
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free carbon dot–microporous graphitic carbon heterojunctions as photocatalysts for CO2 reduction†
Ana
Garcia-Mulero
,
María
Cabrero-Antonino
,
Hermenegildo
García
 * and
Ana
Primo
*
* and
Ana
Primo
*
Instituto de Tecnología Química, Consejo Superior de Investigaciones Científicas, Universitat Politècnica de València, Av. De los naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es; aprimoar@itq.upv.es
First published on 13th February 2024
Abstract
A series of photocatalysts containing various loadings of carbon dots (CD) on microporous graphitic carbons (mpC) was prepared by pyrolysis at 900 °C under inert atmosphere of α-cyclodextrin containing preformed CDs. CDs were obtained by microwave hydrothermal conversion of citric acid and urea, and they were purified by centrifugation and partition between aqueous and organic solvents. The CD/mpC composites exhibit the expected morphology for mpC in TEM with particles of about 20 nm and microporous channels of 0.6 nm. XRD and Raman spectra of CD/mpC were also coincident with those of pristine mpC, but XPS and elemental analysis detected the presence of pyridinic N from the CDs. The optimal CD/mpC sample exhibits higher photocatalytic activity for the simultaneous H2 evolution and CO2 reduction to CH4 than the independent components or than a mechanical CD and mpC mixture reaching a productivity rate of 29 and 5 μmol gcatalyst−1 h−1 for H2 and CH4, respectively. CD/mpC photocatalysts were stable for four consecutive runs.
Introduction
Pyrolysis of carbohydrates can afford turbostratic graphitic carbons that can render defective graphenes upon exfoliation.1–3 In a series of papers, it has been shown that pyrolysis of cyclodextrins, the cyclic glucopyranose oligomers, form microporous graphitic carbons, whose porosity depends on the dimensions of the α, β, or γ cyclic precursor.4–7 It has been proposed that, during the thermal treatment of the cyclic glucose oligomers having a truncated conical shape, the solid first melts at temperatures about 300 °C and then self-assembles, forming tubular structures before being transformed at higher temperatures by dehydration and deoxygenation to the final carbon residue.5 These microporous graphitic carbon structures exhibit activity as catalysts in the aerobic oxidation of hydrocarbons5 and also a photocatalytic activity for overall water splitting7 and photocatalytic CO2 reduction.4 The role of confinement increasing the (photo)catalytic activity was firmly established by comparison of the performance of a series of materials having increasing micropore size or even no micropores.4,5,7 It was observed that the most active microporous graphitic carbon was that derived from α-cyclodextrin having the smaller pore diameter about 0.6 nm.Following this line of research aimed at showing the photocatalytic activity of microporous graphitic carbons derived from cyclodextrin pyrolysis and knowing that heterojunctions and composites of carbonaceous photocatalysts can exhibit an enhanced photocatalytic activity,8,9 it is of interest to determine the performance of these microporous graphitic carbons in combination with carbon dots (CDs). CDs have emerged as highly efficient photocatalytic materials10–13 and, also for these carbon residues, combination with other components can enhance their photocatalytic activity.13–15
Herein, it is reported that the incorporation of CDs on microporous graphitic carbons derived from α-cyclodextrin results in an enhanced photocatalytic activity for the CO2 reduction reaction.
Results and discussion
CDs were prepared from solvothermal treatment in microwaves of an aqueous solution of citrate and urea.16–18 The CDs were purified by removing large particles by ultracentrifugation and subsequent partition between dichloromethane and water/methanol. Increasing amounts of CDs were added to α-cyclodextrin in aqueous solution, and then, as described in Scheme 1, the water was evaporated at low temperature under magnetic stirring.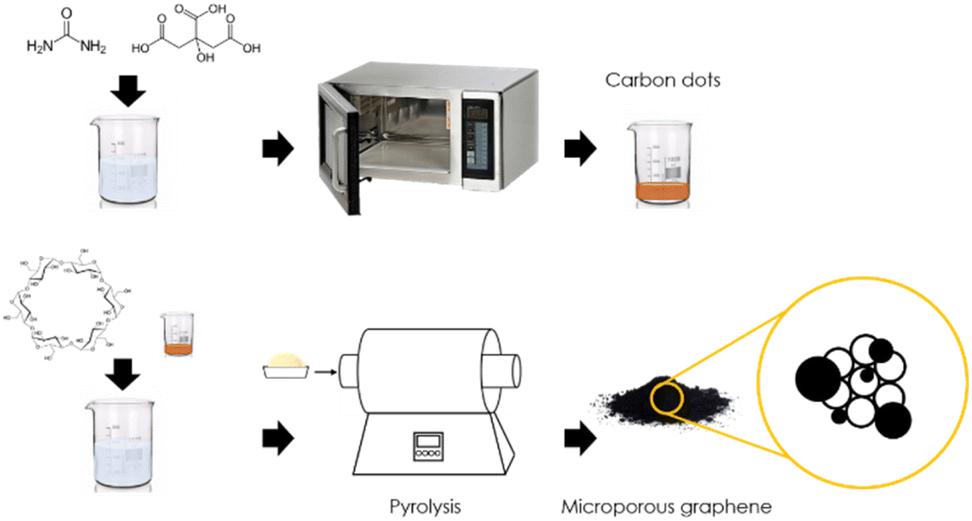 | ||
| Scheme 1 Preparation procedure for CD/mpC heterojunctions. | ||
The dry powder was submitted to pyrolysis at 900 °C under inert atmosphere to obtain the microporous graphitic carbons having CDs (CD/mpC).
Additional samples submitting either CDs or α-cyclodextrin to pyrolysis under the same conditions as those employed in CD/mpC were also prepared as controls.
TEM images of CD/mpC exhibit the expected morphology of small carbon particles about 20 nm mean particle size in which the presence of parallel microporous channels of 0.60 nm diameter was observed. Fig. 1 shows selected images and the pore size measurement based on the contrast change between carbon walls and pores. This indicates that, at the CD loadings used in the preparation of the present composites, there is no significant CD agglomeration and that the self-assembly of α-CD upon melting and their subsequent carbonization process does not become disturbed by the presence of CDs.
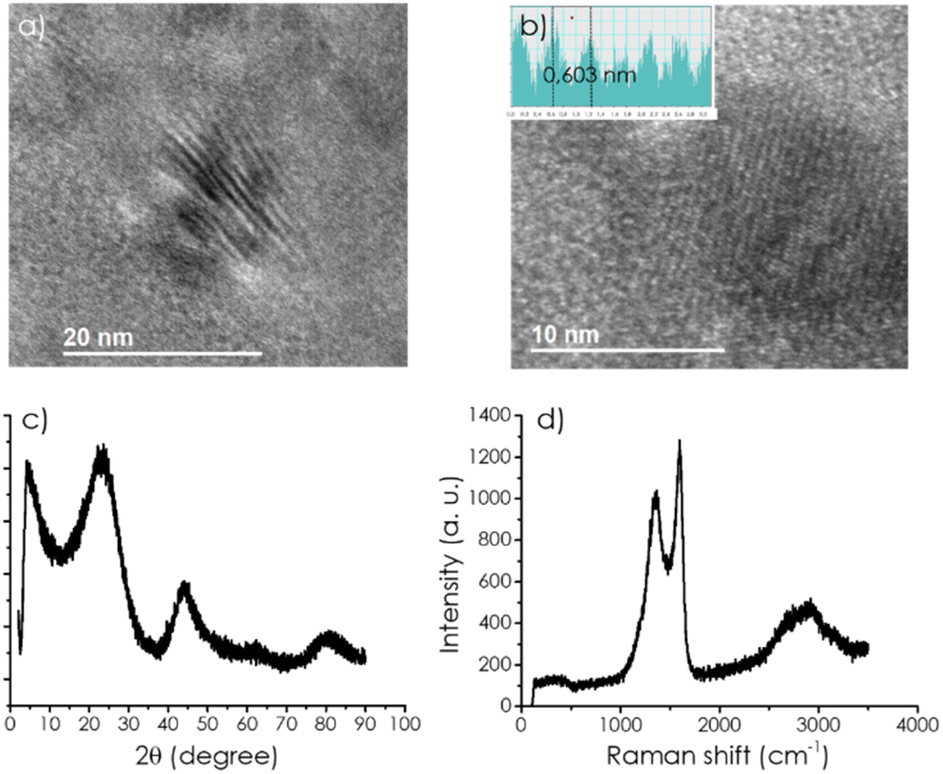 | ||
| Fig. 1 (a) HRTEM images from the sample CD-1/mpC and (b) measurement of the pore size; (c) XRD pattern of CD-1/mpC, and (d) Raman spectra of the same sample. | ||
XRD also confirms the porous and graphitic structure of the CD/mpC series (Fig. 1 and S1†) coincident to that of mpC control. The broad XRD peaks at about 2θ 24 and 44° correspond to the diffraction through the 002 and 101 planes. No influence of the presence of CDs at the loadings employed in the study in the XRD was apparent.
Similarly, Raman spectra of the series of CD/mpC samples (Fig. 1 and S1†) were all coincident and also in accordance with the Raman spectrum reported for pristine mpC, meaning that the presence of CD does not show any signal. It does not influence the Raman spectrum, presenting the characteristic G + D, G, and D bands as broad signals at 2900, 1590, and 1350 cm−1, respectively.
The relative intensity of the G vs. D (IG/ID) peaks is frequently taken as a quantitative indicator of the density of defects in the graphitic material. In the present case, this ratio is 1.19, and it is the same for all the samples, implying again that no further defects are generated on mpC during the pyrolysis by the presence of CDs in the loadings used in the present study.
XPS analysis provides important information on the elements present at the material's surface, their relative abundance, and the distribution of the elements among different families depending on the hybridization and coordination sphere. In the present case, XPS analysis of CDs shows the presence of C, N, and O in its composition with a relative atomic abundance of 68, 13, and 18%, respectively.
Fig. 2 and S2† present the high-resolution XPS peak for these elements and their best deconvolution to individual components. As shown in this Fig. 2, deconvolution of the high-resolution XPS C1s peak shows two components at 284.6 and 287.8 eV attributable to C atoms on graphenic and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O environment. Deconvolution of XPS O1s shows a major component at 531.3 and 533.3 eV ascribed to CO and OH, respectively. In the case of XPS N1s peak, a single component at 399.6 eV is due to pyridinic N atoms.
O environment. Deconvolution of XPS O1s shows a major component at 531.3 and 533.3 eV ascribed to CO and OH, respectively. In the case of XPS N1s peak, a single component at 399.6 eV is due to pyridinic N atoms.
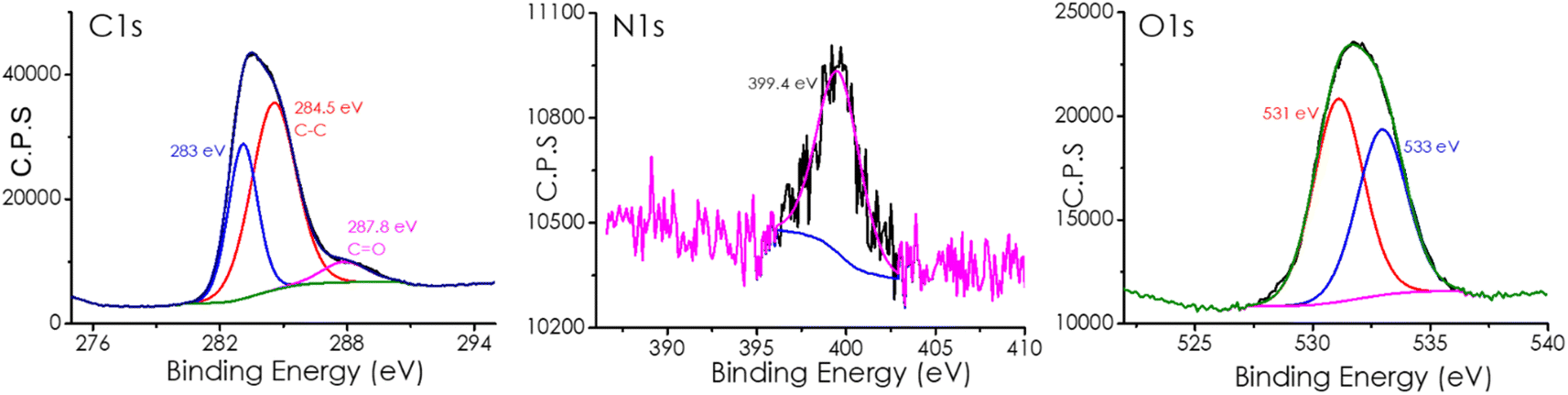 | ||
| Fig. 2 High-resolution XP spectra of the sample CD-1/mpC and the best deconvolution of the different components for C1s (left), N1s (center), and O1s (right). | ||
In comparison, XPS analysis of CD/mpC shows much lesser N percentage. While the high-resolution XPS C1s of CD/mpC exhibited an additional new component at 283.0 eV. The proportion of this other peak varies depending on the CD loading in the composite.
The experimental high-resolution XPS O1s and N1s peaks could fit with the same components as those indicated above for CD. However, the proportion of the O1s component was significantly higher than that of CDs. The number of components and their distribution does not change with the CD loading in the range of proportions used in the present study.
Fig. S2† also presents the XPS peaks of the CD/mpC composites. In any case, it is worth commenting that while no N 1s peak can be detected in XPS of the parent mpC, the CD/mpC composites have detectable N1s peaks at the same binding energy and components as those found in CD. This agrees with CD as a component of the CD/mpC composite with the same structure as pristine CD. Combustion chemical analysis also confirmed the presence of N in the composition of the composites and a significant percentage of O of about 14%. Table 1 gathers these analytical data.
| Sample | Combustion elemental analysis (%) | XPS atomic ratio (%) | |||
|---|---|---|---|---|---|
| N | C | N | C | O | |
| CD-08/mpC | 0.5 | 85.6 | 0.7 | 89 | 11 |
| CD-1/mpC | 0.5 | 86.6 | 0.6 | 90 | 9 |
| CD-12/mpC | 0.6 | 84.3 | 0.7 | 89 | 10 |
Regarding photocatalytic activity, one important point is adsorption capacity and surface area of the material. As previously reported for mpC,5 no measurable N2 adsorption at 77 K was observed for mpC or CD-1/mpC. However, CO2 adsorption at 296 K gives a specific surface area of 725 and 630 m2 g−1, respectively. These values indicate that CO2 can diffuse inside the micropores of mpC, making accessible the photogenerated charge carriers.
Photocatalytic CO2 reduction tests
The photocatalytic reactions were carried out in the batch mode using acetonitrile as solvent containing water as proton source and triethanolamine as the electron donor in a quartz photoreactor with inlet and outlet (51 mL). The total solution volume was 20 mL, and the photocatalyst concentration was 0.5 mg mL−1. Before the reaction, the solution was exhaustively purged with Ar to remove ambient oxygen that could act as electron quencher and, then charged with CO2, which is relatively soluble in this liquid mixture containing amine, water, and acetonitrile. Upon irradiation, simultaneous evolution of H2 and CH4 was observed in all the CD/mpC samples. Controls in the absence of photocatalysts, light, CO2 or electron donors showed no gas evolution, indicating that the three elements are necessary for the process. Using isotopically labeled 13CO2 results in the appearance of 13CH4 as determined by mass spectrometry. Fig. S3† shows the corresponding GC chromatogram, 13CH4 mass spectrum formed in these experiments from 13CO2 in comparison to 12CH4 from 12CO2. Note that 13CH4 cannot be an ion fragment of 13CO2 in the mass spectrometer. Three other samples, namely, mpC, CD, and a mechanical mixture of CD and mpC, were also included as photocatalysts, the three materials giving lesser product evolution than the CD/mpC composites. Fig. 3 and S4† show the temporal profiles for H2 and CH4 evolution of the CD-x/mpC and control samples under study.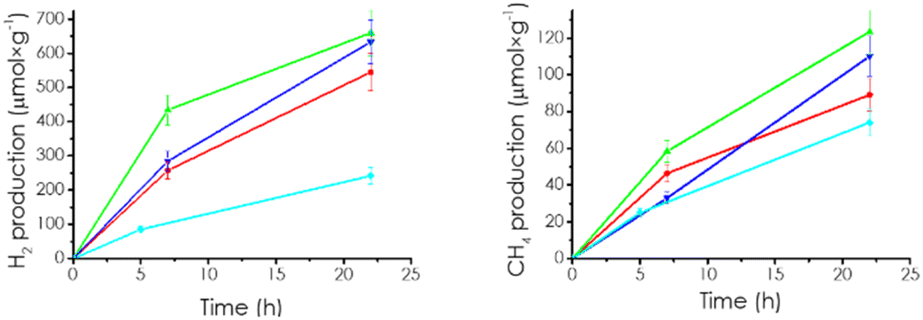 | ||
| Fig. 3 Gas evolution for the samples mpC (cyan), CD-08/mpC (red), CD-1/mpC (green), CD-12/mpC (blue). Reaction conditions: acetonitrile 12 mL, H2O 4 mL, triethanol amine 4 mL, CD-x/mpC at 0.5 mg mL−1, irradiation through quartz with the full output of a 300 W Xe lamp. | ||
Although apparently, H2 evolves preferentially in percentage in the photocatalytic experiments. It should be reminded that considering that CH4 formation from CO2 consumes eight electrons and H2 accounts for 2 electrons, the conduction band electron consumption selectivity was 55 and 45% for H2 and CH4, respectively. It should be noted that the efficiency order for H2 and CH4 generally follows the same order for the series of photocatalysts.
This selectivity is remarkable and deserves further study, particularly regarding the reaction mechanism and the CO2 intermediates on the photocatalyst surface that eventually form CH4.
Regarding the influence of CD loading, an optimal proportion was observed for CD-1/mpC, lower or higher loading resulting in lesser photocatalytic activity in both H2 and CH4 evolution. This optimal in the experimental parameters on the activity is frequently observed in photocatalysis and is attributed to the operation of two opposite effects. On the one hand, increasing CD concentration should benefit the charge carrier separation, enhancing product formation rate. On the other hand, an excessive amount of CD in the system accumulating a certain charge carrier can promote charge carrier recombination when surface coverage is high, decreasing, in this way, the product formation rate. The balance between positive and negative effects is achieved for an optimal CD loading range.
To support the claim that the optimal CD-1/mpC sample corresponds to the one with the highest efficiency in charge separation, transient photocurrent measurements on a transparent conductive electrode containing a thin film of CD/mpC photocatalysts upon irradiation were measured in Na2SO4 as inert electrolyte. The photocurrent of CD-1/mpC was higher in comparison to that of mpC component under the same conditions, indicating that electron extraction is larger in the heterojunction than for mpC. All attempts made to obtain a stable photoelectrode for CD for an analogous comparison were unsuccessful, due to the insufficient adhesion of CD to the FTO electrode. In any case, the transient photocurrent measurements provided the same order for current intensity in the transient photocurrent measurements than the observed photocatalytic activity, meaning that the samples in which higher number of photogenerated electrons per unit of time can be extracted are those for which the photocatalytic activity is the highest. Fig. 4 shows the transient photocurrent measurements for the most active CD-1/mpC sample at various voltage bias.
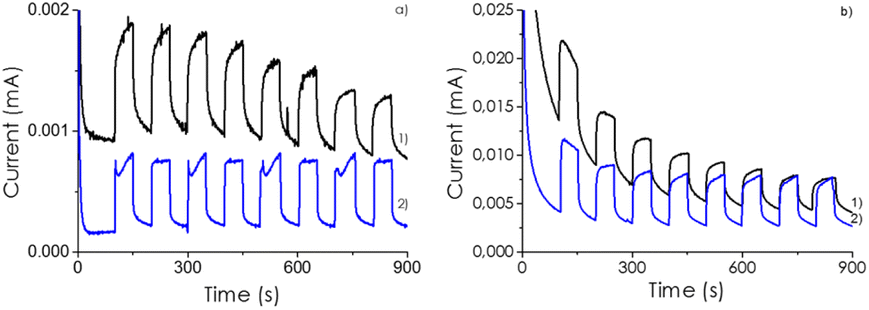 | ||
| Fig. 4 Transient photocurrent measurements for a mpC (1 in blue line) and CD-1/mpC (2 line in black) electrode after 300 s of stabilization in the dark at (a) 0.5 V and (b) 1 V polarization potential. On-off cycles were of 1 min. Measurements conditions: Single cell using Pt and Ag/AgCl as counter and reference electrodes upon irradiation with the full output of a 150 W Xe lamp and 0.5 M Na2SO4 as electrolyte. | ||
Photocatalyst stability was confirmed by performing four consecutive photocatalytic reactions with the same CD-1/mpC sample, outgassing each time the system after 24 h irradiation, washing the photocatalyst by centrifugation and starting the reaction again with the proper amounts of solvents.
As seen in Fig. 5, similar temporal profiles were monitored, indicating photocatalyst stability. The four times reused photocatalyst was characterized by XPS (Fig. S2d†), whereby the same composition as that of the fresh CD-1/mpC was measured.
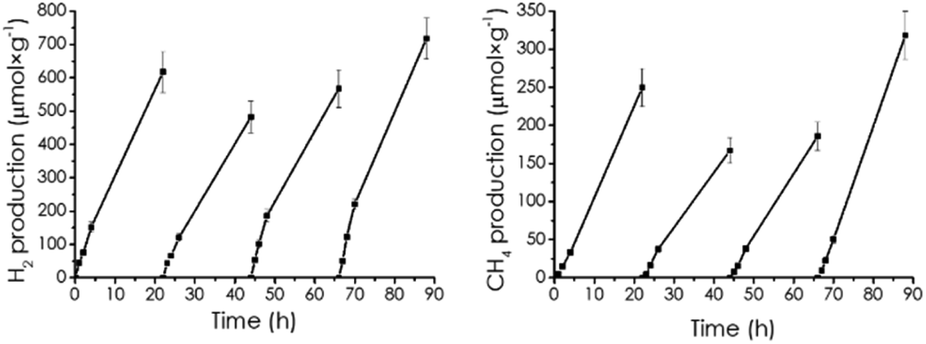 | ||
| Fig. 5 Temporal hydrogen and methane evolution in the photocatalytic irradiation of CD-1/mpC in four consecutive uses. Reaction conditions: acetonitrile 12 mL, H2O 4 mL, triethanol amine 4 mL, CD-1/mpC at 0.5 mg mL−1, irradiation through quartz with the full output of a 300 W Xe lamp. | ||
Valence band maximum energy for mpC was estimated from the onset energy of the first ejected electrons in the XPS measurements after correction by the work function of the instrument, giving a value of −6.65 V vs. vacuum. The conduction band minimum energy was, then, determined from this value and the optical bandgap measured by diffuse reflectance UV-Vis absorption spectroscopy, given a value of −3.55 V vs. vacuum. Similar measurements for the valence and conduction band energy of CD gives values of −6.40 V and −3.35 V, respectively. As shown in Scheme 2, these measurements allow to establish a band energy diagram for the heterojunction, corresponding to a Z-scheme. Scheme 2 also includes the redox potential of the H+/H2 and CO2/CH4 pairs, showing the thermodynamic feasibility of the process.
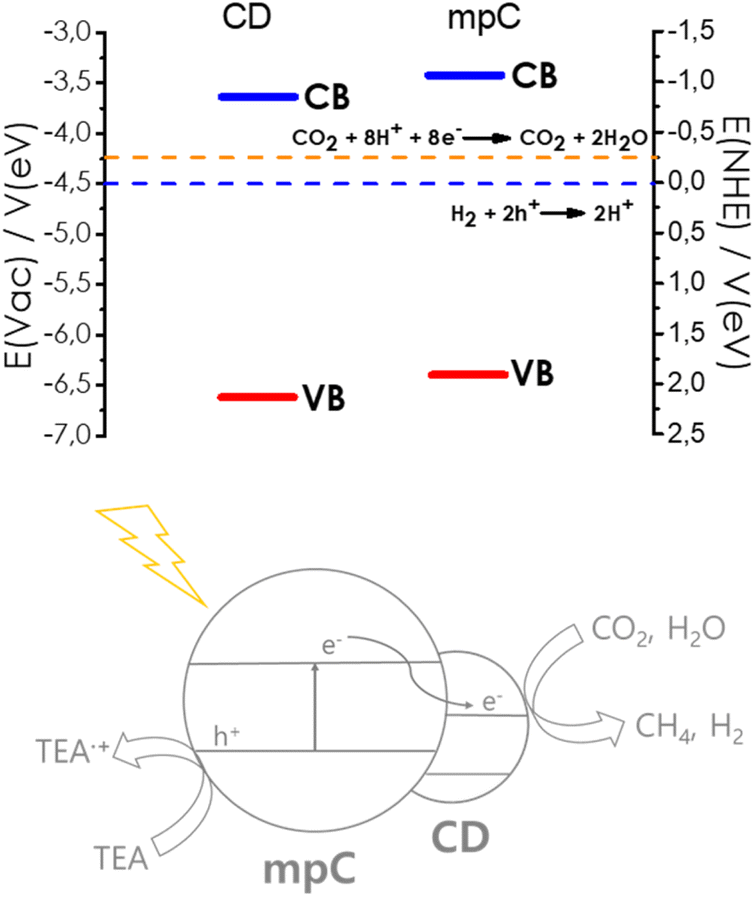 | ||
| Scheme 2 Band energy diagram and proposed reaction mechanism for the photocatalytic CO2 reduction in the presence of sacrificial electron donor by CD/mpC. | ||
Based on the band energy diagram and the previously reported photocatalytic activity of mpC for H2 evolution and CDs,11,19 a reasonable mechanism for CO2 reduction by CD/mpC can be proposed as indicated in Scheme 2. According to it, light absorption by CD and mpC will lead to photoinduced electron/hole separation. Electron migration from photogenerated electron from mpC to CD conduction band will result in an enhancement of the charge separation efficiency through the heterojunction.
Conclusions
This manuscript reports that the photocatalytic activity of metal-free microporous carbons derived from the pyrolysis of α-cyclodextrin can be enhanced using carbon dots to form a heterojunction of metal-free components. The CD/mpC heterojunction exhibits photocatalytic activity in aqueous acetonitrile saturated with CO2 and triethanolamine, evolving simultaneously with H2 and CH4 production. At the optimal carbon dot loading, the intrinsic photocatalytic activity for the microporous graphitic carbon increases by factors of 3.5 and 1.7 for H2 and CH4 evolution, giving at a rate of 29 and 5 μmol gcatalyst−1 h−1 for H2 and CH4 evolution, respectively.This product distribution corresponds to a relative consumption of photogenerated electron conduction of 55 and 45% for H2O and CO2 reduction, respectively.
Transient photocurrent measurements indicate that the enhanced photocatalytic activity of CD-1/mpC derives from a more efficient charge separation in the heterojunction compared to mpC. Thus, the present results show the possibility of forming metal-free photocatalysts' heterojunctions to optimize their efficiency further.
Author contributions
A. G.-M. and M. C.-A. performed the experimental work. H. G. and A. P. designed the study. The manuscript was written with the contribution of all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Spanish Ministry of Science and Innovation (CEX-2021-001230-S and PDI2021-0126071-OB-CO21 funded by MCIN/AEI/10.13039/501100011033) and Generalitat Valenciana (Prometeo 2021/038 and Advanced Materials program Graphica MFA/2022/023 with funding from European Union NextGenerationEU PRTR-C17.I1).References
- A. Primo, P. Atienzar, E. Sanchez, J. M. Delgado and H. García, Chem. Commun., 2012, 48, 9254–9256 RSC
.
- S. Kumar, S. T. Aziz, O. Girshevitz and G. D. Nessim, J. Phys. Chem. C, 2018, 122, 2343–2349 CrossRef CAS
- A. Primo, E. Sánchez, J. M. Delgado and H. García, Carbon, 2014, 68, 777–783 CrossRef CAS
- A. Garcia-Mulero, A. M. Asiri, J. Albero, A. Primo and H. Garcia, Nanoscale, 2022, 14, 11575–11582 RSC
- A. Rendón-Patiño, A. Santiago-Portillo, C. Vallés-Garcia, M. Palomino, S. Navalón, A. Franconetti, A. Primo and H. Garcia, Small Methods, 2020, 4, 1900721 CrossRef
- A. Primo, A. Rendón-Patiño, C. Bucur, A. Jurca, B. Cojocaru, V. I. Parvulescu and H. Garcia, J. Catal., 2022, 405, 355–362 CrossRef CAS
- A. Garcia-Mulero, A. Rendón-Patiño, A. M. Asiri, A. Primo and H. Garcia, ACS Appl. Mater. Interfaces, 2021, 13, 48753–48763 CrossRef CAS PubMed
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC
- L. Xu, J. Zeng, Q. Li, L. Xia, X. Luo, Z. Ma, B. Peng, S. Xiong, Z. Li and L.-L. Wang, Appl. Surf. Sci., 2021, 547, 149207 CrossRef CAS
- Y. Yao, H. Zhang, K. Hu, G. Nie, Y. Yang, Y. Wang, X. Duan and S. Wang, J. Environ. Chem. Eng., 2022, 107336 CrossRef CAS
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CrossRef CAS
- H. Yu, R. Shi, Y. Zhao, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed
- K. Akbar, E. Moretti and A. Vomiero, Adv. Opt. Mater., 2021, 9, 2100532 CrossRef CAS
- H. Luo, Q. Guo, P. Á. Szilágyi, A. B. Jorge and M.-M. Titirici, Trends Chem., 2020, 2, 623–637 CrossRef CAS
- S. Asadzadeh-Khaneghah and A. Habibi-Yangjeh, J. Cleaner Prod., 2020, 276, 124319 CrossRef CAS
- W. Kasprzyk, T. Świergosz, S. Bednarz, K. Walas, N. V. Bashmakova and D. Bogdał, Nanoscale, 2018, 10, 13889–13894 RSC
- X. Yao, Y. Wang, F. Li, J. J. Dalluge, G. Orr, R. Hernandez, Q. Cui and C. L. Haynes, Nanoscale, 2022, 14, 9516–9525 RSC
- J. D. Stachowska, A. Murphy, C. Mellor, D. Fernandes, E. N. Gibbons, M. J. Krysmann, A. Kelarakis, E. Burgaz, J. Moore and S. G. Yeates, Sci. Rep., 2021, 11, 1–14 CrossRef PubMed
- R. M. Sendão, J. C. Esteves da Silva and L. Pinto da Silva, Catalysts, 2023, 13, 179 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00007b |
| This journal is © The Royal Society of Chemistry 2024 |