
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-catalyst design to control charge transfer and product composition for photocatalytic H2 production and biomass reforming†
Van-Can
Nguyen
 a,
Meyta
Sanoe
a,
Novy Pralisa
Putri
a,
Yuh-Lang
Lee
ab and
Hsisheng
Teng
*ab
a,
Meyta
Sanoe
a,
Novy Pralisa
Putri
a,
Yuh-Lang
Lee
ab and
Hsisheng
Teng
*ab
aDepartment of Chemical Engineering, National Cheng Kung University, Tainan 70101, Taiwan. E-mail: hteng@mail.ncku.edu.tw; Fax: +886-6-2344496
bHierarchical Green-Energy Materials (Hi-GEM) Research Center, National Cheng Kung University, Tainan 70101, Taiwan
First published on 3rd February 2024
Abstract
The exploration of co-catalyst effects in photocatalytic H2 production along with biomass reforming is still limited. This study employs S- and N-doped graphene oxide dots (SNGODs) along with Pt and Ag co-catalysts to investigate the photocatalytic reforming of xylose (C5) into its derivatives and gaseous H2. Our study shows that Pt-SNGODs effectively catalyze both the reduction and oxidation steps: water reduction for H2 evolution and oxidative reforming of xylose into C5–C1 species through successive alternation of hydrolysis and photocatalytic oxidation. Ag-SNGODs are less effective in the H2 evolution reaction and the accumulated photogenerated electrons facilitate cleavage of xylose, through the retro-aldol reaction, into C3- and C2-species, which are then reformed into C3–C1 species. In addition to xylose cleavage, Ag-SNGODs are effective in reducing xylose and formate into radicals, which would proceed with C–C coupling to produce a C6 compound. The present study demonstrates how the co-catalyst of photocatalysts can influence the charge transfer dynamics and the product composition in photocatalytic biomass reforming.
Introduction
The environmental issues caused by fossil fuel consumption across the world have urged people to find a new alternative energy source. Hydrogen with a high heating value has emerged as a promising environmentally-benign energy resource.1 Biomass oxidation through photo-reforming, using sunlight as the energy input to produce hydrogen, offers a sustainable green energy solution.1,2 To improve the photocatalytic reduction process for hydrogen generation, a co-catalyst is often used.3 In addition to affecting the hydrogen evolution, the co-catalyst can also influence the organic products derived from biomass oxidation.4 The mechanism by which the co-catalyst affects the photo-reforming products and reaction routes is yet to be fully understood.There has been a growing interest in the direct photocatalytic reforming of biomass into hydrogen and valuable chemicals. The photocatalytic reforming of xylose (C5, C5H10O5) and glucose (C6) is particularly intriguing due to the abundance of these sugars in plant materials.5–8 Most research efforts have focused on glucose and only a limited number of studies reports on the photocatalytic reforming of xylose using semiconductors,9–11 overlooking the potential of xylose. This is notable, considering that hemicellulose, a polysaccharide made up of xylose, constitutes the second most abundant component in lignocellulosic biomass (5–35%).12 Furthermore, xylose production technologies are advancing each year, making it crucial to explore applications for xylose,13 such as its use in biomass reforming. Research on the photocatalytic reforming of xylose into clean fuels and valuable chemicals is essential.
Many metal-oxide catalysts have been used for biomass photo-reforming, especially TiO2,2 which is the most developed photocatalyst due to its advantages of good chemical stability, non-toxicity, and a low cost.3 However, its performance is limited by its ability to only absorb the UV light, which dissipates the abundance of solar light.2,14 CdS photocatalysts that are sensitive to visible light have also been used, but their high charge recombination and toxic cadmium content make them less feasible for the photocatalytic reaction.2,15 Due to the easy method for synthesis and a great physicochemical stability, graphitic carbon nitride (g-C3N4) is widely used for photocatalytic processes.16,17 However, its drawback in visible light absorption and charge recombination needs to be tackled.18 To overcome these shortcomings, graphene-based materials were developed and have been emerged as a promising photocatalyst because of their excellent photo-conductivity, tunable electronic-structure, environmental friendliness, cheapness, and light-absorbing properties.19,20
Notably, graphene oxide dots (GODs) exhibit exceptional property-tunability owing to their quantum size effect and chemical-modulation readiness.21 With sizes typically below 10 nm, GODs showcase unique size-dependent electronic and optical behaviors, enabling precise control over their bandgap and versatile applications.22 The high surface area of GODs provides abundant active sites for catalytic reactions, making them highly efficient in photocatalysis.23 Their excellent photoluminescence, biocompatibility, chemical stability, and low toxicity further enhance their wide appeal, particularly in biomedical and environmental applications.24 Regarding chemical modification of GODs, the introduction of sulfur and nitrogen has been reported to narrow the bandgap, repair vacancy defects, and facilitate charge separation, thereby resulting in enhanced harvesting of solar light, promoted delocalization of photogenerated charges, and improved charge reactivity on the catalyst surface.8,25–27
The challenge of low photocatalytic activity often arises due to the rapid recombination of photogenerated carriers and the inefficient use of surface charges within the photocatalyst.3 To address this issue, incorporating a co-catalyst with the photocatalyst has proven effective. The co-catalyst can enhance the separation of photogenerated electron–hole pairs and serve as an active site, facilitating crucial interfacial reactions.28 Previous studies have highlighted the effectiveness of co-catalyst nanoparticles like Pt, greatly improving photocatalytic H2 production.29 Ag was seldom used as a co-catalyst for photocatalytic H2 production due to the sluggish interfacial H2 evolution reaction.29,30 Nevertheless, Ag enhanced the photocatalytic decomposition of various organic species.31 Regardless of the H2 evolution efficiency, the potential impact of co-catalysts on the photocatalytic biomass reforming into valuable chemicals demands in-depth studies. Therefore, investigating the different effects of Ag and Pt as co-catalysts for the current system is expected to shed light on the transfer patterns of photogenerated charges and the correlation between reductive H2 evolution and oxidative biomass reforming.
In this study, we used sulfur- and nitrogen-doped graphene oxide dots (SNGODs) as the model photocatalyst to examine the influence of the co-catalysts, Pt and Ag, on the photocatalytic reforming of xylose. Under simulated solar irradiation, the co-catalyst-deposited SNGODs, Pt- and Ag-SNGODs, reformed xylose into its biomass derivatives with simultaneous H2 production. Co-catalysts Pt and Ag resulted in different biomass reforming pathways, as a consequence of different charge transfer behaviors between the catalyst and co-catalyst. Detailed reaction mechanisms of xylose conversion into its derivatives were elucidated in this study. Our results indicated that Pt-SNGODs were active in H2 production from the photocatalytic reduction whereas the oxidative xylose reforming mainly produced formate, which is an important commodity in the chemical, agricultural, textile, leather, medicinal, and rubber sectors.32,33 When Ag-SNGODs were used, the xylose reforming system mainly produced gluconic acid and acetic acid, which were important chemicals in food, cleaning, medicine, and agriculture industries.34,35 The present work provides a conceptual interpretation on the product selectivity in photocatalytic biomass reforming based on the selection of the co-catalyst.
Result and discussion
Characterization of the catalyst
The SNGODs were synthesized by annealing graphene oxide (GO) sheets in sulfur and NH3 at 600 °C, oxidizing the annealed product into dots in HNO3 solution, and finally treating the dots in NH4OH solution at 140 °C.20 SNGODs and GO sheets were mixed with H2PtCl6·6H2O or AgNO3 in an aqueous xylose solution to proceed with photo-deposition of Pt or Ag under simulated 1-sun solar irradiation, to produce Pt-SNGODs or Ag-SNGODs, respectively. The GO sheets were used to collect the photogenerated electrons from SNGODs for the deposition of Pt or Ag, which served as H2 generation sites.36 Fig. S1a† presents the transmission electron microscopy (TEM) images of SNGODs and the inset histogram illustrates that SNGODs exhibited a particle distribution around 2–5 nm, with a predominant size of 3–4 nm. A lattice d-spacing of 0.213 nm shown in the high-resolution image (Fig. S1b†) represents the graphene (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 00) planes.37 The TEM images of Pt-SNGODs and Ag-SNGODs along with the GO sheets are shown in Fig. S2 and S3,† respectively. Fig. S2a and S3a† present that both the SNGOD (with a gray color) and co-catalyst (with a darker color) particles were distributed over the GO sheets. Lattice d-spacing of 0.232 and 0.236 nm in the high resolution images of Fig. S2b and S3b† represent the Pt (111) and Ag (111) planes, respectively, indicating successful deposition of the co-catalysts. These TEM images also indicate the high crystallinity of the SNGOD particles, which benefits light absorption and prevents charge recombination, thereby augmenting the photocatalytic activity.21
00) planes.37 The TEM images of Pt-SNGODs and Ag-SNGODs along with the GO sheets are shown in Fig. S2 and S3,† respectively. Fig. S2a and S3a† present that both the SNGOD (with a gray color) and co-catalyst (with a darker color) particles were distributed over the GO sheets. Lattice d-spacing of 0.232 and 0.236 nm in the high resolution images of Fig. S2b and S3b† represent the Pt (111) and Ag (111) planes, respectively, indicating successful deposition of the co-catalysts. These TEM images also indicate the high crystallinity of the SNGOD particles, which benefits light absorption and prevents charge recombination, thereby augmenting the photocatalytic activity.21
The full-range X-ray photoelectron spectroscopy (XPS) spectrum of SNGODs was depicted in Fig. S4a,† which includes S2p (168 eV), S2s (233 eV), C1s (285 eV), N1s (401 eV), and O1s (532 eV) peaks. Sulfur and nitrogen atoms were successfully incorporated into the graphene oxide structure as a result of the treatment. In Table S1,† the atomic ratios of O1s/C1s, N1s/C1s, and S2p/C1s were determined, by the XPS analysis, to be 40%, 11%, and 7%, respectively. Fig. S4b–d† were the focused regimes of Fig. S4a† to clearly show the peaks of C1s, S2p, and N1s, respectively. Using a Gaussian function, the C1s, N1s, and S2p spectra were deconvoluted into their constituting peaks. The C1s peak in Fig. S4b† contained C–C (284.6 eV), C–N (285.9 eV), C–O (286.5 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288 eV), and O–CO (288.7 eV) bonds.38 A nitrogen content due to the nitrogen-doping modification increased the catalyst activity.39 The S2p peak (Fig. S4c†) contained –SH (162.3 eV), C–S–C (163.9 eV), CS (165.1 eV), C–SO2 (168.3 eV), and C–SO3 (170.5 eV) with C–SO2 group give the highest composition (5.7% out of 7%) among the sulfur functionalities.26,27 The sulfur-functionalities at the graphene periphery were highly polar, attractive to photogenerated holes, and functioned as oxidizing sites during photocatalysis.26 The N1s peak (Fig. S4d†) was deconvoluted into pyridinic (398.4 eV), amino (399.2 eV), pyrrolic (399.6 eV), quaternary (400.7 eV), and amide (401.2 eV).40 The quaternary and amide were the major functionality components (Table S1†). The quaternary functionality patched the vacancy defects in the basal plane, while the amide functionality improved orbital conjugation.24
O (288 eV), and O–CO (288.7 eV) bonds.38 A nitrogen content due to the nitrogen-doping modification increased the catalyst activity.39 The S2p peak (Fig. S4c†) contained –SH (162.3 eV), C–S–C (163.9 eV), CS (165.1 eV), C–SO2 (168.3 eV), and C–SO3 (170.5 eV) with C–SO2 group give the highest composition (5.7% out of 7%) among the sulfur functionalities.26,27 The sulfur-functionalities at the graphene periphery were highly polar, attractive to photogenerated holes, and functioned as oxidizing sites during photocatalysis.26 The N1s peak (Fig. S4d†) was deconvoluted into pyridinic (398.4 eV), amino (399.2 eV), pyrrolic (399.6 eV), quaternary (400.7 eV), and amide (401.2 eV).40 The quaternary and amide were the major functionality components (Table S1†). The quaternary functionality patched the vacancy defects in the basal plane, while the amide functionality improved orbital conjugation.24
Moreover, we performed XPS analysis on Pt-SNGODs and Ag-SNGODs to examine the states of the deposited Pt and Ag. Fig. S5a and b† shows the full-range XPS spectra of Pt-SNGODs and Ag-SNGODs, respectively, and depicts the appearance of Pt4f and Ag3d signals from the co-catalysts.41 In Fig. S5c,† the Pt4f spectrum displays two characteristic peaks at 71.7 and 75.1 eV, corresponding to the 4f7/2 and 4f5/2 states of Pt. After deconvolution, the Pt4f spectrum comprises four peaks at 71.7, 72.9, 75.1, and 77.1 eV. The binding energies at 71.7 and 75.1 eV correspond to the Pt0 state whereas those at 72.9 and 77.1 eV correspond to Pt2+ of oxide PtO.41,42 The compositions, calculated based on the peak areas, were 86% Pt and 14% PtO on Pt-SNGODs. In Fig. S5d,† the Ag3d spectrum exhibits two characteristic peaks of the 3d5/2 and 3d3/2 states of Ag and comprises peaks identified as Ag0 states at 367.8 and 373.8 eV and Ag+ states of oxide Ag2O at 367.5 and 373.4 eV.43,44 The compositions are 94% Ag and 6% Ag2O on Ag-SNGODs. This XPS analysis indicated that the deposited Pt and Ag were mainly present in the metallic state on catalyst SNGODs.
The optical absorption spectra of the aqueous suspensions of SNGODs and co-catalyst-deposited SNGODs at 0.18 g L−1 are shown in Fig. 1. All the suspended catalysts exhibited a broad absorption in the UV and visible light regions. The spectra of the suspensions showed a prominent absorption band at ∼300 nm, which corresponded to the energy necessary for the π → π* (the anti-bonding π orbital) transition in the sp2 domains of graphene-based materials.45–47 Heteroatom nonbonding electron states conjugated with the graphitic-π orbital have been recognized as the n-state.23,48,49 An absorption occurred at 300–400 nm, suggesting an n → π* transition due to the O atoms in CO and C–O.45 An n → π* transition also occurred in the light absorption region (wavelengths over 400 nm), suggesting a transition corresponding to CN and CS.24 Doping graphene with O, N, and S heteroatoms leads to enhanced visible light absorption, which is beneficial for using solar irradiation in the photocatalytic reaction. Fig. 1 also indicates that deposition of both co-catalysts (Pt and Ag) increased the catalysts' absorption ability, indicating that the co-catalysts extracted the photogenerated electrons from the conduction band (CB) and enhanced the excitation of electrons on the valence band (VB) through light absorption. The absorption enhancement was slightly stronger when deposited with Pt. We did not detect any surface plasmon resonance signals of Ag nanoparticles from the spectra (normally with absorption at ∼500 nm) of the Ag-SNGOD suspension. Deposition of Ag nanoparticles on the GO sheets, rather than on the SNGOD catalyst surface, might explain the absence of the plasmonic effect. In addition, the small size of the Ag nanoparticles fell outside the range of 10–100 nm, which typically supports surface plasmon resonance.50 When dispersed in aqueous suspensions containing xylose as a hole-scavenger (@xylose), the catalysts' absorbance was significantly increased in the entire wavelength regime (see also Fig. 1). Pt-SNGODs@xylose exhibited higher absorbance in the 350–600 nm regime than did Ag-SNGODs@xylose. A control spectrum for xylose in an aqueous solution (10 g L−1) was provided to elucidate the influence from xylose. Fig. S6† shows the spectrum of the xylose solution, which did not exhibit detectable absorbance from xylose but the absorbance by water at ∼210 nm.
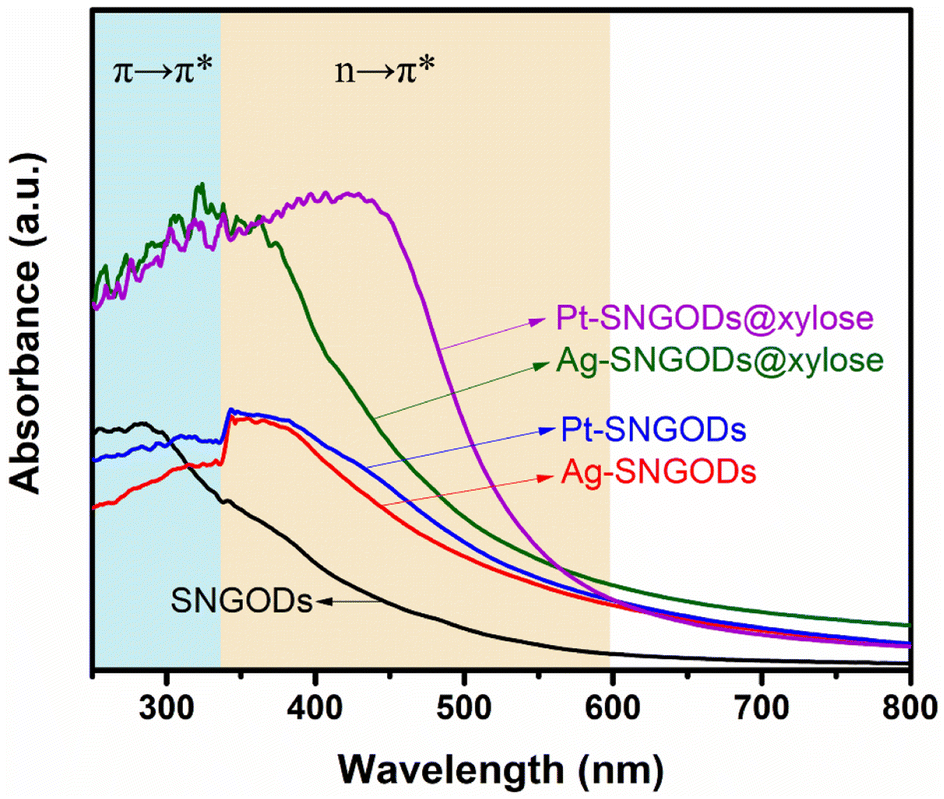 | ||
| Fig. 1 Optical absorption spectra of the aqueous suspensions of SNGODs, Pt-SNGODs, Ag-SNGODs, Pt-SNGODs@xylose, and Ag-SNGODs@xylose. The absorption was arisen from the π → π* and n → π* transitions. | ||
The absorption analysis indicated that, in the bare SNGODs, the dynamics for electron excitation from the VB to CB was governed by the rate of extracting the photogenerated electrons at the CB; the co-catalysts substantially enhanced electron extraction and therefore the light absorbance. When the co-catalysts were effective in extracting electrons from the CB, the electrons availability at the VB would become the bottleneck governing the photo-excitation dynamics. In the presence of the hole-scavenger, xylose that donated electrons to the VB, the Pt- and Ag-SNGODs@xylose exhibited substantially enhanced light absorption due to the high availability of electrons at their VB. The higher absorption of Pt-SNGODs@xylose, relative to that of Ag-SNGODs@xylose, indicated a stronger electron-extracting ability of Pt.
The catalyst samples were also subjected to photoluminescence (PL) spectroscopic analysis with excitation at 405 nm. Fig. S7† shows that the PL intensity decreased with co-catalyst deposition and xylose presence. The decrease in the PL intensity indicated that the electron extraction by the co-catalysts from the CB and the further hole extraction by xylose from the VB had minimized the recombination of charges and thus the PL.39,51–53 The time-resolved photoluminescence (TRPL) spectra of the catalysts under 405 nm excitation were presented in Fig. S8.† The TRPL decay curves were simulated using exponential intensity function of R(t) = B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e−t/τ, where R is the radiative recombination intensity, t is the elapsed time after excitation, B is a constant, and τ is emission lifetime.25 The emission lifetimes of the catalysts were also indicated in Fig. S8 and Table S2.† The Pt and Ag co-catalysts reduced the emission lifetime of the SNGODs because the co-catalysts quickly extracted the photogenerated electrons. Pt exhibited a more effective extraction than did Ag and resulted in a shorter lifetime.52,53 The presence of xylose lowered the τ value of Pt-SNGODs whereas a raised value was detected in Ag-SNGODs@xylose. The lowered value of Pt-SNGODs@xylose was due to the fact that the Pt co-catalyst effectively extracted the electrons donated by xylose (to the VB then to the CB). In contrast, the Ag co-catalyst could not effectively extract the donated electrons, thereby resulting in a prolonged PL emission.
e−t/τ, where R is the radiative recombination intensity, t is the elapsed time after excitation, B is a constant, and τ is emission lifetime.25 The emission lifetimes of the catalysts were also indicated in Fig. S8 and Table S2.† The Pt and Ag co-catalysts reduced the emission lifetime of the SNGODs because the co-catalysts quickly extracted the photogenerated electrons. Pt exhibited a more effective extraction than did Ag and resulted in a shorter lifetime.52,53 The presence of xylose lowered the τ value of Pt-SNGODs whereas a raised value was detected in Ag-SNGODs@xylose. The lowered value of Pt-SNGODs@xylose was due to the fact that the Pt co-catalyst effectively extracted the electrons donated by xylose (to the VB then to the CB). In contrast, the Ag co-catalyst could not effectively extract the donated electrons, thereby resulting in a prolonged PL emission.
Regarding the transfer feasibility of the photogenerated charges, the electronic structure of photocatalysts plays a vital role to govern the redox progress during photocatalytic reactions. We subjected SNGODs, Pt-SNGODs, and Ag-SNGODs to analysis with ultraviolet photoelectron spectroscopy (UPS) to assess their valence band maximum (VBM). By deducting the width of the UPS spectra (Fig. S9†) from the excitation energy of 21.2 eV, the VBM values for SNGODs, Pt-SNGODs, and Ag-SNGODs were determined to be −6.46 eV, −5.84 eV, and −5.95 eV (vs. vacuum), respectively. The absorption spectra of Fig. 1 were converted to the Tauc plots (Fig. S10†) to obtain bandgap energies of 2.7, 2.1, and 2.2 eV for the SNGODs, Pt-SNGODs, and Ag-SNGODs, respectively. The conduction band minimum (CBM) level of the catalysts was determined according to the VBM levels and bandgap energies. Fig. 2 presents the summary of the electronic state levels of the catalysts in comparison with the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) levels in an aqueous solution at pH = 9. The Pt and Ag co-catalysts elevated the CBM levels and decreased the bandgap energy of SNGODs.
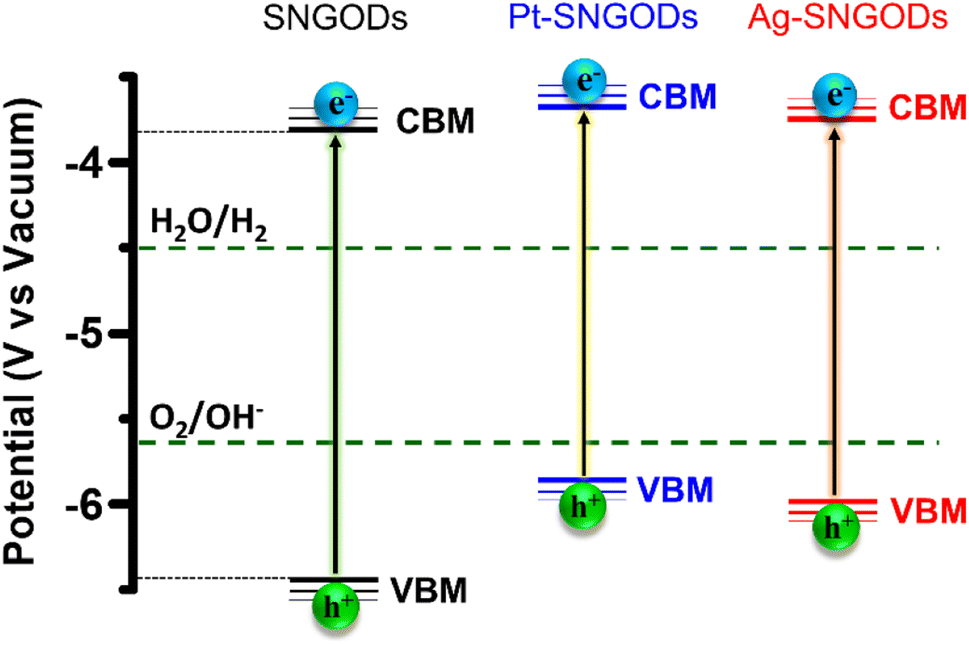 | ||
| Fig. 2 Electronic-energy level diagrams of the SNGODs, Pt-SNGODs, and Ag-SNGODs in relation to the levels for H2 and O2 evolution reactions (HER and OER) in an aqueous solution of pH = 9. | ||
Photocatalytic reforming of xylose
Xylose is highly soluble in water due to its polar hydroxyl functional group.54 In this study, an aqueous solution (250 mL) of xylose (1 g) at pH = 9 was used for the photocatalytic HER and xylose reforming.8,55 The photocatalytic reaction was conducted in a gas-enclosed system with side solar irradiation (AM 1.5 G at 100 mW cm−2). To initiate the reaction, an equivalent amount of 5 wt% co-catalyst (Pt or Ag) precursors was charged into the solution containing SNGODs (0.2 g) and GO sheets (0.006 g). Under irradiation, the photogenerated electrons in SNGODs would be injected onto the GO sheets and therefore the co-catalyst nanoparticles were preferentially deposited on the GO sheets, as indicated in Fig. S2 and S3.† The presence of GO sheets in the system located the HER sites on the GO and protected SNGODs from the damages caused by the H2 gas evolution.53,56–58 The photocatalytic reaction was undergone under a pressure of 75 mmHg.Fig. 3 presents the amounts of H2 produced from the system containing Pt-SNGODs and Ag-SNGODs. The reactions were conducted for 72 h with evacuating interventions every 12 h irradiation, and only H2 was detected as the gas phase product (Fig. S11†). In the first 12 h, the H2 produced was low because the deposition of the co-catalysts on the GO sheets was in progress in the initial period.20 Both catalysts exhibited the highest HER activity after 36 h of the reaction. The total amount of H2 generated over Pt-SNGODs was 7.7 times that over Ag-SNGODs. Although Ag-SNGODs was less active in HER, it was superior in the aspect of long-term stability. An intensive occurrence of HER on the Pt-SNGODs might have destroyed the GO sheets that were deposited with Pt, affecting the long-term stability of the Pt-SNGODs. Direct detection of the GO deterioration during the photocatalysis would be difficult. Nevertheless, we have conducted experiments to explicitly demonstrate the critical role of the GO sheets in the reaction. Fig. S12† presents the influence of the GO incorporation in the H2 evolution from the xylose reforming over Pt-SNGOD catalysts. In the presence of the GO, the H2 evolution was efficient and stable for at least 48 h, whereas the evolution exhibited serious decay without the GO incorporation. The incorporation of the GO sheets has resulted in the relocation of the H2 evolution sites from SNGODs to the GO sheets, thereby minimizing the attack on the SNGODs by the reaction intermediates. This comparison indicated that the declined long-term stability of Pt-SNGODs could be attributed to the destroying GO sheet.
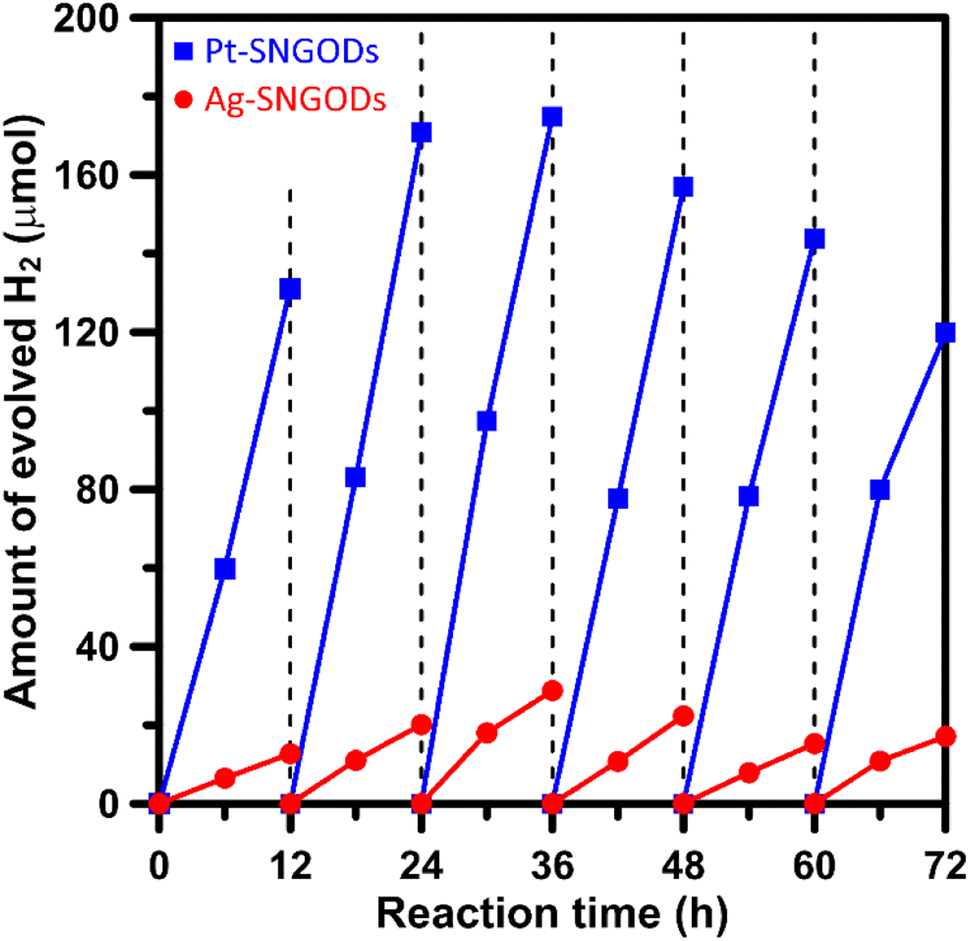 | ||
| Fig. 3 Evolution of H2 from the xylose solutions containing Pt-SNGODs and Ag-SNGODs under simulated solar irradiation (AM 1.5, 100 mW cm−2). | ||
We have calculated the apparent quantum yields (AQYs) for H2 production under 420 nm monochromatic illumination. The AQYs for the Pt-SNGOD and Ag-SNGOD systems were 6.8% and 2.8%, respectively. Table S3† presents the H2 production results reported from literature for photocatalytic reforming of mono-sugar solutions. Our results were comparable to those obtained from other photocatalytic systems. Reporting photocatalytic activity based on per gram of catalyst can be misleading, as the activity does not scale linearly with mass.59,60 The light harvesting efficiency depends on the catalyst dispersion situation and light scattering effects. Therefore, we have provided the AQYs for the H2 production.
The low effectiveness of Ag in donating electrons for HER would lead to the accumulation of photogenerated electrons on the SNGODs. The electron presence can be explored using an electron-trapping agent, 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), which is a radical and its signal in the electron paramagnetic resonance (EPR) spectrum is quenched after trapping an electron from the environment. With the addition of TEMPO, the reacting solutions after irradiation, in the presence or absence of xylose, were analyzed using EPR. Fig. 4a presents the xylose-absent EPR spectra, in which the 1:1:1 signal intensity of the added TEMPO would be reduced with electron trapping.61 Under the xylose-absent condition, the Ag-SNGOD solution exhibited a much lower EPR signal than the Pt-SNGOD solution, indicating that a large number of photogenerated electrons on Ag-SNGODs had interacted with the TEMPO radicals to minimize the EPR signal of TEMPO. Fig. 4b presents the xylose-present EPR spectra. Comparison of Fig. 4a and b depicted that the TEMPO signal with the Ag-SNGODs was substantially increased by the xylose addition, indicating that xylose acted as an acceptor to consume the photogenerated electrons on the Ag-SNGODs and to result in the increased TEMPO signal. As to the Pt-SNGOD solution, Pt is efficient in transferring electrons for the HER. Xylose acts as a sacrificial electron donor to supply the catalyst electrons and promote the electron-trapping probability for TEMPO, thereby leading to a slight decrease in the EPR signal intensity for Pt-SNGOD solution. This EPR analysis indicated that xylose might have accepted the accumulated electrons in the Ag-SNGODs in addition to acting as an electron donor to the VB of the SNGODs.
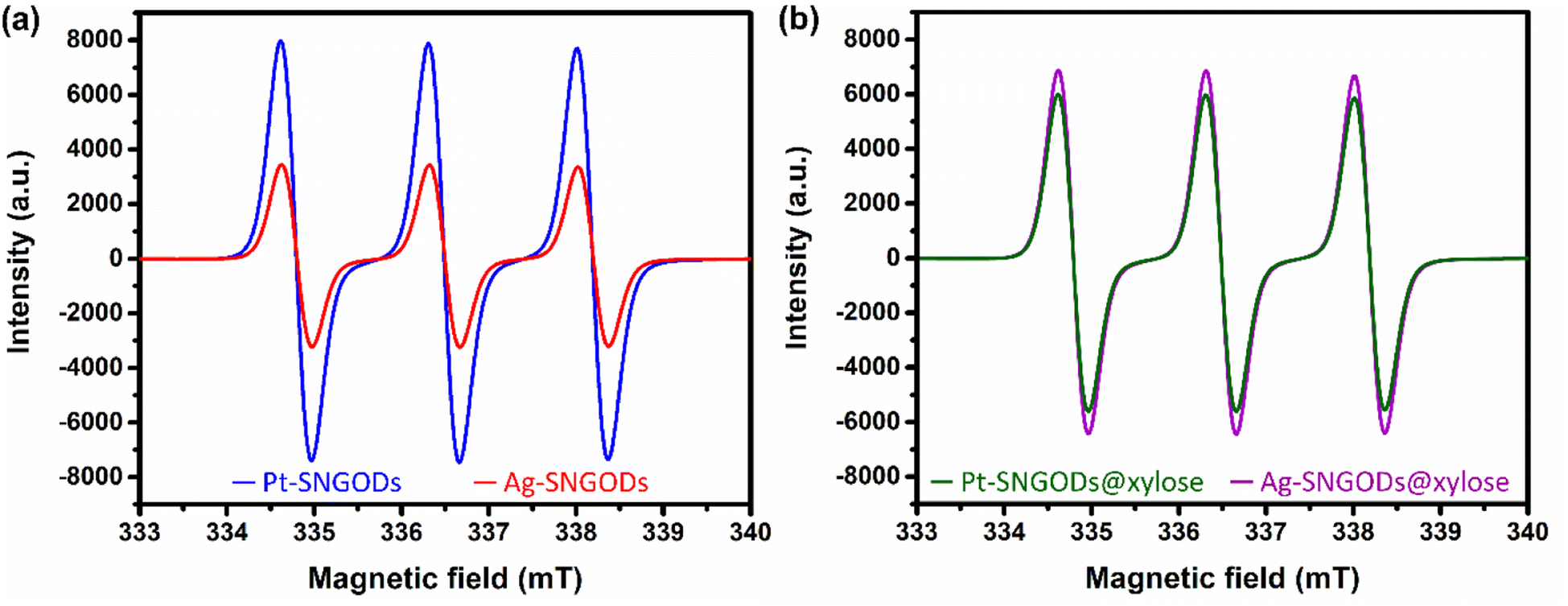 | ||
| Fig. 4 The EPR signals of TEMPO contained in the Pt-SNGOD and Ag-SNGOD reacting solutions in the (a) absence or (b) presence of xylose. The solutions were irradiated with light of 462 nm in wavelength. | ||
Besides the photocatalytic HER, the oxidative photoreforming of xylose into other chemical compounds was investigated. In the liquid phase, ring opening of xylose, after interacting with water, would occur prior to xylose oxidation by the photogenerated holes.62,63 The liquid products from photoreforming over Pt- and Ag-SNGODs were identified by the GC-MS and LC-MS analyses. GC-MS and LC-MS were used to identify products with mass-to-charge ratios (m/z) less than 50 and more than 50, respectively.20 Before analysis, the samples were filtered to remove the catalyst particles. LC-MS analysis was conducted in a negative-ion mode, which created a stable deprotonated ionic molecule, namely, [M–H]−.64 Deprotonation allowed the molecular weight in solution samples to be determined. The precise chemical structure identification of these compounds was then validated using the LC-MS-MS analytical findings, in which the parent compounds were fragmented as seen in Fig. S13–S27.† Meanwhile, the products confirmed from the GC-MS analysis were shown in Fig. S28–S31,† with the MS analysis validated the structures of the GC-MS products (Fig. S29 and S31†). For the quantitative analysis, the amounts of the products were determined based on the prepared standard samples.
After reaction for 72 h, Fig. 5 shows that Pt-SNGODs produced a greater variety of products than Ag-SNGODs, i.e., 14-vs.-9 in the compound type. The LC-MS analyzed products from Pt-SNGODs were C6 (C6H11O7−, gluconate), r-C5 (C5H10O5, xylose), C5 (C5H9O6−, xylonate), r-C4 (C4H8O4, erythrose), C4 (C4H7O5−, 2,3,4-trihydroxybutanoate), r-C3 (C3H6O3, glyceraldehyde), C3 (C3H5O4−, glycerate), C3b (C3H3O2−, 3-hydroxy-2-propenal), r-C2 (C2H4O2, glycolaldehyde), and C2 (C2H3O3−, glycolate). For Ag-SNGODs in the LC-MS analysis, the products were C6, r-C5, r-C3, C3, and r-C2. Meanwhile, in the GC-MS analysis, both catalysts generated the same types of products, namely C2b (C2H3O2−, acetate), C3a (C3H5O2−, propanoate), C1 (CHO2−, formate), and C2a (C2H6O2, 1,2-ethanediol). Fig. 5 shows that C6 was the major product in the liquid phase for Ag-SNGODs, whereas C1 was the main liquid product of photocatalytic reforming with the Pt co-catalyst.
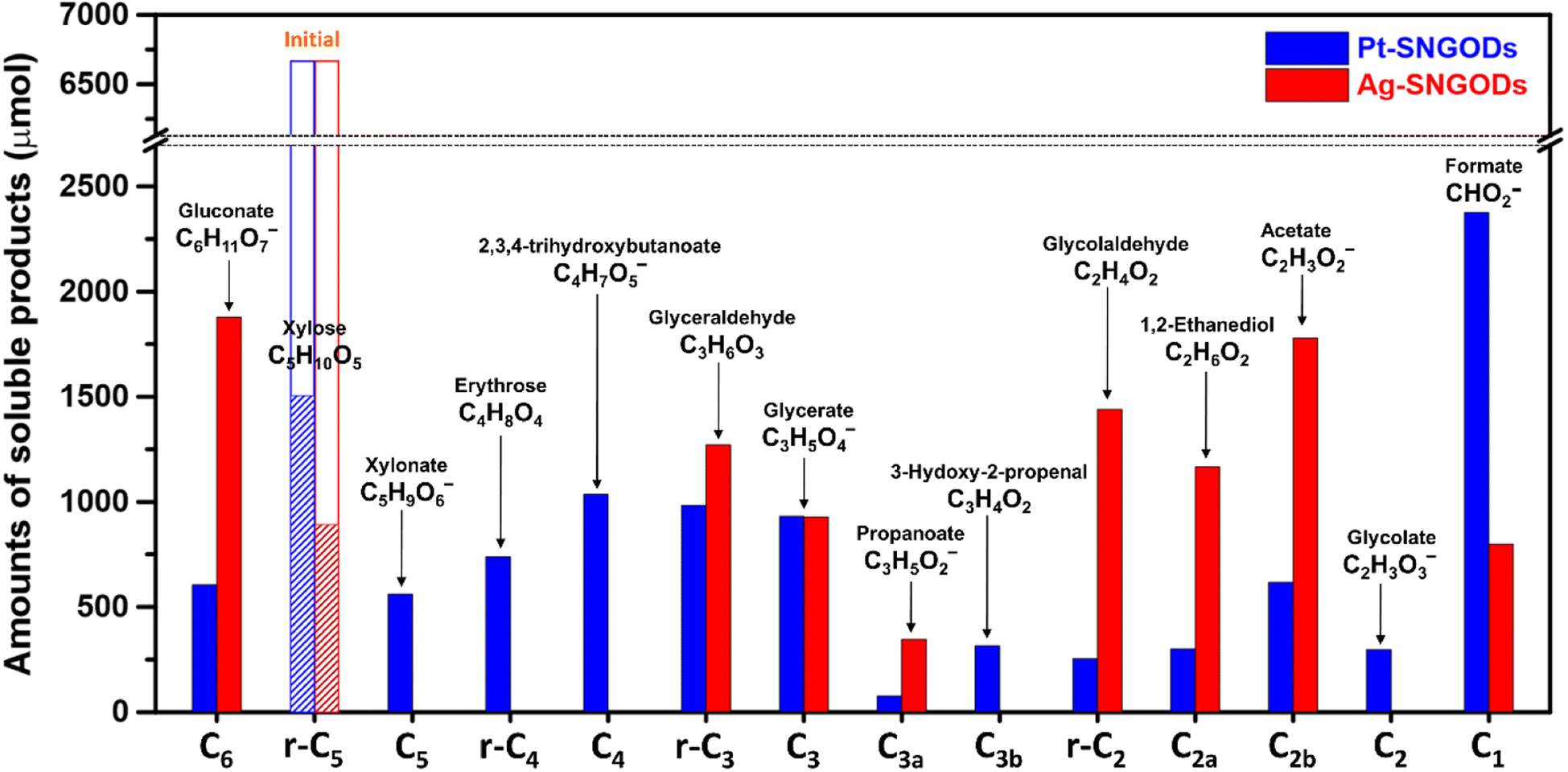 | ||
| Fig. 5 Product compositions in the liquid phase after the photocatalytic reforming of xylose for 72 h. | ||
Mechanism of xylose reforming over Pt- and Ag-SNGODs
The products from photocatalytic xylose reforming over Pt-SNGODs comprised the r-Cx and Cx species with x ranging through 1–5, indicating that the reforming proceeded through successive alternation of C-eliminating hydrolysis and photocatalytic oxidation.20Fig. 6 presents the Pt-SNGODs mechanism proposed based on the products shown in Fig. 5. The ring-form xylose feed was firstly opened into linear-form xylose (r-C5), which was then converted into carboxylate C5 through photocatalytic oxidation. The downgrade reforming of r-C5 to the terminal C1 comprised successive alternating steps: photocatalytic oxidation of the aldoses/aldehydes/alcohols r-Cx to carboxylates (Cx) prior to C-eliminating hydrolysis by removing one formate (C1) molecule.65 Along with the photocatalytic oxidation, water reduction by the photogenerated electrons was taking place to produce H2. Apart from the principal pathway of successive photocatalytic oxidation and hydrolysis, dehydration of r-C3 might have occurred to produce C3H4O2 (C3b), which was detected in the product analysis. Because of the occurrence of photocatalytic HER in the solution, hydrogenolysis of C3 and r-C2 might have occurred to produce C3H5O2 (C3a) and C2H6O2 (C2a), respectively (Fig. 6). C2a might be photocatalytically oxidized to C2H3O2− (C2b). The branched hydrogenolysis pathways must have played an important role in the reforming over Ag-SNGODs because of the high contents of C2a, and C2b in the products (see Fig. 5).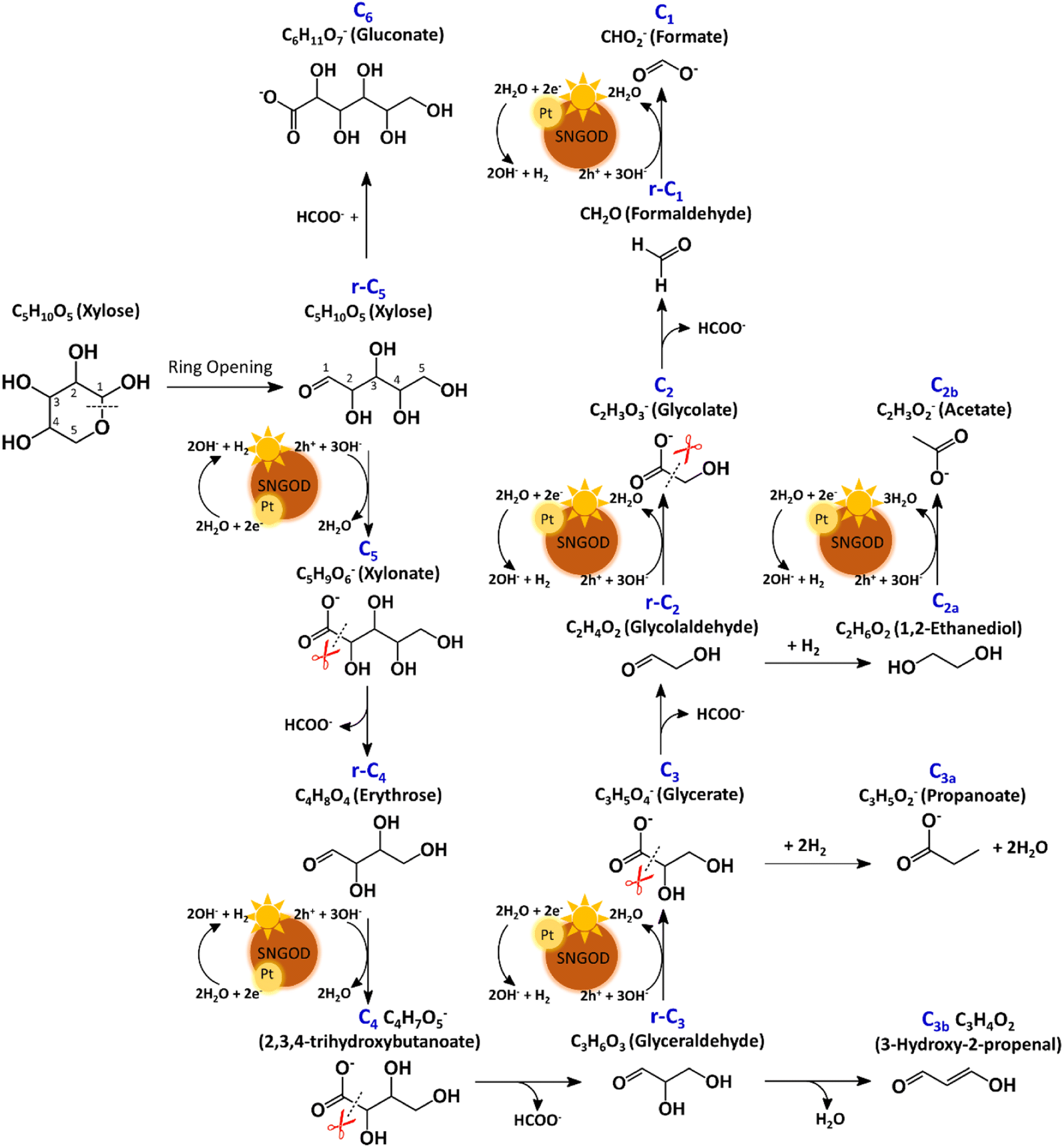 | ||
| Fig. 6 Proposed mechanism of photocatalytic reforming of xylose over Pt-SNGODs. | ||
In the photo-reforming reaction using Ag-SNGODs, Fig. 5 shows that the products in the solution comprised high proportions of C3- and C2-compounds but no C4-compounds, indicating that r-C5 proceeded with cleavage on C2–C3 through the retro-aldol reaction mechanism to produce r-C2 and r-C3, as indicated in Fig. 7.66 The retro-aldol reaction generally takes place in a basic environment and could be catalyzed by a Lewis base.67 The r-C2 and r-C3 from the cleavage were further downgraded to C1 through steps similar to those proposed for the reforming over Pt-SNGODs (see the comparison between Fig. 6 and 7). Notably, the content of C2b (an acetate), derived from C2a (an alcohol), was high over Ag-SNGODs relative to that over Pt-SNGODs. The hydrogenation of r-C2 (an aldehyde) into C2a was more effective than the photocatalytic oxidation of r-C2 into C2 (a carboxylate), attributable to the insufficient photocatalytic activity of Ag-SNGODs. As to the C–C coupling interaction, C6 was profoundly produced from the photoreforming over Ag-SNGODs, with which the contents of r-C5 and C1 in the solution were substantially lower than those in the solution for the reforming over Pt-SNGODs (Fig. 5). The coupling of r-C5 with C1 must have been responsible for the formation of C6. A study reported that C1 reacted with r-C5 through a nucleophilic addition reaction,68 in which the carboxyl group of C1 attached itself to the formyl group on r-C5. The coupling of r-C5 with C1 was effective in the reforming over Ag-SNGODs (Fig. 7).
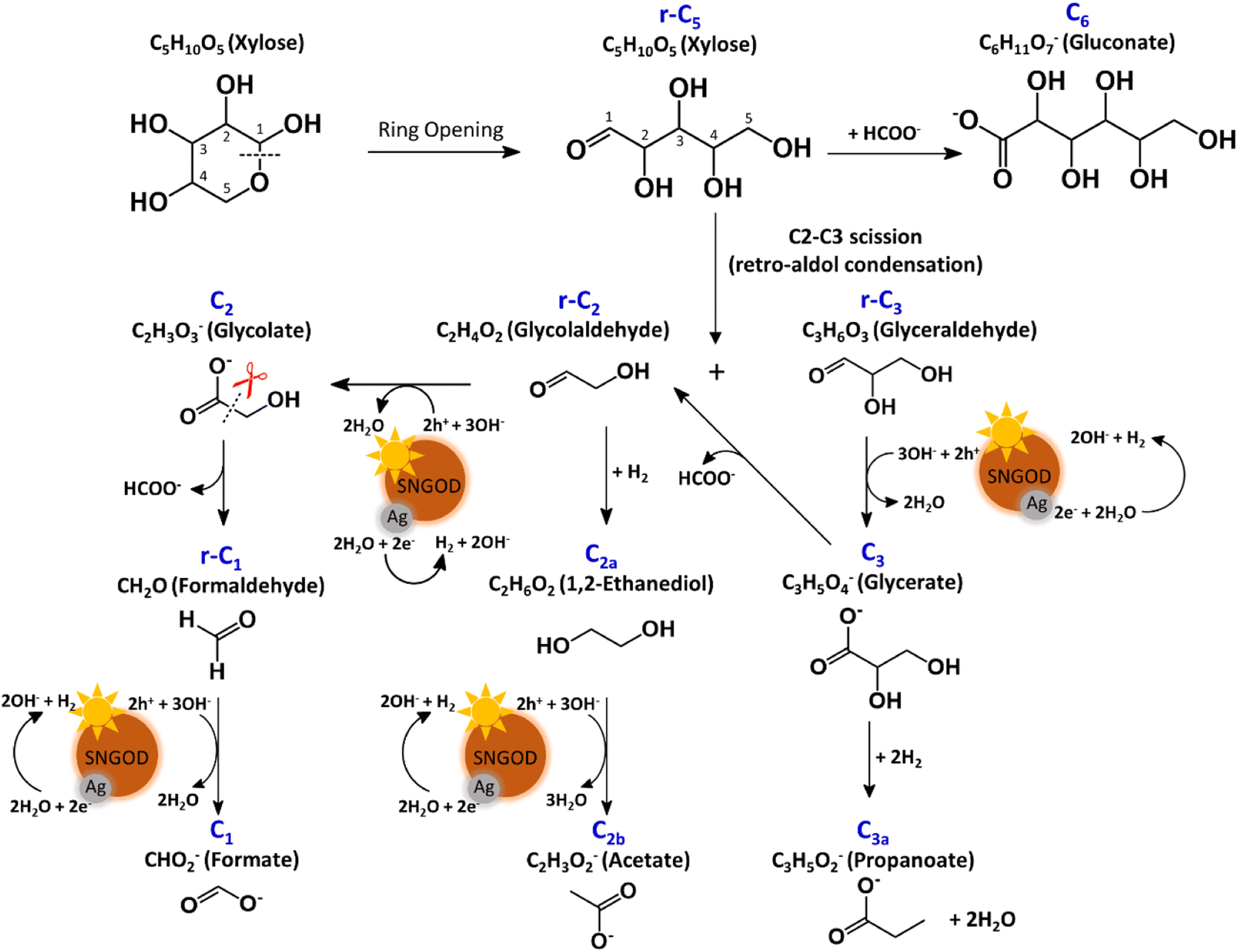 | ||
| Fig. 7 Proposed mechanism of photocatalytic reforming of xylose over Ag-SNGODs. | ||
To test the proposed mechanistic pathways, we randomly performed photocatalytic reforming of intermediates glycolaldehyde and glyceraldehyde (rC2 and rC3, respectively) over Pt-SNGODs and Ag-SNGODs for HER. Fig. S32† shows that both reforming reactions using rC2 and rC3 produced H2, with higher HER rates over Pt-SNGODs. The results of the tests on rC2 and rC3 were consistent with mechanistic pathways proposed in Fig. 6 and 7.
Fig. 8 presents the charge distributions of the irradiated SNGODs when associated with different co-catalysts. The Pt-SNGODs effectively donated electrons into the aqueous solution to produce H2, resulting in the effective oxidation of xylose by the photogenerated holes. The oxidation occurred on the aldehyde end of xylose because the electron-rich oxygen of the formyl group attracted the hole-loaded SNGODs. In contrast, the Ag-SNGODs were less effective in donating electrons for HER, thereby resulting in the accumulation of electrons on the GO sheets and the low efficiency of hole donating to oxidize xylose, even if the holes on the SNGODs (acting as a Lewis acid) still attracted the formyl oxygen of the xylose. With the electron accumulation, the GO sheets would interact with the hydroxyl group on the C3 of xylose, converting xylose into an alkoxide intermediate where the oxygen atom at C3 carried a negative charge. This intermediate would then proceed with the retro-aldol reaction for breaking the C2–C3 bond to produce the r-C2 and r-C3 fragments. The interaction between xylose and the accumulated electrons has been reflected by the EPR analysis shown in Fig. 4.
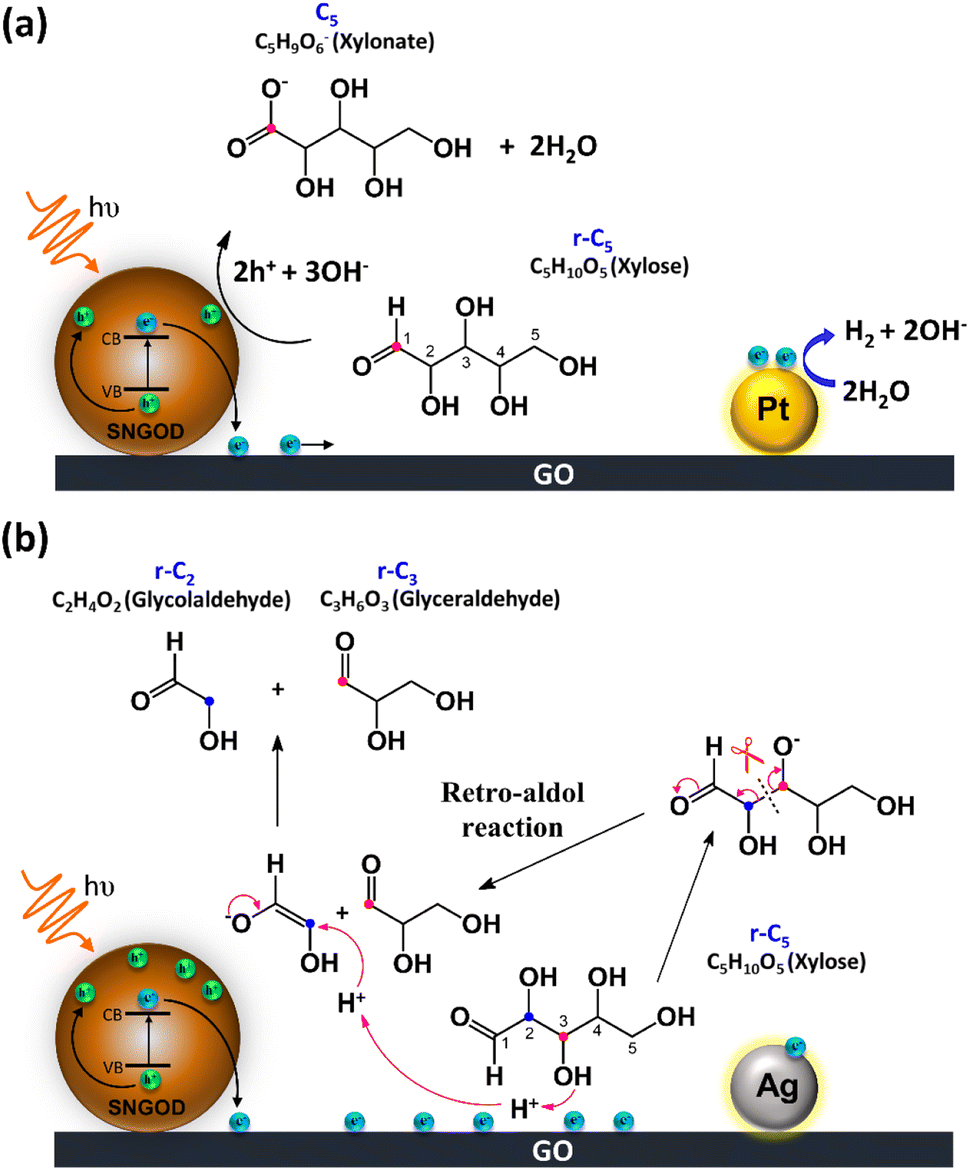 | ||
| Fig. 8 The charge distributions of the SNGODs when associated with different co-catalysts. (a) Pt co-catalyst for oxidative xylose reforming (Pt-SNGODs were active in consuming photogenerated electrons for H2 evolution). (b) Ag co-catalyst for retro-aldol reaction (Ag-SNGODs were less effective in HER). | ||
The lower xylose-oxidizing ability of Ag-SNGODs, relative to that of Pt-SNGODs, also resulted in a higher rate in the C–C coupling of radicals from reductions to produce C6. According to Fig. 3, Ag-SNGODs were not sufficiently effective in coupling H-radicals, derived from photocatalytic reduction of H2O,69 for the occurrence of HER. The abundant xylose and formate in the reacting system would interact with the H-radicals to produce r-C5- and C1-derived radicals, which would subsequently proceed with C–C coupling to form C6. This explains the high C6 content in the products of the photocatalytic xylose reforming over Ag-SNGODs. This study has demonstrated how the co-catalyst property affected the charge accumulation degree and thus the product formation mechanism.
Conclusions
This study explores the impact of co-catalysts (Pt and Ag) when combined with the SNGOD photocatalyst on the photocatalytic reforming of xylose (C5) into H2 and C1–C6 compounds. Pt was more effective than Ag in extracting photogenerated electrons for HER. The accumulation of electrons in Ag-SNGODs was disclosed using EPR analysis. Regarding the oxidative reforming of xylose, Pt-SNGODs effectively converted xylose into C5–C1, through successive alternation of C-eliminating hydrolysis and photocatalytic oxidation, and moderately produced C6 through C–C coupling. With Ag-SNGODs, the accumulated photogenerated electrons interacted with xylose to cleave the r-C5 (xylose), through the retro-aldol reaction, into r-C3 and r-C2, which were then successively decomposed into C3–C1. The EPR analysis evidenced the interaction between xylose and the electrons accumulated in Ag-SNGODs. A profound amount of C6 was produced over Ag-SNGODs, indicating that the HER-inactive Ag-SNGODs provided sufficient H-radicals to generate formate- and xylose-derived radicals for subsequent coupling into C6. Overall, this research has shed light on the selective production of products in photocatalytic biomass reforming, emphasizing the significance of co-catalyst selection.Experimental section
Synthesis of photocatalyst
Using a modified Hummers' method, GO was prepared from natural graphite powder (Bay carbon, SP-1, USA).70 5 g of graphite was mixed with 2.5 g of NaNO3 (≥99%, Merck, Germany) in 150 mL of concentrated H2SO4 (18 M, Wako, Japan). 15 g of KMNO4 (≥99%, J.T. Baker, USA) was gradually added with stirring while maintaining the temperature at 35 °C in an ice bath for 4 h. Then, 250 mL of deionized (D.I.) water was slowly added, and the mixture was stirred for 15 min at 98 °C. Another 800 mL of D.I. water was added and stirred for 30 min to terminate the oxidation process. The GO preparation was completed by adding 10 mL of H2O2. The resulting slurry was washed and filtered six times. GO was obtained by freeze-drying the slurry for 24 h.A ratio of 1:1 between GO and sulfur powder (99.999%, Acros, USA) was used to make the SNGODs by calcining the mixture in an NH3 flow (150 mL min−1) at 600 °C for 3 h. Every 0.2 g of the obtained mixture was oxidized in 60 mL of concentrated HNO3 (60%, Showa, Japan) under sonication for 12 h at room temperature. Then, the HNO3 was removed by evaporating the liquid phase at 140 °C, producing a dot sample. The dot samples were autoclaved in a NH3 solution (30 wt% in water, Sigma-Aldrich, USA) for 5 h at 140 °C. A heating process at 100 °C, along with stirring, was applied to remove the residual NH3 solution. To remove the large dots, the solution obtained was sieved with Sartocon Slice 200 with Hydrosart membranes (Satroius, Germany). Finally, the catalyst named SNGODs was ready to be used.
Photocatalyst characterization
Transmission electron microscopy (TEM, Jeol 2100 F, Japan) was used to determine the microstructure of the catalyst. The chemical composition of the catalysts was quantitatively analyzed using X-ray photoelectron spectroscopy (XPS) with AXIS Ultra DLD (Kratos, UK) equipped with Al Kα radiation. The C1s, N1s, and S2p spectra were split into component peaks using a Gaussian fitting method. The optical absorbance of the catalysts was measured using a Hitachi U-4100 (Japan) spectrophotometer. Suspensions with a concentration of approximately 0.18 mg mL−1 were put in a 1 cm quartz cuvette. The photoluminescence (PL) spectrum of SNGODs in aqueous solution (0.18 mg mL−1) was measured at ambient temperature using a fluorescence spectrophotometer (F-700, Hitachi, Japan). Time-resolved photoluminescence (TRPL) analysis was performed at an excitation wavelength of 405 nm using a spectrofluorometer (FS5, Edinburgh Instruments, UK).Photocatalytic activity measurements
A powder of xylose (Sigma-Aldrich, St. Louis, MO, USA) was used as a substrate or hole scavenger in all the experiments. 1 g of xylose was dissolved in 20 mL of D.I. water, and then 0.2 g of SNGODs, 0.006 g of GO, and 5 wt% of either H2PtCl6·6H2O (26 mg, 99%, Alfa Aesar, USA) or AgNO3 (25 mg, >99%, Acros Organics, UK) were added. The solution was diluted to 250 mL with D.I. water, and the pH was adjusted to 9. During all photocatalytic reactions, the closed glass gas circulation system was connected to a Pyrex reaction vessel with side-irradiation (469.2 mL with a flat illumination window of 4 cm in diameter) for side-irradiation. The source of the irradiation was a solar simulator (Oriel Instruments, 100 mW cm−2, USA) connected to an air mass AM-1.5 G filter. A photodetector (Oriel Instruments, model 71964, USA) was used to determine the intensity of the light irradiated on the system. The gas product was detected by a gas chromatograph (GC, Hewlett-Packard 7890, USA) with a molecular sieve 5A column, thermal conductivity detector, and argon as the carrier gas. The photo-reforming reaction was run for 3 days with evacuating interventions at 75 mmHg every 12 h.The AQY values were calculated according to: AQY (%) = (moles of H2 produced × 2/incident photon flux) × 100%, where the incident photon flux was 49.96 µmol h−1 at 420 nm.59 The bandpass wavelengths of 420 nm correspond to the 20BPF10-420 (Newport, USA) band-pass filters used. The power intensity of monochromatic light was measured using a Si photodiode integrating sphere detector (Newport model 819D-SL-2-CAL2) coupled with an optical power meter (Newport 1919-R). The 420 nm radiation intensity was 0.20 mW cm−2 over an irradiated area of 19.61 cm2.
Electron paramagnetic resonance analysis
We performed the electron paramagnetic resonance (EPR) measurements using a Miniscope MS 5000 EPR spectrometer (Freiberg Instruments, Germany) equipped with a 0.575 W, single wavelength of 462 nm, high-power light-emitting diode. Only 50 µL of sample solution was needed. The sample was placed in a capillary tube and then inserted into the EPR hole. 4-Hydroxy-TEMPO was used as the electron trap. The field modulation, modulation frequency, scan range, and microwave power were 0.2 mT, 100 kHz, 15 mT, and 10 mW, respectively.GC-MS and LC-MS for compounds analysis
The liquid products from photo-reforming were analyzed using a GC system (GC-2010, Shimadzu, Japan) equipped with an HP-INNOWAX column (60 m × 0.25 µm × 0.32 mm; Agilent Technologies, USA) and a mass spectrometer (GCMS-QP2010, Shimadzu, Japan), with helium gas serving as the carrier gas. A sample volume of 1 µL was used for analysis. The operating parameters included a pressure of 91.8 kPa, a total flow rate of 24.7 mL min−1, a column flow rate of 2.2 mL min−1, a linear velocity of 37.9 cm s−1, a purge flow of 5 mL min−1, a split ratio of 10, an oven temperature of 40 °C, and an injection temperature of 150 °C. The GC temperature program involved an initial temperature of 40 °C for 15 min, followed by a ramp of +25 °C min−1 to 200 °C, which was maintained for 20 min. Mass spectrometry parameters included a 70 eV ionization energy, an ion source temperature of 230 °C in electron ionization mode, an interface temperature of 250 °C, a solvent cut time of 1 min, a detector gain of 0.8 kV, a threshold of 5, a start time of 2 min, an end time of 5.58 min, and a selected ion monitoring mode for a mass range of 0 to 100 m/z. The acquired data were analyzed using GC-MS Solution Software (version 4.30, Shimadzu).69Additionally, an UltiMate 3000 HPLC System (Thermo Fisher Scientific, USA) and an EC 100/2 Nucleoshell Hilic column, 2.7 µm (Macherey-Nagel, Germany) were used. The sample was filtered using 0.22 µm membrane filters before analysis. A solvent mixture of 80% acetonitrile and 20% methanol was used, with the column temperature set to 40 °C and the flow rate of 300 µL min−1. The analysis run time was 10 min. Q Exactive Plus Q-Orbitrap MS instruments (Thermo Fisher Scientific, USA) were used in this study. Electrospray-ionization MS was used for analyzing the standard samples and xylose, while high-energy collision dissociation (HCD) was employed for MS-MS analysis. The data acquisition mode targets fragment ions from HCD events with a normalized collision energy (NCE) of 35 eV on eight precursors (195.04967, 165.03900, 149.04402, 119.03340, 105.01778, 89.02254, 89.02287, and 75.00724), detected using an Orbitrap mass analyzer. A negative mode was applied with a 2.5 kV spray voltage, a sheath gas flow rate of 35, an auxiliary gas flow rate of 10, a capillary temperature of 275 °C, and an S-lens level of 50. The resolutions were set at 70000 and 17500 for full MS and MS-MS, respectively. The automatic gain control target was 3 × 106 for full scan and 2 × 105 for MS-MS, with an injection time of 200 ms. Data analysis, including full MS spectra and fragment ions, was conducted using Xcalibur 4.1.31.9.
Author contributions
Van-Can Nguyen: conceptualization, data curation, formal analysis, methodology, writing – original draft, writing – review & editing. Meyta Sanoe: investigation, data curation, formal analysis, methodology, writing – original draft. Novy Pralisa Putri: data curation, formal analysis. Yuh-Lang Lee: data curation, validation. Hsisheng Teng: conceptualization, project administration, resources, supervision, validation, investigation, writing – original draft, writing – review & editing. All authors approved and contributed to the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Science and Technology Council (NSTC) in Taiwan, through projects NSTC 110-2221-E-006-026-MY3, 112-2923-E-006-004, and 112-2223-E-006-008 and by the Hierarchical Green-Energy Materials (Hi-GEM) Research Center, from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.References
- Y. Yao, X. Gao, Z. Li and X. Meng, Catalysts, 2020, 10, 335 CrossRef CAS.
- K. A. Davis, S. Yoo, E. W. Shuler, B. D. Sherman, S. Lee and G. Leem, Nano Convergence, 2021, 8, 1–19 CrossRef PubMed.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1901997 CrossRef CAS PubMed.
- N. Luo, W. Nie, J. Mu, S. Liu, M. Li, J. Zhang, Z. Gao, F. Fan and F. Wang, ACS Catal., 2022, 12, 6375–6384 CrossRef CAS.
- T.-G. Lee, H.-J. Kang, G. A. R. Bari, J.-W. Park, H.-W. Seo, B.-H. An, H. J. Hwang and Y.-S. Jun, Chemosphere, 2021, 283, 131174 CrossRef CAS PubMed.
- L. Lan, H. Daly, R. Sung, F. Tuna, N. Skillen, P. K. Robertson, C. Hardacre and X. Fan, ACS Catal., 2023, 13, 8574–8587 CrossRef CAS PubMed.
- H. Zhao, X. Wang, X. Wu, J. Wang, N. Zhong, A. Seifitokaldani, S. Larter, M. G. Kibria and J. Hu, Resour. Chem. Mater., 2023, 2, 111–116 Search PubMed.
- V.-C. Nguyen, N.-J. Ke, B.-S. Nguyen, Y.-K. Xiao, Y.-L. Lee and H. Teng, J. Mater. Chem. A, 2019, 7, 8384–8393 RSC.
- Y. Lv, W. Shao, Y. Kong, N. Li, X. Huang, Z. Tang, M. Gong, L. Li and W. Wei, J. Environ. Chem. Eng., 2023, 11, 109981 CrossRef CAS.
- L. I. Godina, A. V. Kirilin, A. V. Tokarev, I. L. Simakova and D. Y. Murzin, Ind. Eng. Chem. Res., 2018, 57, 2050–2067 CrossRef CAS PubMed.
- H. A. Duarte, M. E. Sad and C. R. Apesteguia, Int. J. Hydrogen Energy, 2017, 42, 4051–4060 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- J. Satyavolu, J. G. D. Tadimeti and R. Thilakaratne, Energy Convers. Manage.: X, 2021, 10, 100073 CAS.
- V. H. Huong, V.-C. Nguyen, M. N. Ha, D. Van Pham, T. B. Nguyen, Y.-R. Ma, A. B. Ngac and T. T. Loan, Opt. Mater., 2023, 140, 113864 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- L. Wang, K. Wang, T. He, Y. Zhao, H. Song and H. Wang, ACS Sustain. Chem. Eng., 2020, 8, 16048–16085 CrossRef CAS.
- V. H. Huong, T. T. Loan, K.-P. Pham, M. N. Ha, Q. H. Nguyen, Y.-R. Ma, A. B. Ngac and V.-C. Nguyen, J. Alloys Compd., 2024, 972, 172722 CrossRef CAS.
- K. Qi, S.-y. Liu and A. Zada, J. Taiwan Inst. Chem. Eng., 2020, 109, 111–123 CrossRef CAS.
- K.-Q. Lu, Y.-H. Li, Z.-R. Tang and Y.-J. Xu, ACS Mater. Au, 2021, 1, 37–54 CrossRef CAS PubMed.
- V.-C. Nguyen, D. B. Nimbalkar, L. D. Nam, Y.-L. Lee and H. Teng, ACS Catal., 2021, 11, 4955–4967 CrossRef CAS.
- S. Shen, J. Wang, Z. Wu, Z. Du, Z. Tang and J. Yang, Nanomaterials, 2020, 10, 375 CrossRef CAS PubMed.
- T.-F. Yeh, W.-L. Huang, C.-J. Chung, I.-T. Chiang, L.-C. Chen, H.-Y. Chang, W.-C. Su, C. Cheng, S.-J. Chen and H. Teng, J. Phys. Chem. Lett., 2016, 7, 2087–2092 CrossRef CAS PubMed.
- Q. Mei, K. Zhang, G. Guan, B. Liu, S. Wang and Z. Zhang, Chem. Commun., 2010, 46, 7319–7321 RSC.
- H. Tetsuka, R. Asahi, A. Nagoya, K. Okamoto, I. Tajima, R. Ohta and A. Okamoto, Adv. Mater., 2012, 24, 5333–5338 CrossRef CAS PubMed.
- B.-S. Nguyen, Y.-K. Xiao, C.-Y. Shih, V.-C. Nguyen, W.-Y. Chou and H. Teng, Nanoscale, 2018, 10, 10721–10730 RSC.
- S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634–3640 CrossRef CAS.
- D. Qu, M. Zheng, P. Du, Y. Zhou, L. Zhang, D. Li, H. Tan, Z. Zhao, Z. Xie and Z. Sun, Nanoscale, 2013, 5, 12272–12277 RSC.
- Z. Yang, M. Li, S. Chen, S. Yang, F. Peng, J. Liao, Y. Fang, S. Zhang and S. Zhang, Adv. Funct. Mater., 2023, 33, 2212746 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- K. Qi, S. Liu, R. Selvaraj, W. Wang and Z. Yan, Desalin. Water Treat., 2019, 153, 244–252 CrossRef CAS.
- K. Sathishkumar, K. Sowmiya, L. A. Pragasan, R. Rajagopal, R. Sathya, S. Ragupathy, M. Krishnakumar and V. R. M. Reddy, Chemosphere, 2022, 302, 134844 CrossRef CAS PubMed.
- D. A. Bulushev and J. R. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed.
- J.-H. Park, M.-H. Jin, D.-W. Lee, Y.-J. Lee, G.-S. Song, S.-J. Park, H. Namkung, K. H. Song and Y.-C. Choi, Environ. Sci. Technol., 2019, 53, 14041–14053 CrossRef CAS PubMed.
- A. M. Canete-Rodriguez, I. M. Santos-Duenas, J. E. Jimenez-Hornero, A. Ehrenreich, W. Liebl and I. Garcia-Garcia, Process Biochem., 2016, 51, 1891–1903 CrossRef CAS.
- N. Yoneda, S. Kusano, M. Yasui, P. Pujado and S. Wilcher, Appl. Catal., A, 2001, 221, 253–265 CrossRef CAS.
- L.-C. Chen, T.-F. Yeh, Y.-L. Lee and H. Teng, Appl. Catal., A, 2016, 521, 118–124 CrossRef CAS.
- D. C. Elias, R. R. Nair, T. Mohiuddin, S. Morozov, P. Blake, M. Halsall, A. Ferrari, D. Boukhvalov, M. Katsnelson and A. Geim, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- C. Zhang, R. Hao, H. Liao and Y. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS.
- H. Zhao, X. Yu, C.-F. Li, W. Yu, A. Wang, Z.-Y. Hu, S. Larter, Y. Li, M. G. Kibria and J. Hu, J. Energy Chem., 2022, 64, 201–208 CrossRef CAS.
- T. F. Yeh, C. Y. Teng, S. J. Chen and H. Teng, Adv. Mater., 2014, 26, 3297–3303 CrossRef CAS PubMed.
- X. Jiang, X. Yan, W. Ren, Y. Jia, J. Chen, D. Sun, L. Xu and Y. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 31076–31082 CrossRef CAS PubMed.
- K. Kumar, B. Nandan, P. Formanek and M. Stamm, J. Mater. Chem., 2011, 21, 10813–10817 RSC.
- S. Dutta, C. Ray, S. Sarkar, M. Pradhan, Y. Negishi and T. Pal, ACS Appl. Mater. Interfaces, 2013, 5, 8724–8732 CrossRef CAS.
- Z. Jiang, X. Lv, D. Jiang, J. Xie and D. Mao, J. Mater. Chem. A, 2013, 1, 14963–14972 RSC.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- F. Liu, M. H. Jang, H. D. Ha, J. H. Kim, Y. H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657–3662 CrossRef CAS PubMed.
- L. C. Chen, C. Y. Teng, C. Y. Lin, H. Y. Chang, S. J. Chen and H. Teng, Adv. Energy Mater., 2016, 6, 1600719 CrossRef.
- D. Qu, Z. Sun, M. Zheng, J. Li, Y. Zhang, G. Zhang, H. Zhao, X. Liu and Z. Xie, Adv. Opt. Mater., 2015, 3, 360–367 CrossRef CAS.
- L.-C. Chen, Y.-K. Xiao, N.-J. Ke, C.-Y. Shih, T.-F. Yeh, Y.-L. Lee and H. Teng, J. Mater. Chem. A, 2018, 6, 18216–18224 RSC.
- M. Duval Malinsky, K. L. Kelly, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. B, 2001, 105, 2343–2350 CrossRef.
- H. Zhao, C.-F. Li, X. Yu, N. Zhong, Z.-Y. Hu, Y. Li, S. Larter, M. G. Kibria and J. Hu, Appl. Catal., B, 2022, 302, 120872 CrossRef CAS.
- I. F. Teixeira, N. V. Tarakina, I. F. Silva, G. A. A. Diab, N. L. Salas, A. Savateev and M. Antonietti, J. Mater. Chem. A, 2022, 10, 18156–18161 RSC.
- D. B. Nimbalkar, V.-C. Nguyen, C.-Y. Shih and H. Teng, Appl. Catal., B, 2022, 316, 121601 CrossRef CAS.
- U. Nwosu, A. Wang, B. Palma, H. Zhao, M. A. Khan, M. Kibria and J. Hu, Renewable Sustainable Energy Rev., 2021, 148, 111266 CrossRef CAS.
- X. Fu, J. Long, X. Wang, D. Y. Leung, Z. Ding, L. Wu, Z. Zhang, Z. Li and X. Fu, Int. J. Hydrogen Energy, 2008, 33, 6484–6491 CrossRef CAS.
- B. Pan, Y. Wang, Y. Liang, S. Luo, W. Su and X. Wang, Int. J. Hydrogen Energy, 2014, 39, 13527–13533 CrossRef CAS.
- B. Li, W. Wang, J. Zhao, Z. Wang, B. Su, Y. Hou, Z. Ding, W.-J. Ong and S. Wang, J. Mater. Chem. A, 2021, 9, 10270–10276 RSC.
- T. Shan, Y. Li, S. Ke, B. Su, L. Shen, S. Wang, X. Yang and M.-Q. Yang, J. Mater. Sci. Technol., 2024, 179, 155–165 CrossRef.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant and A. I. Cooper, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- S. Cao and L. Piao, Angew. Chem., Int. Ed., 2020, 59, 18312–18320 CrossRef CAS PubMed.
- J. Ma, Y. Li, D. Jin, Z. Ali, G. Jiao, J. Zhang, S. Wang and R. Sun, Green Chem., 2020, 22, 6384–6392 RSC.
- J. P. Ona, R.-M. Latonen, N. Kumar, M. Peurla, I. Angervo and H. Grénman, Electrochim. Acta, 2022, 426, 140754 CrossRef CAS.
- L. Chen, Y. Huang, R. Zou, J. Ma, Y. Yang, T. Li, M. Li, Q. Hao, H. Xie and X. Peng, Green Chem., 2021, 23, 1382–1388 RSC.
- K. Li, X. An, K. H. Park, M. Khraisheh and J. Tang, Catal. Today, 2014, 224, 3–12 CrossRef CAS.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2013, 15, 17–45 RSC.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. M. Alonso, R. Mariscal and M. L. Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- M. S. Holm, Y. J. Pagán-Torres, S. Saravanamurugan, A. Riisager, J. A. Dumesic and E. Taarning, Green Chem., 2012, 14, 702–706 RSC.
- D. K. Tran, A. Z. Rashad, D. J. Darensbourg and K. L. Wooley, Polym. Chem., 2021, 12, 5271–5278 RSC.
- V.-C. Nguyen, D. B. Nimbalkar, V. H. Huong, Y.-L. Lee and H. Teng, J. Colloid Interface Sci., 2023, 649, 918–928 CrossRef CAS PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01544k |
| This journal is © The Royal Society of Chemistry 2024 |