired using a HITACHI F-7000 fluorescence spectrophotometer. The X-ray photoelectron spectroscopy (XPS) characterization was performed using a Thermo SCIENTIFIC ESCALAB 250Xi Spectrometer instrument with C 1s (binding energy of 284.6 eV) as a reference. An Ultima IV multipurpose X-ray diffractometer was used to record powder X-ray diffraction (XRD) patterns in the 2θ range of 5° to 90° (Cu-Kα radiation, 3 kW). Fourier transform infrared (FT-IR) spectra were obtained using a Nicolet IS5 spectrometer. Thermogravimetric (TGA) measurements were performed using a TA TGA55 instrument. In situ infrared (IR) spectra were acquired on a Nicolet IS50 IR spectrometer.Electrochemical measurements
All the electrochemical performance tests were conducted via a typical standard three-electrode system that included a graphite electrode as a counter electrode and a glassy carbon electrode (GCE, 3 mm diameter, 0.07065 cm2) and a saturated calomel electrode (SCE, in acidic) or Hg/HgO (in alkaline) as reference electrodes. 10 μL of catalyst ink was loaded onto a 3 mm diameter GCE for HER testing. The catalyst ink was made by dispersion of 10 mg catalyst in 1 mL of 0.1% Nafion isopropanol solution and sonicating for 30 min. The linear sweep voltammetry (LSV) curve was recorded at a scan rate of 5 mV s−1 at a constant temperature of 25 °C (water bath holding). All potentials in this study were converted to reversible hydrogen electrode (RHE) potentials using: ERHE = EHg/HgO + 0.0591pH + 0.098 (in 1.0 M KOH) and ERHE = ESCE + 0.0591pH + 0.242 (in 0.5 M H2SO4). All the LSV curves were corrected with a 95% iR compensation. Electrochemical impedance spectroscopy (EIS) measurements were performed at 0.1 to 105 Hz with an AC amplitude of 5 mV. Cyclic voltammetry (CV) at a scan rate of 100 mV s−1 and chronoamperometry (i–t) measurements were conducted to test long-term stability. The copper underpotential deposition (UPD) method was performed to determine the electrochemical active site density of the as-prepared catalysts. The catalysts were electrochemically cleaned using a 0.5 M H2SO4 + 20 mM CuSO4 + 60 mM NaCl solution, followed by copper deposition at various underpotentials. Following that, an LSV test (5 mV s−1) was carried out from the specified underpotential to the point that the UPD copper had all been removed. When the polarization potential fluctuates, the charge map of the stripped peak reveals a plateau. The charge on the plateau was employed to compute the density of electrochemically active areas. The turnover frequency (s−1) can be obtained using the formula: TOF = I/2nF, where I is the current corresponding to the LSV curve (A), F is the Faraday constant (C mol−1), n is the number of active sites (mol), and the factor 1/2 is an assumption that two electrons are required for the formation of a hydrogen molecule.Preparation of Ru/NCDs
Ru3+–NCDs precursors were first synthesized by a hydrothermal process. Specifically, 3 g melamine, 1.5 g chitosan and 0.45 g RuCl3·3H2O were homogeneously dispersed in 20 mL DI to form a slurry. The prepared slurry was transferred to a reactor containing a Teflon liner and reacted at 200 °C for 5 h to prepare Ru3+–NCDs. After this, the reactants were freeze-dried to finally obtain Ru3+–NCDs powder. The collected brown powder was pyrolyzed in a tube furnace under an argon atmosphere at a heating rate of 2 °C min−1 for 2 h at 700 °C. The Ru/NCDs were obtained after cooling to ambient temperature.Preparation of Ru/NC and Ru NPs
Ru/NC and Ru NPs were also prepared as comparisons. Specifically, 3 g melamine, 1.5 g chitosan, and 0.45 g RuCl3·3H2O were homogeneously dispersed in 20 mL DI. Then the dispersed homogeneous slurry was freeze-dried to obtain a dark powder. The collected powder was heated to 700 °C at a rate of 2 °C min−1 in an argon environment for 2 h. The Ru/NC powder was formed after cooling to ambient temperature. The Ru NPs were obtained by pyrolysis of RuCl3·3H2O under the same conditions.DFT computational details
Thermodynamic calculation of hydrogen adsorption according to density functional theory (DFT) was carried out using the Vienna Ab initio Simulation Package (VASP, version 5.4.4). The valence electron was described using a plane-wave basis with an energy cutoff of 450 eV, and the valence–core interactions were described using the projection augmented wave (PAW) approach. Correlation terms were exchanged using the Perdew–Burke–Ernzerhof (PBE) functional predicated on the generalized gradient approximation (GGA). For the structural optimization process, the k-points in the Brillouin zone are sampled as 2*2*1 grid points by the Monkhorst–Pack method. The energy convergence threshold for the electronic iteration is 10−5 eV and the geometric convergence criterion is that the force on each atom is less than 0.02 eV Å−1.The free energies of hydrogen adsorption were calculated using the formulae as follows:
| G = E + ZPE − TS |
The d-band centers were calculated using the formulae as follows:

where I(z) is the charge displacement curve, which represents the cumulative charge from z to z0, p(x,y,z) is the change of the charge density in the 3d charge density difference diagram, z0 is the starting point of integration in the z direction, a is the length of the lattice in the x direction and b is the length of the lattice in the y direction.
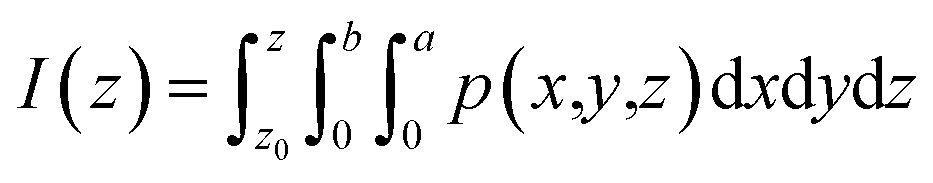
Author contributions
Zonglin Liu: investigation, writing-original draft, validation. Honglei Zhang: software, methodology. Dongyue Liu: data curation. Yujie Feng: validation. Dechang Jia: supervision. Caicai Li: methodology, supervision, writing-reviewing and editing. Qingfeng Sun: resources. Yu Zhou: project administration. Zhenhui Kang: conceptualization, writing-reviewing and editing. Baoqiang Li: supervision, writing-reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (52061135204 and 51972086), Interdisciplinary Research Foundation of HIT (IR2021105), Heilongjiang Provincial Key R&D Projects (2022ZX02C24), Russian Science Foundation (21-43-00020), State Key Laboratory of Urban Water Resource and Environment of Harbin Institute of Technology (2022TS21), Ministry of Science and Higher Education of the Russian Federation (075-15-2022-1114), Key Laboratory of Bio-based Material Science & Technology (Northeast Forestry University) Ministry of Education (SWZ-ZD202103), Zhejiang Provincial Natural Science Foundation for Exploring Program Q (LQ20C160004) and Scientific Research Foundation of Zhejiang A&F University (2019FR009).References
- T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2022, 10, 2205077 CrossRef PubMed.
- J. Mao, J. Fang, Q. Chen, Z. Li and Y. Li, Chem. Commun., 2023, 55, 166 Search PubMed.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- X. Huang, K. Zhang, B. Peng, G. Wang, M. Muhler and F. Wang, ACS Catal., 2021, 11, 9618–9678 CrossRef CAS.
- Z. Chen, Q. Li, H. Xiang, Y. Wang, P. Yang, C. Dai, H. Zhang, W. Xiao, Z. Wu and L. Wang, Inorg. Chem. Front., 2023, 10, 1493–1500 RSC.
- Z. Wu, P. Yang, Q. Li, W. Xiao, Z. Li, G. Xu, F. Liu, B. Jia, T. Ma, S. Feng and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300406 CrossRef CAS PubMed.
- J. Zhang, Q. Zhang and X. Feng, Adv. Mater., 2019, 31, 1808167 CrossRef PubMed.
- M. Song, Z. Zhang, Q. Li, W. Jin, Z. Wu, G. Fu and X. Liu, J. Mater. Chem. A, 2019, 7, 3697–3703 RSC.
- R. Sharma, S. Gyergyek, J. Chamier, P. Morgen and S. M. Andersen, ChemElectroChem, 2021, 8, 1183–1195 CrossRef CAS.
- F. Y. Yu, Z. L. Lang, L. Y. Yin, K. Feng, Y. J. Xia, H. Q. Tan, H. T. Zhu, J. Zhong, Z. H. Kang and Y. G. Li, Nat. Commun., 2020, 11, 490 CrossRef PubMed.
- L. Zhao, Y. Zhang, L. B. Huang, X. Z. Liu, Q. H. Zhang, C. He, Z. Y. Wu, L. J. Zhang, J. Wu, W. Yang, L. Gu, J. S. Hu and L. J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- B. Tang, X. Yang, Z. Kang and L. Feng, Appl. Catal., B, 2020, 278, 119281 CrossRef CAS.
- Y. Gao, Z. Chen, Y. Zhao, W. Yu, X. Jiang, M. He, Z. Li, T. Ma, Z. Wu and L. Wang, Appl. Catal., B, 2022, 303, 120879 CrossRef CAS.
- Y. Sun, Z. Xue, Q. Liu, Y. Jia, Y. Li, K. Liu, Y. Lin, M. Liu, G. Li and C.-Y. Su, Nat. Commun., 2021, 12, 1369 CrossRef CAS PubMed.
- Y. Liu, N. Chen, W. Li, M. Sun, T. Wu, B. Huang, X. Yong, Q. Zhang, L. Gu, H. Song, R. Bauer, J. S. Tse, S.-Q. Zang, B. Yang and S. Lu, SmartMat, 2022, 3, 249–259 CrossRef CAS.
- H. Wang, P. Yang, X. Sun, W. Xiao, X. Wang, M. Tian, G. Xu, Z. Li, Y. Zhang, F. Liu, L. Wang and Z. Wu, J. Energy Chem., 2023, 87, 286–294 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, 379 CrossRef PubMed.
- Y. Yang, Y. Yu, J. Li, Q. Chen, Y. Du, P. Rao, R. Li, C. Jia, Z. Kang, P. Deng, Y. Shen and X. Tian, Nano-Micro Lett., 2021, 13, 160 CrossRef CAS PubMed.
- M. Lao, G. Zhao, P. Li, T. Ma, Y. Jiang, H. Pan, S. X. Dou and W. Sun, Adv. Funct. Mater., 2021, 31, 2100698 CrossRef CAS.
- H. Zhang, W. Zhou, X. F. Lu, T. Chen and X. W. Lou, Adv. Energy Mater., 2020, 10, 2000882 CrossRef CAS.
- M. Xiong, Z. Gao and Y. Qin, ACS Catal., 2021, 11, 3159–3172 CrossRef CAS.
- D. Zhao, Z. Li, X. Yu, W. Zhou, Q. Wu, Y. Luo, N. Wang, A. Liu, L. Li and S. Chen, Chem. Eng. J., 2022, 450, 138254 CrossRef CAS.
- W. Jin, H. Wu, W. Cai, B. Jia, M. Batmunkh, Z. Wu and T. Ma, Chem. Eng. J., 2021, 426, 130762 CrossRef CAS.
- W. Li, Y. Zhao, Y. Liu, M. Sun, G. I. N. Waterhouse, B. Huang, K. Zhang, T. Zhang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 3290–3298 CrossRef CAS PubMed.
- Y. Jiang, T.-W. Huang, H.-L. Chou, L. Zhou, S.-W. Lee, K.-W. Wang and S. Dai, J. Mater. Chem. A, 2022, 10, 17730–17739 RSC.
- Z. Liu, B. Li, Y. Feng, D. Jia, C. Li, Q. Sun and Y. Zhou, Small, 2021, 17, e2102496 CrossRef PubMed.
- T. Feng, G. Yu, S. Tao, S. Zhu, R. Ku, R. Zhang, Q. Zeng, M. Yang, Y. Chen, W. Chen, W. Chen and B. Yang, J. Mater. Chem. A, 2020, 8, 9638–9645 RSC.
- Y. Zhai, B. Zhang, R. Shi, S. Zhang, Y. Liu, B. Wang, K. Zhang, G. I. N. Waterhouse, T. Zhang and S. Lu, Adv. Mater., 2022, 12, 2103426 CAS.
- J. Li, J. Hu, M. Zhang, W. Gou, S. Zhang, Z. Chen, Y. Qu and Y. Ma, Nat. Commun., 2021, 12, 3502 CrossRef CAS PubMed.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS PubMed.
- A. Janotti and C. G. Van de Walle, Nat. Mater., 2007, 6, 44–47 CrossRef CAS PubMed.
- Y. Tan, Y. Zhu, X. Cao, Y. Liu, J. Li, Z. Chen and J. Hu, ACS Catal., 2022, 12, 11821–11829 CrossRef CAS.
- J. Wei, K. Xiao, Y. Chen, X.-P. Guo, B. Huang and Z.-Q. Liu, Energy Environ. Sci., 2022, 1, 800 Search PubMed.
- K. Wei, H. Nie, Y. Li, X. Wang, Y. Liu, Y. Zhao, H. Shi, H. Huang, Y. Liu and Z. Kang, J. Colloid Interface Sci., 2022, 616, 769–780 CrossRef CAS PubMed.
- T. Wu, J. Hong, Z. Lu, H. Wu, C. Wu, Z. Tang, X. Liu, B. Zeng, Y. Xu, G. Chen, C. Yuan and L. Dai, Appl. Catal., B, 2021, 285, 119795 CrossRef CAS.
- L. Cai, Z. Lin, M. Wang, F. Pan, J. Chen, Y. Wang, X. Shen and Y. Chai, J. Mater. Chem. A, 2017, 5, 24091–24097 RSC.
- Y. Zong, T. Yang, P. Hao, B. Shan, B. Peng, X. Hu, R. Tao, X. Chen, P. Wu and K. Zhang, Microporous Mesoporous Mater., 2020, 302, 110229 CrossRef CAS.
- J. Liu, Y. Geng, D. Li, H. Yao, Z. Huo, Y. Li, K. Zhang, S. Zhu, H. Wei, W. Xu, J. Jiang and B. Yang, Adv. Mater., 2020, 32, 1906641 CrossRef CAS PubMed.
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed.
- J. Dai, Y. Zhu, Y. Chen, X. Wen, M. Long, X. Wu, Z. Hu, D. Guan, X. Wang, C. Zhou, Q. Lin, Y. Sun, S. C. Weng, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2022, 13, 1189 CrossRef CAS PubMed.
- J. Li, Y. Tan, M. Zhang, W. Gou, S. Zhang, Y. Ma, J. Hu and Y. Qu, ACS Energy Lett., 2022, 7, 1330–1337 CrossRef CAS.
- L. Zhu, H. Lin, Y. Li, F. Liao, Y. Lifshitz, M. Sheng, S.-T. Lee and M. Shao, Nat. Commun., 2016, 7, 12272 CrossRef CAS PubMed.
- M. Sheng, B. Jiang, B. Wu, F. Liao, X. Fan, H. Lin, Y. Li, Y. Lifshitz, S.-T. Lee and M. Shao, ACS Nano, 2019, 13, 2786–2794 CrossRef CAS PubMed.
- Y. Cheng, S. Lu, F. Liao, L. Liu, Y. Li and M. Shao, Adv. Funct. Mater., 2017, 27, 1700359 CrossRef.
- A. Damian and S. Omanovic, J. Power Sources, 2006, 158, 464–476 CrossRef CAS.
- F. Li, G. F. Han, H. J. Noh, I. Ahmad, I. Y. Jeon and J. B. Baek, Adv. Mater., 2018, 30, e1803676 CrossRef PubMed.
- X. Yang, A.-Y. Lu, Y. Zhu, M. N. Hedhili, S. Min, K.-W. Huang, Y. Han and L.-J. Li, Nano Energy, 2015, 15, 634–641 CrossRef CAS.
- J. Wang, Z. Wei, S. Mao, H. Li and Y. Wang, Energy Environ. Sci., 2018, 11, 800–806 RSC.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Y. Xu, N. Jiang, F. Pan, Q. Wang, Z. Gao and G. Chen, Energy Convers. Manage., 2017, 133, 535–547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07016f |
| This journal is © The Royal Society of Chemistry 2024 |