
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization, and biomedical evaluation of ethylene-bridged tetra-NHC Pd(II), Pt(II) and Au(III) complexes, with apoptosis-inducing properties in cisplatin-resistant neuroblastoma cells†
Wolfgang R. E. Büchele‡
 a,
Tim P. Schlachta‡a,
Andreas L. Gebendorfera,
Jenny Pamperincd,
Leon F. Richtera,
Michael J. Sauera,
Aram Prokop*bcd and
Fritz E. Kühn*a
a,
Tim P. Schlachta‡a,
Andreas L. Gebendorfera,
Jenny Pamperincd,
Leon F. Richtera,
Michael J. Sauera,
Aram Prokop*bcd and
Fritz E. Kühn*a
aTechnical University of Munich, School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Molecular Catalysis, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: fritz.kuehn@ch.tum.de; Tel: +49 89 289 13477
bDepartment of Pediatric Hematology/Oncology, Children's Hospital Cologne, Amsterdamer Straße 59, 50735 Cologne, Germany
cDepartment of Pediatric Oncology/Hematology, Helios Clinics Schwerin, Wismarsche Straße 393-397, 19055 Schwerin, Germany. E-mail: aram.prokop@helios-gesundheit.de
dDepartment of Human Medicine, MSH Medical School Hamburg, Am Kaiserkai 1, 20457 Hamburg, Germany
First published on 27th March 2024
Abstract
Synthesis and characterization of the first two cyclic ethylene-bridged tetradentate NHC ligands, with an unsaturated (imidazole) and saturated backbone (2-imidazoline), are described. Complexes of both ligands containing palladium(II) have been obtained. For platinum(II) and gold(III), only the unsaturated tetracarbene complexes could be isolated. The attempts to synthesize a methylene-bridged 2-imidazoline macrocycle are also described. Furthermore, a novel bisimidazolinium ligand precursor and its open-chain PdII and PtII tetracarbene complexes are obtained. Finally, it is shown that the unsaturated gold(III) tetracarbene is able to induce apoptosis in malignant SK-N-AS neuroblastoma cells via the mitochondrial and ROS pathway and overcomes resistance to cisplatin in vitro.
Introduction
N-heterocyclic carbenes (NHCs), first described in 1991,1 have found many applications.2 There are several structural features that allow the tuning of their electronic properties. Ring size, the adjacent heteroatoms, N-substituents, and the backbone can be modified. Changing one or more structural properties of a NHC ligand can lead to significantly different reactivities and stabilities of the resulting complexes.3 Often several NHC units are combined in multidentate ligands, making use of the chelating effect, and a plethora of multidentate NHC metal complexes has been reported.4,5Our group has developed several bidentate and cyclic tetradentate NHC ligands. The respective transition metal complexes have been applied e.g. in medicinal chemistry6,7 and epoxidation catalysis.3 While the bidentate ligands can form open-chain tetracarbene complexes,8,9 the tetradentate ligands give cyclic tetracarbene compounds. Most commonly applied in our recent examinations is the calix[4]imidazolium ligand precursor (a, Fig. 1).10 Its iron complex (c) can be used as olefin epoxidation catalyst achieving unprecedented activity.3 Coinage metal tetracarbene complexes (and metal NHC complexes in general11–17) have been investigated regarding their antiproliferative activity and selectivity against cancer cells (b, d–f, Fig. 1).6,7
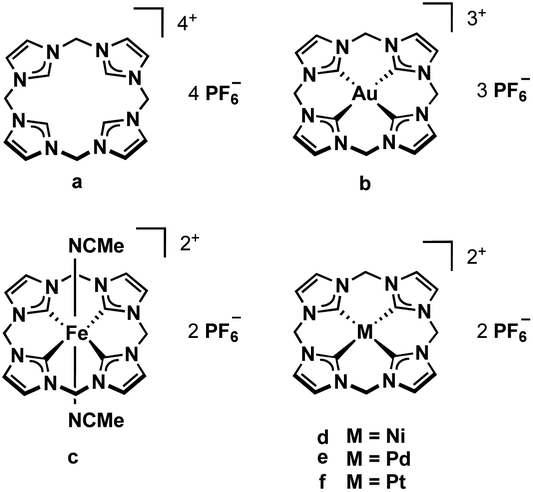 | ||
| Fig. 1 Tetracarbene ligand precursor a and derived transition metal complexes b–f. | ||
In this study, the scope of multidentate NHC ligands is extended with an ethylene-bridged bisimidazolinium ligand precursor and two cyclic ethylene-bridged tetradentate NHC ligands, with an unsaturated (imidazole) and saturated backbone (2-imidazoline). PdII, PtII and AuIII tetracarbene complexes containing the novel ligands are synthesized, characterized and applied in preliminary medicinal studies regarding their activity in inducing apoptosis in malignant cells. Finally, the synthetic attempts to a calix[4]imidazolinium macrocycle (structurally analog to c but with a saturated backbone) are described, because there is an increasing demand for reliable training data, including data on negative outcomes, for machine learning systems in chemistry.18
Especially the two new macrocyclic ligand precursors are intended to lay the foundation for electronic comparisons induced by the different backbone in future studies. The unsaturated backbone of the imidazole moiety causes partial aromaticity, increasing NHC stability by ca. 100 kJ mol−1.19–21 A saturated backbone, in turn, can lead to higher basicity because the electron density is more concentrated on the C2 carbene carbon atom due to the lack of π-interactions.22
Results and discussion
The synthesis of a saturated macrocyclic ligand precursor similar to c, but containing 2-imidazoline moieties instead of imidazole, calix[4]imidazolinium, was pursued parallel to the synthesis of the other ligand precursors. However, the synthesis was not successful with the chosen synthetic approaches as described in the ESI.†Synthesis and characterization of H2L3
H2L3 is based on the literature known ethylene-bridged imidazoline moiety (1).23 Alkylation of 1 with MeI in MeCN at 82 °C, followed by an anion exchange with NH4PF6 in water, gives H2L3 in 91% yield (Fig. 2).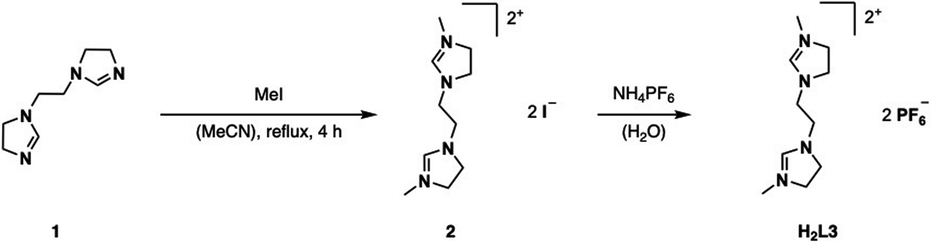 | ||
| Fig. 2 Synthesis of ligand precursor H2L3 via alkylation and anion exchange. | ||
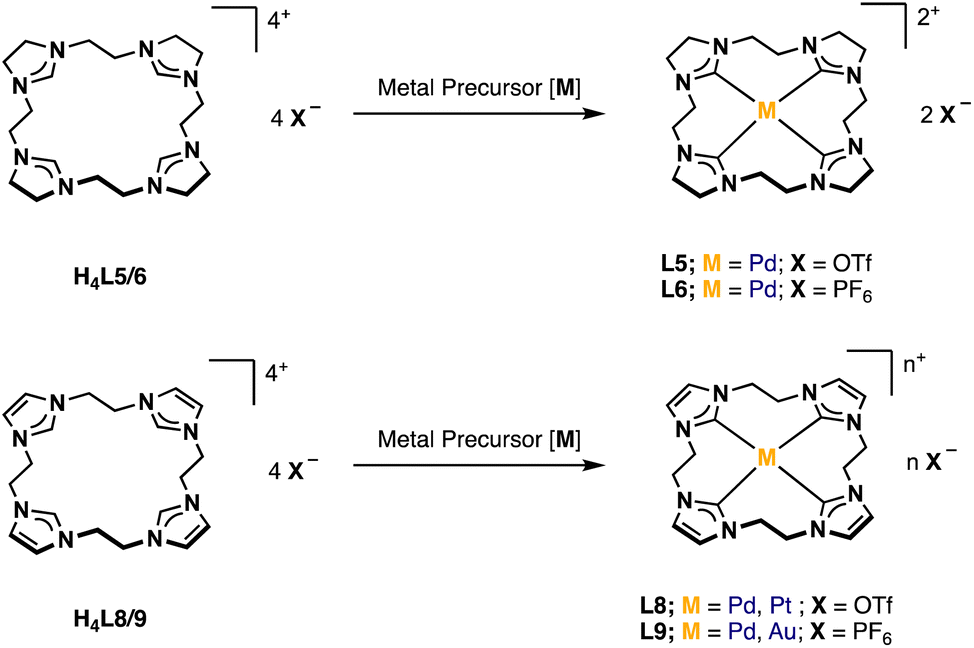
Synthesis and characterization of H4L5/6 and H4L8/L9
For the preparation of H4L5/6 and H4L8/9, a slightly modified literature procedure for similar macrocycles was used (Fig. 3).10 Ring closure to form the macrocyclic imidazolium salt a is commonly achieved with CH2(OTf)2,3 but also CH2Br2 is reported.24 Here the ethylene-bridged imidazoline 1 (ref. 23) and the ethylene-bridged imidazole 7 (ref. 25) are reacted with ethylene bistriflate (4) under dry conditions at −45 °C over a period of 5 h in dry MeCN for H4L5 and H4L8 (see ESI†).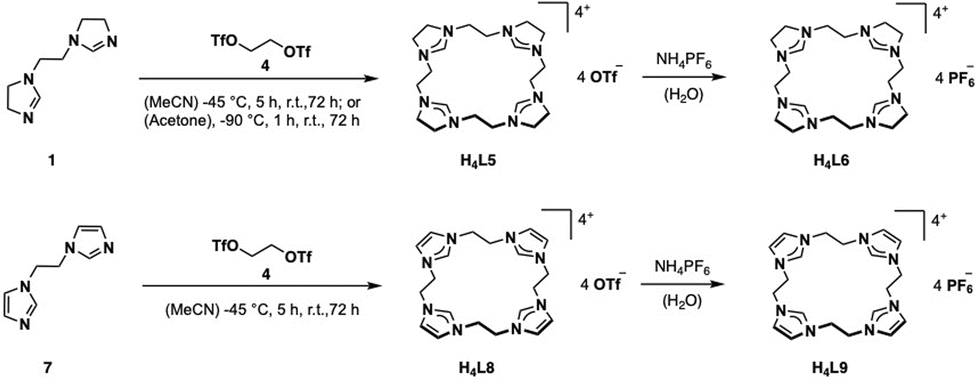 | ||
| Fig. 3 Synthesis of ligand precursors H4L5/6 and H4L8/9 via ring closure with ethylene bistriflate and anion exchange. | ||
The synthesis of H4L5 and H4L8 yields a mixture of a 20-membered macrocycle (87% H4L5 and 90% H4L8, as determined by NMR), consisting of four imidazole (C[4]) units, and a 30-membered macrocycle (13% H4L5, 10% H4L8) consisting of six imidazole (C[6]) units (see ESI†). Separation attempts of C[4] and C[6] via column chromatography, precipitation or sublimation were not successful. However, by increasing the cooling period during the addition of the ethylene bistriflate at −45 °C to a total of 5 h, the purity of the kinetically preferred C[4] unit could be easily increased up to 98% C[4] for H4L5 and in case of H4L8 an increase up to 100% (ESI†). Due to the absence of similar macrocyclic imidazolinium compounds, H4L5 is compared to a and H4L8 in the following.3
Relative to a, all signals of H4L5 and H4L8 are upfield shifted, indicating a higher electronic density due to the +I effect of the ethylene bridge leading to an increased shielding effect in the NMR.10,26 The higher upfield shift of H4L5 compared to H4L8 can be explained by the electronic inducing effect of the saturated bond.20,26,27
Unlike in 1H-NMR, each individual 13C signal of H4L8 in DMSO-d6 is in the same range as the signals obtained for the macrocyclic compound a.10 However, in case of H4L5, opposite to the 1H-NMR, the C2 carbon resonance at 159.16 ppm is downfield shifted compared to H4L8 and a (H4L8, Δδ ≤ 22.08 ppm, a, Δδ ≤ 22.0 ppm), thus contradicting expectations. According to literature and as described by H. V. Huynh, the hypothetical free carbene of the imidazoline ligand H4L5 should be a stronger σ-donor than H4L8, so an enhanced upfield shift of the C2 signal of H4L5 should have been detectable.20,28,29 Interestingly this expectation is not met here, and apparently other factors play a role. Every other resonance in the 13C-NMR is upfield shifted.10
Salt metathesis of the formed macrocyclic salts can be performed with NH4PF6 to increase the solubility in organic solvents and as additional purification step.3,30 Thus, an anion exchange in water towards PF6− is conducted with H4L5 and H4L8, resulting in H4L6 (81%) and H4L9 (88%).
Synthesis and characterization of complexes (Pd/PtL3, PdL5/6, Pd/PtL8, Pd/AuL9)
A well-established route to obtain NHC complexes is to convert the corresponding imidazolium salts with group 10 metal acetates. In this reaction, the acetate serves as an internal base capable of deprotonating imidazolium- and imidazolinium salts to form NHCs, which subsequently coordinate to the metal.6,31–33 An alternative route is via a silver transmetalation.34 In the first approaches, attempts were made to synthesize the respective AgI complex with H2L3 to obtain a dinuclear structure similar to already published open chain bis-NHC-complexes.9,25,35 However, no product formation was observed in our case. Either no reaction took place or complex signals were observed in the aliphatic region of δ = 1.9–4.4 ppm in the 1H-NMR after purification, indicating the decomposition of H2L3. Several other conditions with different AgI-salts and addition of sodium acetate as internal base at different temperatures were tested without success. A possible problem might be the stability of the AgI-complex. Another issue might be hydrolysis of imidazolines under acidic and basic conditions.36,37 It has been proposed in literature that the moisture in the solvent can react with sodium acetate to generate hydroxide ions which can attack the electrophilic center of the C2 carbon and lead to ring-opening products, rather than nucleophilic attacking the acidic proton at the C2 carbon.38 Therefore, the next attempts were conducted under moisture-free reaction conditions by using dried solvents. Even the direct metalation with palladium(II) acetate or palladium(II) chloride under dry reaction conditions did not lead to the desired product. The focus was then shifted to a combination of the transmetalation route using Ag2O in situ with the direct metalation, by applying the respective metal precursor and sodium acetate as a mild base in dry solvents (Fig. 4).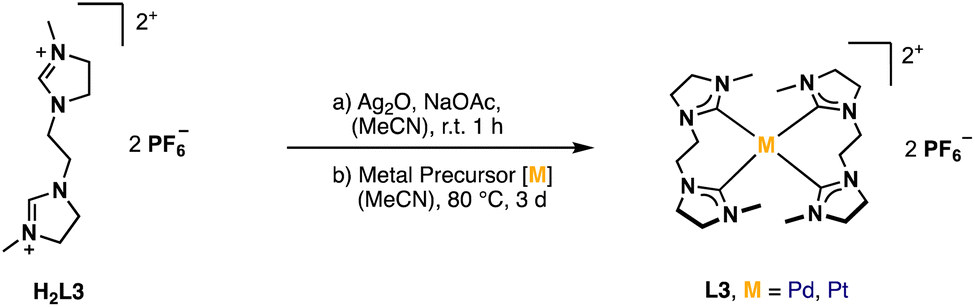 | ||
| Fig. 4 General synthesis of Pd/PtL3. | ||
The absence of the acidic imidazolinium proton signal and appearance of characteristic carbene carbon signals confirms the successful formation of PdL3 and PtL3. Unfortunately, despite several attempts, a clean elemental analysis for PtL3 could not be obtained. Also, the 1H-NMR of PtL3 shows some impurities, which could not be identified and no 195Pt isotope coupling phenomena was observed in the 13C-NMR.
The carbene carbon signal of PdL3 at 194.29 ppm in DMSO-d6 [PtL3; 188.38 ppm in CD3CN], is surprisingly downfield shifted compared to other Pd(II) bis-NHCs reported in literature.39,40 Due to the theoretically stronger σ-donation of the imidazolinylidene ligand H2L3 compared to its unsaturated analog, an upfield shift of the 13CNHC signal was expected. Literature indicates that the significant downfield shift of the carbene carbon resonance from imidazolium to imidazolinium compounds is a general phenomenon.38,41,42 Another interesting fact is that the analytic data, including HR-ESI-MS and elemental analysis, are not supporting a dinuclear complex or a mono-carbene complex as expected, but indicate that PdL3 has rather a [Pd(L3)2](PF6)2 structure similar to e. This is further confirmed by single-crystal X-ray diffraction (SC-XRD). The PdL3 complex displays a distorted square planar structure. Two L3 ligands coordinate to the Pd center, resulting in an open-chain tetracarbene complex of similar geometry like the cyclic complex e.34 The Pd–C (2.039 Å, 2.038 Å) distances are in good accord with palladium(II) NHC complexes reported in literature.34,39 The alkyl groups of the ligand L3 adopt a syn conformation in the solid state, while the imidazole rings are tilted by 55.30° out of the palladium square plane (Fig. 5).
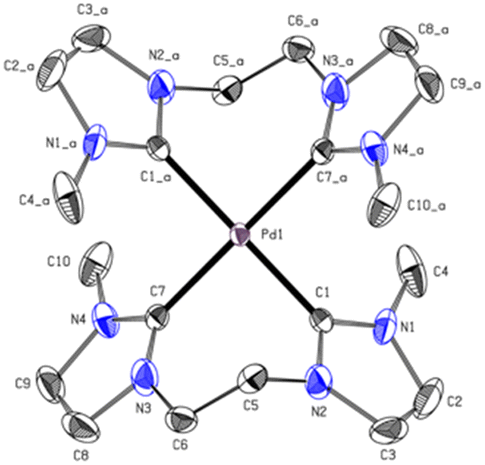 | ||
| Fig. 5 ORTEP-style representation of the cationic fragment of complex PdL3. Hydrogen atoms and hexafluorophosphate anions are omitted for clarity. Thermal ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): C1–Pd1 2.039(2); C7–Pd1 2.038(2); C7_a–Pd1–C7 180.0; C7–Pd1–C1_a 91.64(9); C7–Pd1–C1 88.36(9), C7_a–Pd1–C1_a–N2_a 55.30. | ||
The PtL3 complex exhibits a similarly distorted square planar structure compared to PdL3. The Pt–C (2.033 Å, 2.039 Å) distances are comparable to similar literature known group 10 NHC compounds.43–47 The alkyl groups of L3 also adopt a syn conformation, while the imidazole rings are tilted by 50.53° out of the palladium square plane as in PdL3 (Fig. 6).
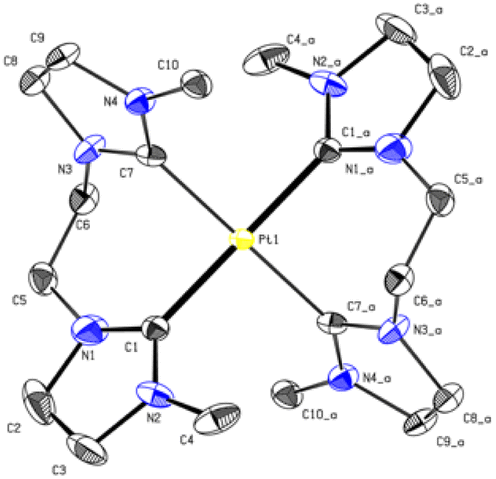 | ||
| Fig. 6 ORTEP-style representation of the cationic fragment of complex PtL3. Hydrogen atoms and hexafluorophosphate anions are omitted for clarity. Thermal ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): Pt1–C1 2.0337 (18); Pt1–C7 2.039 (6), C1_a–Pt1–C1 180.00(7); C1–Pt1–C7_a 91.2(5); C1–Pt1–C7 88.8(5), C7_a–Pd1–C1_a–N2_a 50.53°. | ||
Complex PdL5/6 and PdL8/9
Since H4L5 and H4L8 are quite similar to other macrocycles (a), it seemed suitable to synthesize PdL5 and PdL8 according to alike compounds via the direct metalation route.32 Therefore, H4L8 was first converted with Pd(OAc)2 in a mixture of dry DMSO/MeCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 40 °C for 16 h.34 However, no product formation was observed after work-up. Several other conditions such as increasing temperature and reaction time led to the absence of the imidazolium protons and the formation of new product signals in the 1H-NMR after 4 d at 80 °C. Still these intensities were very low, and no product could be isolated. Another approach was tried via the transmetalation route with AgI salts, but this also led to no product formation. Finally, both PdL8 (50%, Fig. 7) and PdL5 (3%) could be obtained by applying the same reaction conditions as for the already synthesized complexes PdL3 and PtL3. The yield of the imidazolinylidene tetracarbene complex could be increased to 46%, by using H4L6 instead of H4L5, resulting in PdL6.
:1) at 40 °C for 16 h.34 However, no product formation was observed after work-up. Several other conditions such as increasing temperature and reaction time led to the absence of the imidazolium protons and the formation of new product signals in the 1H-NMR after 4 d at 80 °C. Still these intensities were very low, and no product could be isolated. Another approach was tried via the transmetalation route with AgI salts, but this also led to no product formation. Finally, both PdL8 (50%, Fig. 7) and PdL5 (3%) could be obtained by applying the same reaction conditions as for the already synthesized complexes PdL3 and PtL3. The yield of the imidazolinylidene tetracarbene complex could be increased to 46%, by using H4L6 instead of H4L5, resulting in PdL6.
 | ||
| Fig. 7 General synthesis of PdL5/6, Pd/PtL8 and Pd/AuL9. | ||
Again, the absence of the acidic position 2 proton signals in the 1H-NMR and appearance of the carbene carbon peaks confirm the formation of Pd(II) carbene complexes. The observed chemical shift of PdL8 is in the typical range of Pd(II) tetra-NHC compounds and indicates the formation of a complex with similar coordination sphere as e.6,34,45 The 1H-NMR of PdL5 in CD3CN shows three signals, with two of them in a similar range to PdL8 and one upfield shifted signal of the backbone protons. As already mentioned in the discussion of H4L5, the 13C-NMR of PdL5 is contrary to expectations. The carbene carbon of PdL5 (191.30 ppm) is surprisingly strong downfield shifted compared to PdL8 (165.84 ppm) and in a similar range to the carbene carbon of PdL3 (195.6 ppm in CD3CN). Literature indicates that the significant downfield shift of the carbene carbon resonance from imidazole to imidazoline compounds is a general phenomenon.38,41,42 The uncertainty of a 13C-NMR measurement is expected to be below 0.1 ppm; by using three times the weighted standard deviation, a difference of >0.4 ppm is required for a significant difference that exceeds the statistic uncertainty.3,48 Therefore, PdL5 (191.30 ppm in CD3CN) and PdL8 (165.84 ppm in CD3CN) show a sufficiently different chemical shift to allow its discussion. In general, the normal NHC unit (without any modification) of the tetracarbene ligands is in a range of rather low to negligible π-backdonation, hence the changes in electronic properties are dominated by the σ-donation of the tetracarbenes.3,29 According to literature, the imidazoline ligand L5 should be in general a stronger σ-donor than L8, so an enhanced upfield shift of the carbene signal would have been detectable.3,20,28,29 Interestingly, this expectation is also not met here, and apparently other factors may play a role, as already observed with PdL3 and PtL3. Therefore, further investigations on this subject, e.g. by means of DFT calculations, have to be carried out, since only conjectures can be made with the present analytical data. The elemental analysis and HR-ESI-MS for PdL8 are in accord with a composition [Pd(L8)](OTf)2 similar to e. It needs to be noted that no clean elemental analysis of PdL6 could be obtained. However, the elemental analysis and HR-ESI-MS of PdL6 are in accordance with the composition [PdL6](PF6)2. Due to unsatisfying results in crystallization of PdL8, an anion exchange in water towards PF6− was conducted, resulting in PdL9 (41%). Single crystals suitable for SC-XRD were obtained by slow diffusion of Et2O into MeCN solution of PdL9. As expected, the Pd(II) ion is coordinated in a nearly square planar fashion with C–Pd–C angles deviating from 180° by ∼8°, thus lifting the metal slightly above the carbene carbon atom plane (Fig. 8). However, due to the C2-bridge, the ligand is strongly bent (C5–Pd1–C5_b = 98.97°) and adopts a crisp-shape, while tilting the imidazole rings 53.62° in an alternating pattern out of the palladium square plane.49 The Pd–C distance (2.019 Å) is comparable to those of other cyclic Pd(II) tetracarbene compounds reported in literature.6,34,43,45–47
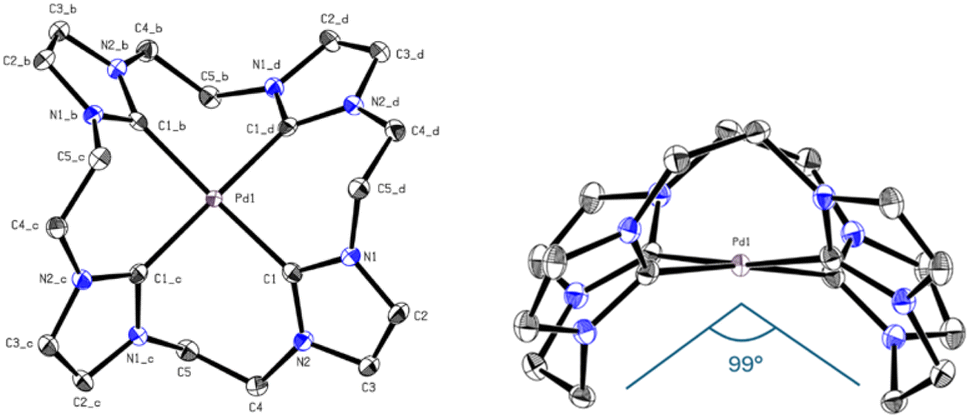 | ||
| Fig. 8 ORTEP-style representation of the cationic fragment of complex PdL9. Hydrogen atoms and hexafluorophosphate anions are omitted for clarity. Thermal ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): C1–Pd 2.019(2), C1–Pd1–C1_b 172.03(12), C1–Pd1–C1_d–N2_d 53.62, C5–Pd1–C5_b 98.97°. | ||
In the following Table 1 the M–Ccarbene bond lengths [Å], the Ccarbene–M–Ccarbene angle [°], the tilt of the NCN unit [°] of the complexes Pd/PtL3 and PdL9 and additionally the Cbridge–M–Cbridge angle [°] for PdL9 are summarized.
| Compound | PdL3 | PtL3 | PdL9 |
|---|---|---|---|
| M–Ccarbene [Å] | 2.038|2.039 | 2.033|2.039 | 2.019 |
| Ccarbene–M–Ccarbene [°] | 180 | 180 | 172.03 |
| Tilt NCN unit [°] | 55.30 | 50.53 | 53.62 |
| Cbridge–M–Cbridge [°] | — | — | 98.97 |
Complex PtL8
Applying the same reaction conditions and work-up methods to Pt(MeCN)2Cl2 instead of Pd(OAc)2 results in the formation of PtL8 (25%,Fig. 7). The absence of acidic proton signals in the 1H-NMR and the appearance of the carbene 13C-peak at 159.39 ppm in CD3CN confirms the formation of the respective Pt(II) complex. The chemical shift of the carbene carbon is in accordance with Pt(II) tetra-NHC complexes previously reported in literature and is slightly shifted to the upfield compared to PdL8 (13CNHC in CD3CN at 165.84 ppm) by 6.45 ppm.43,46,50 No 195Pt isotope coupling was observed. The 1H-NMR in CD3CN shows similar signals compared to PdL8, where the bridge protons also split into two multiplets at 5.01 and 4.44 ppm. In addition, HR-ESI-MS is in accordance to a similar composition as PdL8. Despite multiple attempts, no single crystals suitable for SC-XRD were obtained. However, the discussed analytical data strongly support a similar structure compared to PdL8 and similarly structured tetracarbene ligand.6,34Complex AuL9
For the synthesis of AuL9 (Fig. 7), the same reaction conditions were applied as reported in the literature for similar complexes.51 Therefore, H4L8 was converted with KAuCl4 and NaOAc in dry DMSO under exclusion of light at 100 °C for 5 h. After the work-up, including an ion exchange to PF6− as a purification step, AuL9 (47%) was obtained. The absence of acidic proton signals in the 1H-NMR and the appearance of a new 13C-peak at 146.03 ppm in CD3CN confirm the formation of the respective Au(III) complex. The chemical shift of the carbene carbon is in accord with Au(III) tetracarbene complex (e) previously reported in literature and slightly downfield shifted by 1.79 ppm when compared to e.51 Furthermore, the backbone carbons are also slightly downfield shifted by 0.68 ppm. The 1H-NMR in CD3CN shows similar signals compared to complex PdL8 and PtL8 with the backbone protons at 7.47 ppm and the bridge protons as two multiplets in close proximity at 4.83 and 4.71 ppm. Both elemental analysis52 and HR-ESI-MS are in agreement with the composition [Au(L15)](PF6)3. Although no single crystals suitable for SC-XRD were obtained, the discussed analytical data strongly support the coordination of one tetracarbene ligand similar to PdL9.Biological evaluation
Induction of apoptosis as cell death type
PdL3, PdL8, AuL9 and their respective protonated ligand precursors were tested for their apoptotic effects on Nalm-6 cells (human B cell precursor leukemia cell line) and SK-N-AS cells (human neuroblastoma cell line) at different concentrations and quantified by the nuclear DNA fragmentation by flow cytometry analysis. PdL3 and PdL8 as well as the ligand precursors do not show any apoptosis inducing effects in Nalm-6 cells and SK-N-AS cells (see ESI†). AuL9 shows no apoptotic effect in Nalm-6 cells, but significant apoptosis induction by AuL9 is detected in SK-N-AS cells (Fig. 9A); therefore, the effect of AuL9 in SK-N-AS cells was further characterized.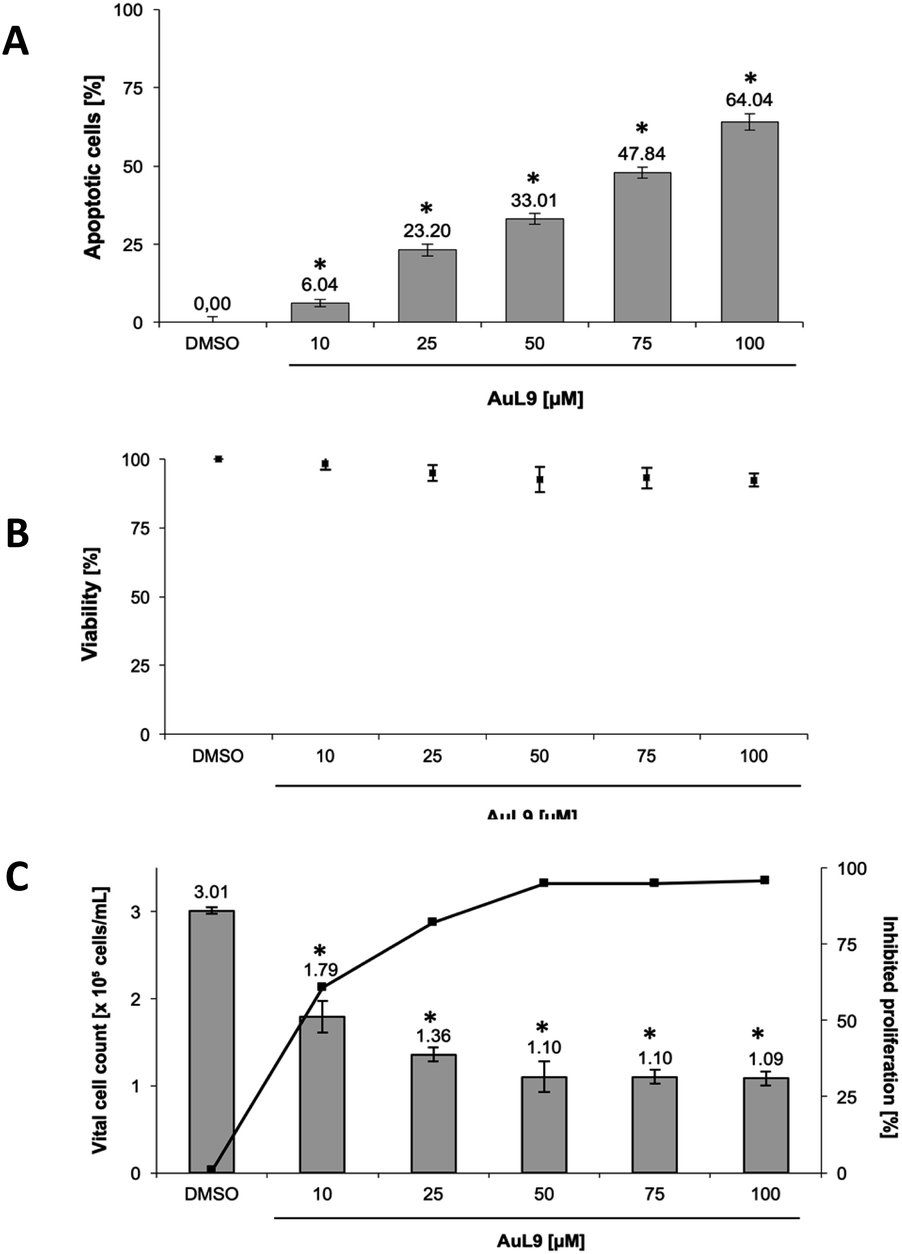 | ||
| Fig. 9 (A) AuL9 induces apoptosis in SK-N-AS cells. The cells were treated with different concentrations of AuL9 and incubated for 96 h. Nuclear DNA fragmentation was analyzed. (B) To exclude unspecific cytotoxic effects, such as necrotic cell death, the viability of SK-N-AS cells was determined by measurement of LDH release into the medium after 2 h of incubation with different concentrations of AuL9. No significant LDH release could be detected in cells treated with AuL9 up to a concentration of 100 μM. Values are given as mean% of DMSO control ± SD (n = 3). (C) The inhibition of proliferation of AuL9 treated SK-N-AS cells was measured after 48 h using the CASY Cell-Counter System. A significant inhibition of cell growth was observed at concentrations as low as 10 μM. Inhibition of proliferation is given in mean% of control ± SD (n = 3); *: p < 0.05 vs. DMSO, t-test. | ||
To exclude necrotic effects of AuL9, lactate dehydrogenase (LDH) leakage from SK-N-AS cells after 2 h incubation with AuL9 was measured. LDH is released from the cell in case of necrosis and can be detected in the cell culture medium in case of loss of cell integrity and thus serves as a necrosis indicator.53 AuL9 shows no significant non-specific cytotoxic effects on SK-N-AS cells in the relevant concentration range up to 100 μM (Fig. 9B).
In addition to apoptosis induction, it was tested whether AuL9 can inhibit the proliferation of malignant cells. For this purpose, SK-N-AS cells were incubated with different concentrations of AuL9 for 48 hours. The proliferation inhibition was determined by comparing the total cell number of vital cells of the DMSO control with the total cell number of vital cells of the treated cells. The results show that AuL9 inhibits cell proliferation of SK-N-AS cells in a dose-dependent manner (Fig. 9C). A concentration of 50 μM AuL9 causes nearly 100% inhibition of proliferation, indicating G1 arrest.
For the investigation of the mechanism of action of AuL9, the mitochondrial membrane potential of SK-N-AS cells was measured after 48 h incubation with AuL9. It was shown that the mitochondrion and thus the intrinsic apoptosis pathway plays at least a partial role in the effect of AuL9 (Fig. 10A).
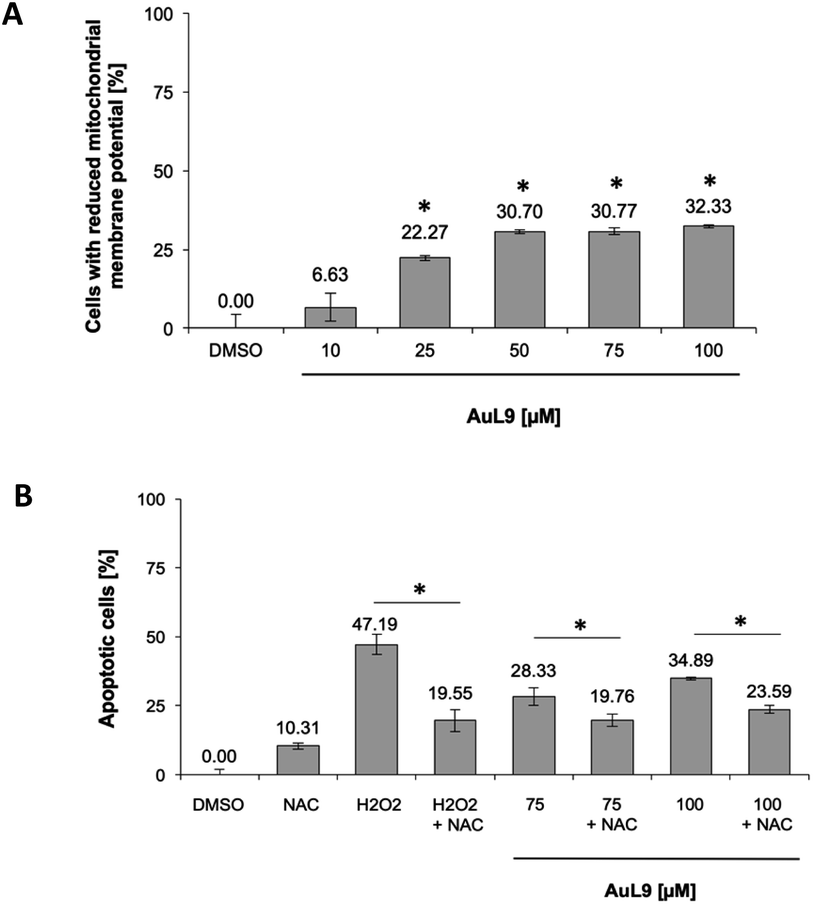 | ||
| Fig. 10 (A) The mitochondrial membrane potential in SK-N-AS cells was impaired by AuL15 treatment, which implicates mitochondrial pathway involvement in apoptosis induction. The mitochondrial membrane potential was measured by flow cytometric analysis in SK-N-AS cells after 48 h of incubation with different concentrations of AuL15 and staining with the cationic dye JC-1. Values are mean% of cells with low mitochondrial membrane potential ± SD (n = 3); *: p < 0.05 vs. DMSO, t-test. B The induction of apoptosis in SK-N-AS cells in response to AuL15 treatment was shown to be dependent on the ROS mediated pathway. The cells were incubated for 72 h with 50 μM H2O2 as a positive control or different concentrations of AuL15 with or without pretreatment of the cells with the ROS inhibitor N-acetylcysteine (NAC, 5 mM) 1.5 h prior to substance addition. Nuclear DNA fragmentation was analyzed by flow cytometric analysis. Values are mean% of apoptotic cells ± SD (n = 3); *: p < 0.05 vs. DMSO, t-test. | ||
To further characterize the role of mitochondria in AuL9-induced apoptosis, the apoptosis pathway mediated by reactive oxygen species (ROS) was investigated. Therefore, N-acetylcysteine (NAC) as a known ROS inhibitor and H2O2, which belongs to the ROS, as a positive control was investigated. It was shown that apoptosis induction could be significantly inhibited by NAC. It can therefore be concluded that the generation of ROS plays a role in the AuL9-induced apoptosis (Fig. 10B). However, it is not possible in the present state to be sure how the ROS are generated and whether AuL9 directly leads to an increased ROS production or triggers pathways that result in the generation of ROS.
Overcoming cisplatin resistance
Cisplatin is a well-known chemotherapeutic agent for the treatment of many different types of cancer.54 The development of resistance in tumor cells is a major problem in therapy and is usually the limiting factor in the cure of cancer patients.55 Therefore, it is of great importance for drug development that new agents are able to overcome cytostatic drug resistance. In addition to SK-N-AS cells, AuL9 was tested on cisplatin resistant SK-N-AS cells and cisplatin resistance overcoming could be demonstrated (Fig. 11). In a previous characterization of the cisplatin resistant SK-N-AS cells, procaspase-8 under expression was shown.56 The cisplatin resistance overcoming of SK-N-AS cells indicates that procaspase-8 has a minor role in AuL9-induced apoptosis.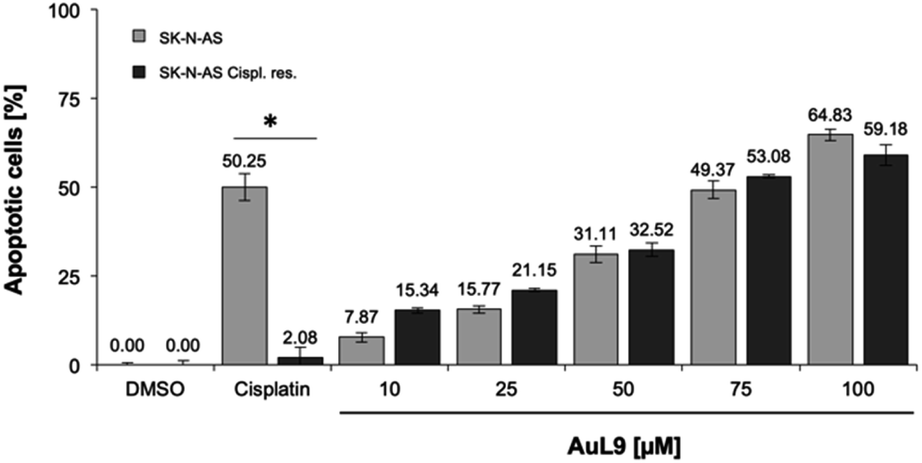 | ||
| Fig. 11 SK-N-AS and SK-N-AS cisplatin resistant cells were treated with different concentrations of AuL15 and incubated for 96 h. It is shown that AuL9 was also effective in inducing apoptosis in cisplatin resistant cells, thus overcoming resistance. 8.25 μM cisplatin has been used as a positive control to prove resistance. Nuclear DNA fragmentation was analyzed by flow cytometric analysis. Values are mean% of apoptotic cells ± SD (n = 3); *: p < 0.05 vs. DMSO, t-test. | ||
Conclusion and outlook
A synthetic approach to a calix[4]imidazolinium macrocycle as saturated analog to a is presented. The synthesis of two new macrocyclic ligand systems, being bridged by ethylene groups and containing imidazoline (H4L5/6) and imidazole moieties (H4L8/9) are discussed. In addition, a novel bisimidazolinium ligand precursor (H2L3) is described. All complexes (Pd/PtL3, PdL5/6, Pd/PtL8, Au/PdL9) with their respective ligands synthesized in this work are not accessible via the direct metalation of the respective ligand, due to irreproducible or unreliable results, except for AuL9. Even the route via the silver salt transmetalation does not lead to reliable results. The silver complexes of the respective ligands could not be isolated, probably due to instability of the respective complexes. Therefore, a modified synthetic method has been established. Here, in situ transmetalation with silver oxide is used in combination with the metal precursor and an excess of sodium acetate as a mild base, resulting in the corresponding complexes. Furthermore, the complexes PdL3, PdL8, AuL9 and their respective ligands were tested for their ability to induce apoptosis on Naml-6 and SK-N-AS cells. According to the experiments performed, the data suggest that AuL9 is capable of inducing apoptosis in malignant cells via the mitochondrial and ROS pathway. However, so far, an effect could only be observed on SK-N-AS neuroblastoma cells. In addition, a relatively high dose of AuL9 is required to induce apoptosis in neuroblastoma cells, which could be challenging for clinical applicability. AuL9 is able to overcome resistance to cisplatin in neuroblastoma cells (SK-N-AS) in vitro. Further characterization experiments would be required to determine the exact mechanism of action of AuL9, for example identification of molecular targets that are involved in the AuL9 induced apoptosis, as well as the selectivity for cancer cells.Experimental section
General procedures and analytical methods
Unless otherwise stated, all manipulations were performed under normal atmosphere without dried and degassed chemicals. All syntheses regarding the complexes were conducted under the exclusion of light. Every work-up was performed under normal atmosphere without dried and degassed chemicals; the complexes' work-ups were conducted in addition under the exclusion of light unless otherwise stated. Purification, in case of the Pt and Pd complexes, is achieved by dissolving the crude product in MeCN and filtering it through basic aluminum oxide to remove impurities. Acidic aluminum oxide promotes the decomposition of the complexes while pH-neutral aluminum oxide leads in smaller yields.6 All obtained complexes are air- and water stable; however, PtL3, PtL8 and AuL9 decompose after extensive exposition to light. Solvents were obtained water-free from a MBraun solvent purification system and stored over molecular sieves (3 Å). The procedures for novel compounds obtained during the synthetic approaches to the saturated macrocyclic ligand precursor, containing 2-imidazoline moieties instead of imidazole, calix[4]imidazolinium, (2-imidazoline, N-benzyl-2-imidazoline, 3,3′-methylenebis(1-benzyl-2-imidazolinium)dibromide, N1,N1,N2,N2-tetrabenzylethane-1,2-diamine, tert-butyl (2-aminoethyl)carbamate, tert-butyl 2-imidazoline-1-carboxylate) are stated in the ESI.† N-Benzylethylenediamine (12),57–59 ethylenebis(trifluoromethanesulfonate) (4),60 1,1′-ethylene-di-2-imidazoline (1)23 and 1,1′-ethylenebis-1H-imidazolyl (7)25,61 were synthesized according to literature procedures. All other reagents were purchased from commercial suppliers and used without further purification. NMR spectra were recorded on a Bruker Avance DPX 400 (1H-NMR, 400 MHz; 13C-NMR, 100 MHz; 19F-NMR, 376 MHz) and chemical shifts are given in δ values in ppm (parts per million) relative to TMS (tetramethylsilane) and reported relative to the residual signal of the deuterated solvent.62 Elemental analysis (C/H/N) were obtained by the Microanalytical Laboratory at Technische Universität München. Electrospray ionization mass spectrometry (ESI-MS) data were acquired on a Thermo Fisher Ultimate 3000 and with higher resolution (HR-ESI-MS) on Exactive Plus Orbitrap from Thermo Fisher.Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 121.80 (q, 1J19F–13C = 320 Hz, OTf−), 50.14 (CH2–CH2). 19F-NMR (376 MHz, CD3CN) δ (ppm) = −79.32 (CF3). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 9.00 (t, 4J = 1.7 Hz, 4H, N–CH–N), 7.57 (d, 4J = 1.6 Hz, 8H, CH), 4.74 (s, 16H, CH2). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 137.08 (N–CH–N), 123.28 (HCCH), 120.66 (q, 1J19F–13C = 320 Hz, OTf−), 49.24 (CH2–CH2). Elemental analysis for C24H28N8O12F12S4 (%) anal. calc.: C 29.54; H 2.84; N 11.54; S 13.13 found: C 29.54; H 2.84; N 11.54; S 13.25. ESI-MS: m/z [H4L8–4OTf−]4+ calc.: 95.06, found: 94.91, [H4L8–3OTf−]3+ calc.: 176.39, found 176.36, [H4L8–2OTf−]2+ calc.: 339.07, found: 339.22, [H4L8–1OTf−]1+ calc.: 827.03, found 826.93.CH), 49.44 (CH2–CH2). Elemental analysis for C20H28N8F24P4 (%) anal. calc.: C 25.01; H 2.94; N 11.67; S 0.00 found: C 25.08; H 2.90; N 11.31; S 0.00.:1) and stirred for 1 h at ambient temperature, followed by the addition of Pd(OAc)2 (71.8 mg, 320 μmol, 1.05 eq.). The resulting reaction mixture is heated to 80 °C for 3 d and is filtered, after cooling to ambient temperature, over a short plug of basic aluminum oxide. The filter column is eluted with MeCN (100 mL) and all volatile compounds are removed in vacuo. The resulting oily solution (still approx. 6 mL of DMSO remaining) is resuspended in MeCN (6 mL) and centrifuged. Upon the addition of Et2O (25 mL) to the supernatant, a brown/black solid is precipitated. After another addition of Et2O (120 mL) a white solid is precipitated. The white crude material is collected via centrifugation, washed with Et2O (3 × 5 mL) and redissolved in MeCN (5 mL). After purification [3 times dissolving in MeCN (4 mL) and precipitating with Et2O (∼15 mL)] and removing all volatile compounds in vacuo, the titled compound PdL5 is obtained as an off-white solid (7.00 mg, 8.87 μmol, 3%). 1H-NMR (400 MHz, CD3CN): δ (ppm) = 4.10–4.00 (m, 8H, CH2,(bridge)), 3.76–3.54 (m, 16H, CH2,(backbone)), 3.52–3.45 (m, 8H, CH2,(bridge)). 13C-NMR (101 MHz, CD3CN): δ (ppm) = 191.3 (N–C–N), 51.0 (CH2,(bridge)/CH2,(backbone)), 47.4 (CH2,(backbone)/CH2,(bridge)). 19F-NMR (376 MHz, CD3CN): δ (ppm) = −79.33 (CF3). ESI-MS: m/z = calc. for [PdL5–OTf−]+: 639.13 ([PdL5–OTf−]+); found: 639.44; calc. for [PdL5–2OTf−]+: 245.09 ([M–2OTf−]+); found: 245.20.CH), 48.86 (CH2–CH2). 19F-NMR (376 MHz, CD3CN) δ (ppm) = −79.27 (CF3). HR-ESI-MS: m/z [PtL8–2OTf−]2+ calc.: 285.5881, found: 285.5864, [PtL8–OTf−]+ calc.: 720.1287, found: 720.1264.CH), 48.58 (CH2–CH2). Elemental analysis for C20H24AuF18N8P3 × 0.1 MeCN (%) anal. calc.: C 24.62; H 2.70; N 11.11; S 0.00 found: C 24.82; H 2.78; N 11.15; S 0.57. HR-ESI-MS: m/z [AuL9–3PF6−]3+ calc.: 191.0591, found: 191.0587, [AuL9–PF6−]+ calc.: 863.1068, found: 863.1038.
CH), 121.80 (q, 1J19F–13C = 320 Hz, OTf−), 50.14 (CH2–CH2). 19F-NMR (376 MHz, CD3CN) δ (ppm) = −79.32 (CF3). 1H-NMR (400 MHz, DMSO-d6) δ (ppm) = 9.00 (t, 4J = 1.7 Hz, 4H, N–CH–N), 7.57 (d, 4J = 1.6 Hz, 8H, CH), 4.74 (s, 16H, CH2). 13C-NMR (101 MHz, DMSO-d6) δ (ppm) = 137.08 (N–CH–N), 123.28 (HCCH), 120.66 (q, 1J19F–13C = 320 Hz, OTf−), 49.24 (CH2–CH2). Elemental analysis for C24H28N8O12F12S4 (%) anal. calc.: C 29.54; H 2.84; N 11.54; S 13.13 found: C 29.54; H 2.84; N 11.54; S 13.25. ESI-MS: m/z [H4L8–4OTf−]4+ calc.: 95.06, found: 94.91, [H4L8–3OTf−]3+ calc.: 176.39, found 176.36, [H4L8–2OTf−]2+ calc.: 339.07, found: 339.22, [H4L8–1OTf−]1+ calc.: 827.03, found 826.93.CH), 49.44 (CH2–CH2). Elemental analysis for C20H28N8F24P4 (%) anal. calc.: C 25.01; H 2.94; N 11.67; S 0.00 found: C 25.08; H 2.90; N 11.31; S 0.00.:1) and stirred for 1 h at ambient temperature, followed by the addition of Pd(OAc)2 (71.8 mg, 320 μmol, 1.05 eq.). The resulting reaction mixture is heated to 80 °C for 3 d and is filtered, after cooling to ambient temperature, over a short plug of basic aluminum oxide. The filter column is eluted with MeCN (100 mL) and all volatile compounds are removed in vacuo. The resulting oily solution (still approx. 6 mL of DMSO remaining) is resuspended in MeCN (6 mL) and centrifuged. Upon the addition of Et2O (25 mL) to the supernatant, a brown/black solid is precipitated. After another addition of Et2O (120 mL) a white solid is precipitated. The white crude material is collected via centrifugation, washed with Et2O (3 × 5 mL) and redissolved in MeCN (5 mL). After purification [3 times dissolving in MeCN (4 mL) and precipitating with Et2O (∼15 mL)] and removing all volatile compounds in vacuo, the titled compound PdL5 is obtained as an off-white solid (7.00 mg, 8.87 μmol, 3%). 1H-NMR (400 MHz, CD3CN): δ (ppm) = 4.10–4.00 (m, 8H, CH2,(bridge)), 3.76–3.54 (m, 16H, CH2,(backbone)), 3.52–3.45 (m, 8H, CH2,(bridge)). 13C-NMR (101 MHz, CD3CN): δ (ppm) = 191.3 (N–C–N), 51.0 (CH2,(bridge)/CH2,(backbone)), 47.4 (CH2,(backbone)/CH2,(bridge)). 19F-NMR (376 MHz, CD3CN): δ (ppm) = −79.33 (CF3). ESI-MS: m/z = calc. for [PdL5–OTf−]+: 639.13 ([PdL5–OTf−]+); found: 639.44; calc. for [PdL5–2OTf−]+: 245.09 ([M–2OTf−]+); found: 245.20.CH), 48.86 (CH2–CH2). 19F-NMR (376 MHz, CD3CN) δ (ppm) = −79.27 (CF3). HR-ESI-MS: m/z [PtL8–2OTf−]2+ calc.: 285.5881, found: 285.5864, [PtL8–OTf−]+ calc.: 720.1287, found: 720.1264.CH), 48.58 (CH2–CH2). Elemental analysis for C20H24AuF18N8P3 × 0.1 MeCN (%) anal. calc.: C 24.62; H 2.70; N 11.11; S 0.00 found: C 24.82; H 2.78; N 11.15; S 0.57. HR-ESI-MS: m/z [AuL9–3PF6−]3+ calc.: 191.0591, found: 191.0587, [AuL9–PF6−]+ calc.: 863.1068, found: 863.1038.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors thank Prof. Dr T. Simon (University Cologne) for providing the SK-N-AS cell line, Dr Seeger for providing the Nalm-6 cell line. A. Prokop thanks the Dr. Kleist Foundation (Berlin), Foundation David (Cologne), Koch Foundation (Berlin) and Blankenheimerdorf e.V. (Eifel) for the financial support in the biological studies. Further thanks go to A. Gradenegger, F. Tschernuth from Prof. Dr S. Inoue, and C. Hoefer for synthetic support of the compound H4L8 and Patrick Mollik for HR-ESI-MS measurements. Thanks also go to Rea Sangiovanni for the graphical abstract.Notes and references
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363, DOI:10.1021/ja00001a054.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem, 2021, 5, 711–725, DOI:10.1038/s41570-021-00321-1.
- T. P. Schlachta and F. E. Kühn, Chem. Soc. Rev., 2023, 52, 2238–2277, 10.1039/D2CS01064J.
- A. Biffis, M. Baron and C. Tubaro, in Adv. Organomet. Chem., ed. P. J. Pérez, Academic Press, 2015, vol. 63, pp. 203–288, DOI:10.1016/bs.adomc.2015.02.002.
- V. Charra, P. de Frémont and P. Braunstein, Coord. Chem. Rev., 2017, 341, 53–176, DOI:10.1016/j.ccr.2017.03.007.
- M. A. Bernd, E. B. Bauer, J. Oberkofler, A. Bauer, R. M. Reich and F. E. Kühn, Dalton Trans., 2020, 49, 14106–14114, 10.1039/D0DT02598D.
- E. B. Bauer, M. A. Bernd, M. Schütz, J. Oberkofler, A. Pöthig, R. M. Reich and F. E. Kühn, Dalton Trans., 2019, 48, 16615–16625, 10.1039/C9DT03183A.
- J. F. Schlagintweit, C. H. G. Jakob, K. Meighen-Berger, T. F. Gronauer, A. Weigert Muñoz, V. Weiß, M. J. Feige, S. A. Sieber, J. D. G. Correia and F. E. Kühn, Dalton Trans., 2021, 50, 2158–2166, 10.1039/D0DT04114A.
- C. H. G. Jakob, B. Dominelli, J. F. Schlagintweit, P. J. Fischer, F. Schuderer, R. M. Reich, F. Marques, J. D. G. Correia and F. E. Kühn, Chem.–Asian J., 2020, 15, 4275–4279, DOI:10.1002/asia.202001104.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804, DOI:10.1021/ic503043h.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Angew. Chem., Int. Ed., 2023, 62, e202218000, DOI:10.1002/anie.202218000.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462, 10.1039/C8CS00570B.
- M. G. Karaaslan, A. Aktaş, C. Gürses, Y. Gök and B. Ateş, Bioorg. Chem., 2020, 95, 103552, DOI:10.1016/j.bioorg.2019.103552.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284, 10.1039/C2DT32617E.
- I. Ott, in Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells, ed. K. K.-W. Lo, Academic Press, 2017, pp. 147–179, DOI:10.1016/B978-0-12-803814-7.00005-8.
- I. Ott, in Adv. Inorg. Chem., ed. P. J. Sadler and R. van Eldik, Academic Press, 2020, vol. 75, pp. 121–148, DOI:10.1016/bs.adioch.2019.10.008.
- S. Nayak and S. L. Gaonkar, ChemMedChem, 2021, 16, 1360–1390, DOI:10.1002/cmdc.202000836.
- Nature Editorial, Nature, 2023, 617, 438, DOI:10.1038/d41586-023-01612-x.
- C. Heinemann, T. Müller, Y. Apeloig and H. Schwarz, J. Am. Chem. Soc., 1996, 118, 2023–2038, DOI:10.1021/ja9523294.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496, DOI:10.1038/nature13384.
- T. P. Schlachta, Master's thesis, Technical University of Munich, Garching, Germany, 2020.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956, DOI:10.1021/ol990909q.
- P. S. Athey and G. E. Kiefer, J. Org. Chem., 2002, 67, 4081–4085, DOI:10.1021/jo016111d.
- Y. Chun, N. J. Singh, I. C. Hwang, J. W. Lee, S. U. Yu and K. S. Kim, Nat. Commun., 2013, 4, 1797, DOI:10.1038/ncomms2758.
- Z. Li, E. R. R. Mackie, P. Ramkissoon, J. C. Mather, N. Wiratpruk, T. P. Soares da Costa and P. J. Barnard, Dalton Trans., 2020, 49, 12820–12834, 10.1039/D0DT02225J.
- J. W. Emsley, J. Feeney and L. H. Sutcliffe, High Resolution Nuclear Magnetic Resonance Spectroscopy, Elsevier, 2013, vol. 2 Search PubMed.
- R. J. Abraham, M. Canton and L. Griffiths, Magn. Reson. Chem., 2001, 39, 421–431 CrossRef CAS.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892, DOI:10.1016/j.ccr.2008.05.018.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492, DOI:10.1021/acs.chemrev.8b00067.
- T. P. Schlachta, G. G. Zámbó, M. J. Sauer, I. Rüter, C. A. Hoefer, S. Demeshko, F. Meyer and F. E. Kühn, J. Catal., 2023, 426, 234–246, DOI:10.1016/j.jcat.2023.07.018.
- W. A. Herrmann, G. Gerstberger and M. Spiegler, Organometallics, 1997, 16, 2209–2212 CrossRef CAS.
- P. De Fremont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef CAS.
- J. F. Schlagintweit, L. Nguyen, F. Dyckhoff, F. Kaiser, R. M. Reich and F. E. Kühn, Dalton Trans., 2019, 48, 14820–14828 RSC.
- P. J. Altmann, D. T. Weiss, C. Jandl and F. E. Kühn, Chem.–Asian J., 2016, 11, 1597–1605, DOI:10.1002/asia.201600198.
- B. Dominelli, G. M. Roberts, C. Jandl, P. J. Fischer, R. M. Reich, A. Pöthig, J. D. G. Correia and F. E. Kühn, Dalton Trans., 2019, 48, 14036–14043, 10.1039/C9DT03035B.
- M. M. Watts, J. Am. Oil Chem. Soc., 1990, 67, 993–995 CrossRef CAS.
- M. A. Kalam, K. Haraguchi, S. Chandani, E. L. Loechler, M. Moriya, M. M. Greenberg and A. K. Basu, Nucleic Acids Res., 2006, 34, 2305–2315, DOI:10.1093/nar/gkl099.
- C.-Y. Liao, K.-T. Chan, C.-Y. Tu, Y.-W. Chang, C.-H. Hu and H. M. Lee, Chem.–Eur. J., 2009, 15, 405–417, DOI:10.1002/chem.200801296.
- W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew Chem. Int. Ed. Engl., 1995, 34, 2371–2374, DOI:10.1002/anie.199523711.
- D. T. Weiss, P. J. Altmann, S. Haslinger, C. Jandl, A. Pöthig, M. Cokoja and F. E. Kühn, Dalton Trans., 2015, 44, 18329–18339, 10.1039/C5DT02386F.
- R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495, DOI:10.1021/ja0438821.
- M. Süßner and H. Plenio, Chem. Commun., 2005, 5417–5419, 10.1039/B512008J.
- F. E. Hahn, V. Langenhahn, T. Lügger, T. Pape and D. Le Van, Angew. Chem., Int. Ed., 2005, 44, 3759–3763, DOI:10.1002/anie.200462690.
- R. McKie, J. A. Murphy, S. R. Park, M. D. Spicer and S.-z. Zhou, Angew. Chem., Int. Ed., 2007, 46, 6525–6528, DOI:10.1002/anie.200702138.
- Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081–3087, 10.1039/C2SC20628E.
- C. S. t. Brinke and F. Ekkehardt Hahn, Dalton Trans., 2015, 44, 14315–14322, 10.1039/C5DT02115D.
- H. M. Bass, S. A. Cramer, A. S. McCullough, K. J. Bernstein, C. R. Murdock and D. M. Jenkins, Organometallics, 2013, 32, 2160–2167, DOI:10.1021/om400043z.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404, DOI:10.1021/om900667d.
- P. J. Altmann and A. Pöthig, Chem. Commun., 2016, 52, 9089–9092, 10.1039/C6CC00507A.
- H. M. Bass, S. A. Cramer, J. L. Price and D. M. Jenkins, Organometallics, 2010, 29, 3235–3238, DOI:10.1021/om100625g.
- E. B. Bauer, M. A. Bernd, M. Schütz, J. Oberkofler, A. Pöthig, R. M. Reich and F. E. Kühn, Dalton Trans., 2019, 48, 16615–16625 RSC.
- It needs to be noted that the elemental analysis still includes MeCN as an impurity.
- F. K.-M. Chan, K. Moriwaki and M. J. De Rosa, in Immune Homeostasis: Methods and Protocols, ed. A. L. Snow and M. J. Lenardo, Humana Press, Totowa, NJ, 2013, pp. 65–70, DOI:10.1007/978-1-62703-290-2_7.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378, DOI:10.1016/j.ejphar.2014.07.025.
- N. Vasan, J. Baselga and D. M. Hyman, Nature, 2019, 575, 299–309, DOI:10.1038/s41586-019-1730-1.
- J. F. Schlagintweit, C. H. G. Jakob, N. L. Wilke, M. Ahrweiler, C. Frias, J. Frias, M. König, E.-M. H. J. Esslinger, F. Marques, J. F. Machado, R. M. Reich, T. S. Morais, J. D. G. Correia, A. Prokop and F. E. Kühn, J. Med. Chem., 2021, 64, 15747–15757, DOI:10.1021/acs.jmedchem.1c01021.
- A. Frost and A. Carlson, J. Org. Chem., 1959, 24, 1581–1582, DOI:10.1021/jo01092a614.
- D. E. Goldberg and K. C. Patel, J. Inorg. Nucl. Chem., 1972, 34, 3583–3584, DOI:10.1016/0022-1902(72)80260-4.
- L. W. Jenneskens, J. Mahy, E. M. M. de Brabander-van den Berg, I. van der Hoef and J. Lugtenburg, Recl. Trav. Chim. Pays-Bas, 1995, 114, 97–102, DOI:10.1002/recl.19951140305.
- E. Lindner, G. von Au and H. J. Eberle, Chem. Ber., 1981, 114, 810–813 CrossRef CAS.
- Z. Li, E. R. Mackie, P. Ramkissoon, J. C. Mather, N. Wiratpruk, T. P. S. da Costa and P. J. Barnard, Dalton Trans., 2020, 49, 12820–12834 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179, DOI:10.1021/om100106e.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, biological studies, analytical data and crystallographic data. CCDC 2299372–2299374. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra01195c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |