
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ambipolar PEDOT-perfluorinated porphyrin electropolymer: application as an active material in energy storage systems†
Elizabeth Bermúdez Prieto‡
a,
Edwin J. González López‡ b,
Claudia A. Solis‡a,
Jhair C. Leon Jaramillo‡a,
Lorena P. Macora,
Rodrigo E. Domínguezc,
Yohana B. Palaciosb,
Silvestre Bongiovanni Abeld,
Edgardo N. Durantinib,
Luis A. Oteroa,
Miguel A. Gervaldo*a and
Daniel A. Heredia*b
b,
Claudia A. Solis‡a,
Jhair C. Leon Jaramillo‡a,
Lorena P. Macora,
Rodrigo E. Domínguezc,
Yohana B. Palaciosb,
Silvestre Bongiovanni Abeld,
Edgardo N. Durantinib,
Luis A. Oteroa,
Miguel A. Gervaldo*a and
Daniel A. Heredia*b
aIITEMA-CONICET, Departamento de Química, Facultad de Ciencias Exactas, Físico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Agencia Postal Nro. 3, X5804BYA Río Cuarto, Córdoba, Argentina. E-mail: mgervaldo@exa.unrc.edu.ar; Fax: +54 358 76233; Tel: +54 358 4676538
bIDAS-CONICET, Departamento de Química, Facultad de Ciencias Exactas, Físico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Agencia Postal Nro. 3, X5804BYA Río Cuarto, Córdoba, Argentina. E-mail: dheredia@exa.unrc.edu.ar
cINFIQC-CONICET, Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba X5000HUA, Argentina
dINTEMA-CONICET, Facultad de Ingeniería, Universidad Nacional de Mar del Plata, B7606WV Mar del Plata, Buenos Aires, Argentina
First published on 16th May 2024
Abstract
The development of functional organic materials is crucial for the advancement of various fields, such as optoelectronics, energy storage, sensing, and biomedicine. In this context, we successfully prepared a stable ambipolar perfluoroporphyrin-based polymeric film by electrochemical synthesis. Our strategy involved the synthesis of a novel tetra-pentafluorophenyl porphyrin covalently linked to four 3,4-ethylenedioxythiophene (EDOT) moieties. The resulting monomer, EDOT-TPPF16, was obtained through a straightforward synthetic approach with a good overall yield. The unique molecular structure of EDOT-TPPF16 serves a dual function, with EDOT moieties allowing electropolymerization for polymeric film formation, while the electron-acceptor porphyrin core enables electrochemical reduction and electron transport. The electrochemical polymerization permits the polymer (PEDOT-TPPF16) synthesis and film formation in a reproducible and controllable manner in one step at room temperature. Spectroelectrochemical experiments confirmed that the porphyrin retained its optoelectronic properties within the polymeric matrix after the electrochemical polymerization. The obtained polymeric material exhibited stable redox capabilities. Current charge–discharge cycles and electrochemical impedance spectroscopy of the electrochemically generated organic film demonstrated that the polymer could be applied as a promising active material in the development of supercapacitor energy storage devices.
1. Introduction
Structures based on porphyrins hold significant biological and technical importance as they have crucial roles in light absorption, oxygen transportation, redox reactions, and electron transfer processes.1,2 Porphyrins possess exceptional characteristics, including optical, magnetic, redox, and photophysical properties, making them highly valuable for a wide range of applications. Chemically modified porphyrins with different functionalities can be tailored for use in solar cells,3,4 sensors,5,6 medical imaging,7,8 photodynamic therapy,9–11 supercapacitors,1,12,13 and other applications. Porphyrins are characterized by their highly stable aromatic macrocyclic nature, abundant nitrogen atoms, and an extensive π-conjugated system. In recent years, research on organic polymers based on aromatic macrocycles has shown rapid and diverse advancements.14–17 In particular, porphyrin-based polymers have received considerable attention because these polymers usually retain the original outstanding optical and electrical properties of the constituent porphyrin monomers, as well as their structural adaptability.18 These polymers are widely used in diverse fields, such as energy storage,19,20 adsorption,16,21 separation,16,17 sensing,22 and optoelectronics,23,24 among others. Specially designed porphyrin monomers can be used to produce polymers with highly branched structures, primarily through modifications at the four meso positions with polymerizable groups.25,26 Tetradentate porphyrin-based polymers can improve substance transport through the void spaces in the polymeric matrix.21 Additionally, porphyrin-based conducting polymers can be designed for diverse purposes for the development of electronic and optoelectronic devices, as either hypercrosslinked fully conjugated polymers or non-conjugated redox polymers.16,27,28Powdered polymers derived from porphyrins can exhibit high porosity and a crystalline structure; however, they are often difficult to manage and dissolve, limiting their practical applications. Traditional coating techniques such as spin coating, dip coating, or layer-by-layer self-assembling encounter challenges in creating uniform films from these powdered porphyrin polymers. Additionally, chemical polymerization typically involves complex or multi-step procedures and challenging purification steps due to low solubility.29,30 An innovative approach to face this situation is to incorporate electropolymerizable groups into porphyrin macrocycles. These moieties participate in electrochemically induced coupling reactions, producing novel polymeric materials with connected macrocycles.31,32 Electrochemical methods enable the direct polymer synthesis and the fabrication of polymeric films on conductive substrates in a single step, circumventing the challenges associated with the use of polymer powders obtained from chemical polymerization.33 Electrochemical polymerization is a versatile methodology used to create organic films over metallic contacts or transparent conducting oxides. The electrochemical process is simple, reproducible, and has clear advantages over chemical polymerization and other deposition techniques. Depending on the electrochemical method used, it is possible to achieve precise control over film thickness.34–36 Similarly, the morphology of the resultant films can be tailored by adjustment of the experimental conditions and electrochemical parameters.36–38 Unlike chemical synthesis, electropolymerization is a one-step process, eliminating the need for subsequent purification as only the monomer is present in the solution. There is also a significant reduction in waste production.39 In addition, electropolymerization is conducted at room temperature, providing an advantage over thermal evaporation methods that require materials with high thermal stability. In many cases, polymers tend to decompose before reaching the sublimation stage in the thermal evaporation processes.
On the other hand, currently, many reports related to the development of organic functional materials40 involve the synthesis of new compounds through complex synthetic pathways that require multiple steps, high-cost reagents, complex reaction conditions, and/or reactions that result in low yields. Although these processes frequently produce materials with remarkable properties that are essential for the advancement of state-of-the-art technologies,41–43 it is critical to recognize the difficulties associated with this situation. Practical implementation, large-scale production, and commercialization of the reported materials may be hindered, impeding the transition from laboratory to marketplace. Consequently, in pursuit of practical applications, there is a pressing need to pivot toward more straightforward, cost-effective synthetic approaches that take into account both scalability and feasibility. In response to this demand, we have developed a simple synthetic procedure of two steps for the monomeric tetradentate porphyrin that utilizes readily accessible, affordable, and commercially available starting materials.
In this work, we report for the first time the monomer synthesis of a tetra-ethylenedioxythiophene (EDOT) modified fluorinated porphyrin (EDOT-TPPF16) and the subsequent electrochemical synthesis of polymeric films (PEDOT-TPPF16). The design of this monomer combines the highly robust perfluorinated porphyrin, which has extraordinary photo and chemical stability, with the versatile and widely used electropolymerizable EDOT group, which is an adaptable matrix to generate electrodeposited polymers that show excellent stability and redox properties.44–46 The presence of fluorine atoms in the porphyrin macrocycle is known to enhance photostability, influence redox characteristics, and increase singlet oxygen generation.47,48 In this sense, fluorinated porphyrin-based polymers have been utilized as oxygen carriers to enhance photodynamic therapy, particularly in alleviating tumor hypoxia.47,49,50 Additionally, perfluorinated porphyrins have found applications in developing active materials for oxygen sensing,51,52 and fluorinated porous conjugated polyporphyrins were applied in heterogeneous catalysis.53 The electrochemical polymerization of EDOT-TPPF16 led to the synthesis and deposition of electroactive polymeric films that were characterized by UV-visible spectroscopy, cyclic voltammetry (CV), atomic force microscopy (AFM), scanning electron microscopy (SEM) and spectroelectrochemistry. PEDOT-TPPF16 films were evaluated as pseudocapacitive polymers through galvanostatic charge–discharge (GCD) experiments and electrochemical impedance spectroscopy (EIS). Our outcomes showed that these polymeric films are promising materials that can be used in energy storage devices. To the best of our knowledge, this work represents the first electropolymerization of a perfluorinated porphyrin substituted with EDOT, showcasing promising and potential optoelectronic properties.
2. Experimental section
2.1. Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to give the desired macrocycle TPPF20 (1.25 g, 1.28 mmol, 67%) as a purple solid. The NMR data for this porphyrin were consistent with the literature. 1H NMR (500 MHz, CDCl3) δ 8.92 (s, 8H, pyrrole-H), −2.91 (s, 2H, pyrrole N–H). 19F NMR (500 MHz, CDCl3) δ −136.44 to −136.56 (m, 8F), −151.21 (t, J = 20.8 Hz, 4F), −161.32 (td, J = 22.7, 7.5 Hz, 8F).:1) to afford the desired product EDOT-TPPF16 (539 mg, 0.34 mmol, 74%). 1H NMR (500 MHz, CDCl3) δ 8.91 (s, 8H), 6.49 (d, J = 3.6 Hz, 4H), 6.47 (d, J = 3.6 Hz, 4H), 4.85–4.81 (m, 4H), 4.81–4.73 (m, 8H), 4.54 (d, J = 12.0 Hz, 4H), 4.46 (dd, J = 11.7, 6.2 Hz, 4H), −2.90 (s, 2H). 19F NMR (500 MHz, CDCl3) δ −138.01 (dd, J = 21.2, 5.8 Hz, 8F), −156.71 (dd, J = 22.3, 7.5 Hz, 8F). MALDI-TOF [m/z] 1583.172 [M + H]+ (1583.119 calculated for C72H39F16N4O12S4).
:3) to give the desired macrocycle TPPF20 (1.25 g, 1.28 mmol, 67%) as a purple solid. The NMR data for this porphyrin were consistent with the literature. 1H NMR (500 MHz, CDCl3) δ 8.92 (s, 8H, pyrrole-H), −2.91 (s, 2H, pyrrole N–H). 19F NMR (500 MHz, CDCl3) δ −136.44 to −136.56 (m, 8F), −151.21 (t, J = 20.8 Hz, 4F), −161.32 (td, J = 22.7, 7.5 Hz, 8F).:1) to afford the desired product EDOT-TPPF16 (539 mg, 0.34 mmol, 74%). 1H NMR (500 MHz, CDCl3) δ 8.91 (s, 8H), 6.49 (d, J = 3.6 Hz, 4H), 6.47 (d, J = 3.6 Hz, 4H), 4.85–4.81 (m, 4H), 4.81–4.73 (m, 8H), 4.54 (d, J = 12.0 Hz, 4H), 4.46 (dd, J = 11.7, 6.2 Hz, 4H), −2.90 (s, 2H). 19F NMR (500 MHz, CDCl3) δ −138.01 (dd, J = 21.2, 5.8 Hz, 8F), −156.71 (dd, J = 22.3, 7.5 Hz, 8F). MALDI-TOF [m/z] 1583.172 [M + H]+ (1583.119 calculated for C72H39F16N4O12S4).2.2. Spectroscopic properties
The UV-visible absorption spectra of EDOT-TPPF16 and PEDOT-TPPF16 were obtained in a diluted acetonitrile (ACN) solution and a thin film electrodeposited on indium tin oxide (ITO), respectively. The spectrum of EDOT-TPPF16 was recorded and compared with that of TPPF20 at room temperature using a quartz cuvette with a 10 mm path. The absorption spectrum corresponding to PEDOT-TPPF16 on ITO was acquired by placing the electrode in the spectrometer cell holder. For background correction, a bare ITO electrode was utilized. Fluorescence emission spectra of EDOT-TPPF16 and TPPF20 were recorded in diluted ACN solutions at room temperature, exciting the sample at the maximum of the Soret band (Abs = 0.1).2.3. Electrochemistry and electrodeposition
Electrochemical measurements were carried out as described previously.26,35,54,55 CV, differential pulse voltammetry (DPV), GCD tests, and EIS were performed at room temperature with a Palm Sense 4 potentiostat. Electrochemical studies were performed in a deaerated (with high purity argon) monomer solution containing 0.1 M of the supporting electrolyte or only with electrolyte—the last solution for the electrochemical responses of the electropolymerized films. A three-electrode system composed of a glassy carbon (GC) or ITO coated glass working electrode, a platinum wire counter electrode, and a silver wire quasi-reference electrode were utilized for electrochemistry experiments. Ferrocene was used as internal standard for reference potential calibration in organic media (vs. Saturated Calomel Electrode, SCE). Fc/Fc+ = 0.40 V vs. SCE.36 The ITO electrode substrates (Delta Technologies, nominal resistance 8–12 Ω per square) were cleaned sequentially in an ultrasonic bath with acetone, ethanol, and deionized water and dried before use.
The quantification of the electropolymerized material on the ITO electrode was determined following the methodology outlined by Zhang et al. and other researchers.56 This method assumes that, after electropolymerization, any alterations in the absorption coefficient of the porphyrin chromophore are negligible.26 Adopting the above-mentioned methodology ensures a reliable estimation of the quantity of electropolymerized material by considering the minimal changes in the absorption characteristics of the porphyrin chromophore.
2.4. X-ray diffraction (XRD)
XRD scanning of PEDOT-TPPF16 electrodeposited on ITO was measured using a Panalytical X'Pert PRO X-ray Diffractometer (30 mA and 40 kV) equipped with a Cu-Kα radiation source (λ ∼ 1.54 Å) at room temperature. The scans were performed in a 2θ range of 10–60° at 1° min−1.2.5. Fourier transform infrared spectroscopy (FTIR)
FTIR spectroscopic analysis of EDOT-TPPF16, and PEDOT-TPPF16 was performed in attenuated total reflectance mode (ATR-FTIR) using a Nicolet 6700, Thermo Scientific Inc. device. Spectra were recorded in the range of 400–4000 cm−1, performing 64 scans with a resolution of 4 cm−1 at room temperature.2.6. Spectroelectrochemical studies
Spectroelectrochemical measurements were carried out as described previously.35,57 In situ UV-visible spectroelectrochemical measurements under potentiostatic control were obtained using a Hewlett Packard UV-vis diode array spectrophotometer at kinetic measurement mode by utilizing a three-electrode configuration of an ITO modified with polymeric film (see Fig. S1†). A naked ITO electrode was used as a blank for absorption background correction.2.7. Surface characterization
2.8. GCD and EIS measurements
In order to evaluate the performance of the porphyrin films as supercapacitive materials, GCD and EIS experiments were carried out in a three-electrode configuration using a GC working electrode modified with polymeric film, in TBAP/ACN solution.GCD-test was performed over a cell cycled between 0–(+1.0) and 0–(−1.0) V at various current densities with chronopotentiometry method. Before and after charge–discharge experiments, PEDOT-TPPF16 film was evaluated by CV in the supporting electrolyte solution until obtained repetitive profiles.
The specific capacitance was calculated with respect to the mass of the activated polymeric material using eqn (1).
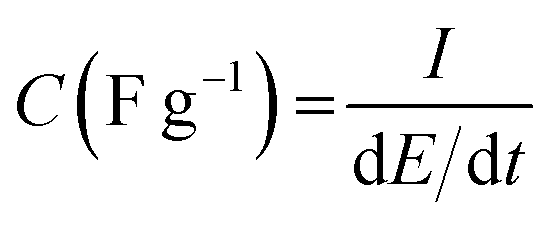 | (1) |
EIS was performed using a range of 0.1–105 Hz with a perturbation amplitude of 5 mV at an applied potential of 0.75 V.59,60
3. Results and discussion
3.1. Design, synthesis, and characterization of EDOT-TPPF16 monomer
As mentioned above, the development of simple synthetic processes is paramount for advancing materials used in the field of optoelectronics. The implementation of organic materials in real practical applications necessitates a transition to less complex and more economical synthetic procedures. Thus, we implemented a straightforward two-step synthesis approach, utilizing readily accessible and affordable initial materials.The synthesis of EDOT-TPPF16 was carried out as shown in Scheme 1. The first step involved an acid-catalyzed condensation reaction between pyrrole and pentafluorobenzaldehyde in DCM at room temperature for 44 hours. After this period, the hydrogenated tetrapyrrolic macrocycle was oxidized with DDQ in a one-pot reaction, affording the tetra-aryl porphyrin TPPF20 with a good yield of 67%.61 This macrocycle is a versatile building block that can be used to construct more complex and elaborated materials by attaching additional moieties with the desired properties.62–64 Pentafluorophenyl (PFP) groups located around the porphyrin ring can be regioselectively functionalized with a wide range of nucleophiles (alcohols, azide, thiols, amines, etc.) through nucleophilic aromatic substitution (SNAr) reactions.61–65
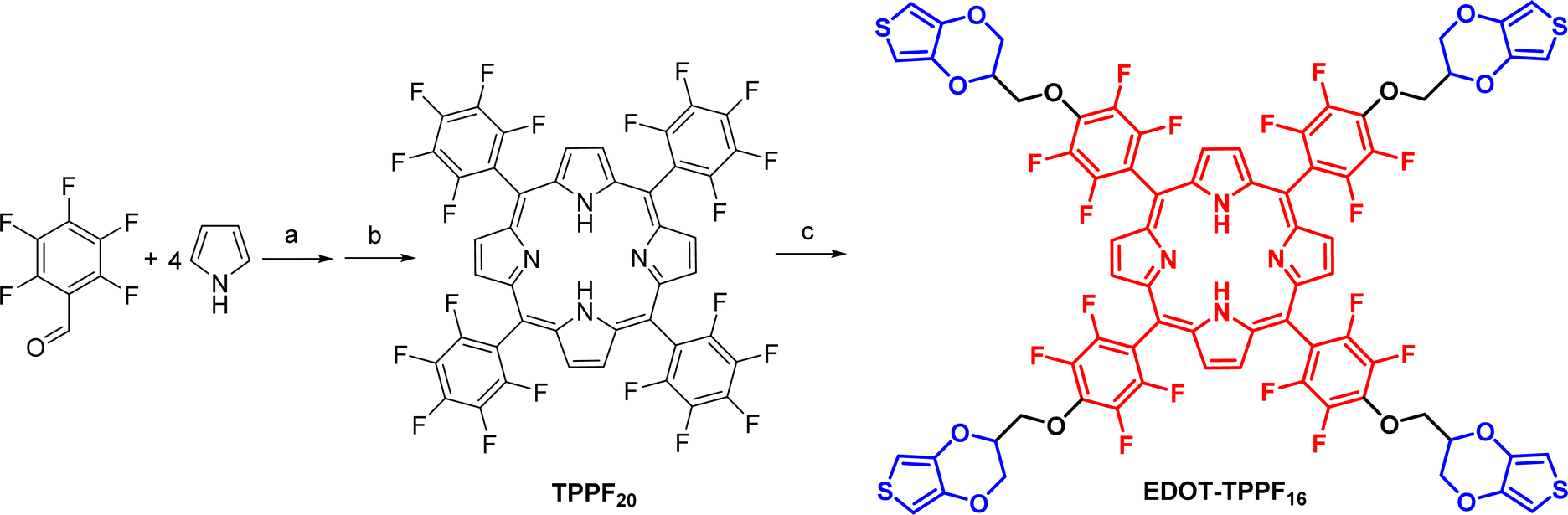 | ||
| Scheme 1 Synthesis of EDOT-TPPF16. Reagents and conditions: (a) BF3·OEt2, DCM, r.t., 44 h; (b) DDQ, DCM, r.t., 1.5 h, 67%; (c) EDOT-OH, DMF, Cs2CO3, 70 °C, 4 h, 74%. | ||
In a second step, the fluorinated template TPPF20 was subjected to a regioselective SNAr with EDOT-OH. For this kind of transformation using alcohols, it is common to employ KOH in dry tetrahydrofuran, using an excess of alcohol to avoid the formation of hydroxide.65 However, we opted for different conditions. The functionalization of the flexible matrix TPPF20 was carried out in DMF and using Cs2CO3.66 Thus, after one hour of reaction, the alcohol was converted into the alkoxide, and the substitution of the four para-fluoro atoms of the PFP groups occurred. The EDOT-substituted porphyrin (EDOT-TPPF16) was obtained with yields as high as 74% and good reproducibility.
The molecular structure of EDOT-TPPF16 was fully elucidated and determined by Fourier-transform infrared spectroscopy (FT-IR), nuclear magnetic resonance (NMR) spectroscopy, and matrix assisted laser desorption ionization – time of flight (MALDI-TOF) spectrometry (Fig. S2–S8†). The tetrasubstituted tetrapyrrolic macrocycle was confirmed in the first instance by MALDI-TOF, which disclosed the incorporation of the four EDOT moieties. The 1H NMR spectrum of EDOT-TPPF16 combined the characteristic resonances of the EDOT unit, together with the diagnostic protons corresponding to the porphyrin ring. In the aromatic region, due to the symmetric substitution of the tetrapyrrolic macrocycle, the eight β-pyrrolic protons gave rise to a singlet at δ = 8.91 ppm. The thiophene protons appeared as two doublets at δ = 6.49 and δ = 6.47 ppm (J = 3.6 Hz) due to the asymmetric substitution on the EDOT ring. The integrals of these two signals were four each, confirming the incorporation of four EDOT units, and consistent with the cruciform architecture of the porphyrin-core. In the aliphatic region, the methylene and methine resonances belonging to the dioxin ring are overlapped in the region between 4.40 and 4.90 ppm, giving rise to complex multiples. Finally, in the upfield region, a singlet at δ = −2.90 ppm that integrated for two is assigned to the inner NH protons of the macrocycle. On the other hand, 19F spectrum of EDOT-TPPF16 was used not only to confirm the tetra-substitution but also to demonstrate the regioselectivity of the SNAr. The spectrum of the tetra-substituted porphyrin exhibits a complete disappearance of the triplet at δ = −151.21 ppm corresponding to the para-fluorine atoms. Furthermore, the doublet belonging to meta-fluorine atoms exhibits a shift from δ = −161.32 ppm in TPPF20 to δ = −156.71 ppm in EDOT-TPPF16. These fluorine atoms near the nucleophile are more sensitive than the doublet in the ortho position, which only presents a slight shift.66
At this point, we synthesized a meso-pentafluorophenyl-substituted tetrapyrrole system that combines an electron-withdrawing porphyrin core with four electropolymerizable EDOT moieties in the periphery. We chose the TPPF20 nucleus because the highly robust perfluorinated porphyrin exhibits extraordinary chemical stability, water repulsion properties, and undergoes two reversible reduction processes.61,67,68 The attachment of EDOT moieties into this macrocycle offers an innovative approach to participate in electrochemical coupling reactions, creating a new polymeric material with interconnected porphyrin cores. EDOT polymers are characterized by their electrochemical stability, ultrafast electron-transfer rates, and good hole-transporting and capacitive properties.45,46 Thus, EDOT units in EDOT-TPPF16 can be employed for the electrochemical production of polymeric films directly onto conductive substrates in a single step, with the porphyrin core serving as the n-type material and the PEDOT chain as the p-type material.
3.2. Electrochemical characterization of EDOT-TPPF16
Comparative electrochemical studies of TPPF20 and EDOT-TPPF16 were carried out using CV and DPV measurements in order to determine their electrochemical properties. The analysis was performed on GC (Fig. 1a and b). TPPF20 CV presents two quasireversible oxidations (DPV peaks at 1.38 and 1.63 V vs. SCE) and two quasireversible reductions (DPV peaks at −0.89 and −1.31 V vs. SCE). These results are typical for free base porphyrins, which usually show two well-defined reversible single-electron macrocycle oxidations and reductions in nonbinding solvents,69 related to the formation of the corresponding radical cation and dication, and radical anion and dianion, respectively.70–72 However, it is known that peripheral substitution in tetraphenyl porphyrins has a strong effect in modulating their electron donor–acceptor capabilities. In the present case, we observed that the TPPF20 oxidation occurs at more positive (anodic) and the reduction at less negative (cathodic) potentials compared to the values reported for tetraphenyl porphyrins without substituents. This is a consequence of the electron-withdrawing effects characteristic of the perfluorinated benzene rings.69,70 On the other hand, EDOT-TPPF16 exhibits similar quasireversible reductions but only one irreversible oxidation peak at around 1.25 V (cyan voltammogram in Fig. 1a), with a peak current value around four times higher than the first reduction peak. It is known that EDOT is oxidized at around 1.2 V vs. SCE, showing an irreversible peak in the first anodic scan;73,74 thus, the irreversible redox process can be assigned to oxidation of the four EDOT units bounded to the macrocycle in EDOT-TPPF16.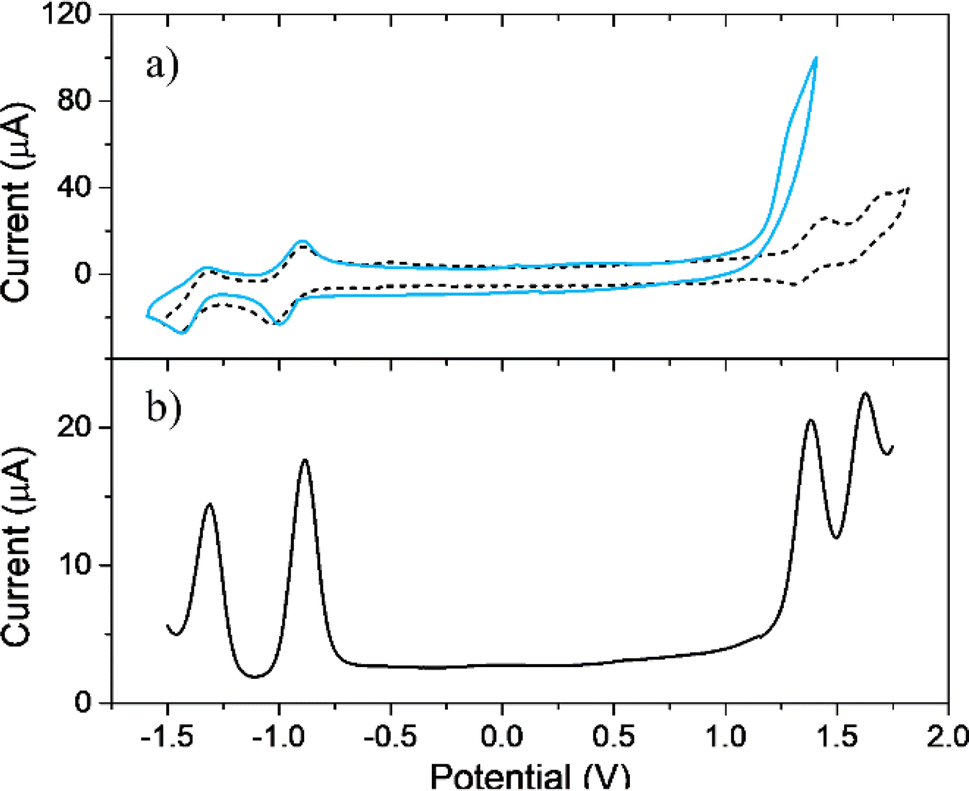 | ||
| Fig. 1 (a) Cyclic voltammograms of TPPF20 (black dashed line) and EDOT-TPPF16 (cyan continuous line) in a solution of DCE containing supporting electrolyte (TBAPF6, 0.1 M). GC electrode, scan rate 0.100 V s−1. (b) DPV of TPPF20 clearly exhibiting the four redox processes. | ||
3.3. Electrodeposition of PEDOT-TPPF16 polymeric film
It is well-known that upon oxidation EDOT and its derivatives give rise to unstable radical cations that subsequently react to produce EDOT dimers, exhibiting these dimers easier oxidations than the corresponding EDOT monomers.54,75 Furthermore, EDOT dimers, trimers, and oligomers have the potential to undergo radical coupling, leading to the formation of PEDOT chains (Scheme S1†). Fig. 2a shows twenty repetitive CV anodic scans for EDOT-TPPF16 in the −0.30 to 1.25 V potential range, where increments in the oxidation–reduction currents are detected with each new cycle, typical of the growing of an electroactive film over the GC electrode.75–77 To confirm the formation of a film, the electrochemical response of the electrode was obtained in a solution containing only supporting electrolyte (Fig. 2b). The electrochemical response of PEDOT-TPPF16 film is characterized by the presence of two reduction processes (−0.85 and −1.40 V) and one broad oxidation between 0.20 and 1.10 V with a peak potential at around 0.50 V (Table 1). The two redox systems at negative potentials can be associated with the reductions of the porphyrin macrocycle, generating the corresponding radical anion and dianion.69–72 The broad oxidation can be due to the oxidation of new EDOT dimer, trimer, or long chains which were formed by the coupling of EDOT radical cations, generated during continuous cycling.75,76 Superimposed in this broad oxidation might be also involved the oxidation of the porphyrin macrocycle.70,73 A similar electrochemical response has been reported for a film obtained after electropolymerization of a EDOT-trimer mono substituted porphyrin monomer.78 The peak currents for the redox systems present a linear relationship with the scan rate (Fig. 2c), which indicates an electroactive product irreversibly adsorbed over the electrode surface. The assessment of the stability of the polymeric material during oxidation and reduction processes was evaluated through the application of continuous CV cycles. Fig. 2d and e show one hundred superimposed cycles for both oxidation and reduction processes, respectively. The noteworthy outcome is the absence of any substantial evidence indicating a marked decrease in the current values over the cycles. This observation shows that the organic material remains resilient, exhibiting no signs of dissolution or structural alterations throughout the CV, thereby maintaining its electrochemical activity intact.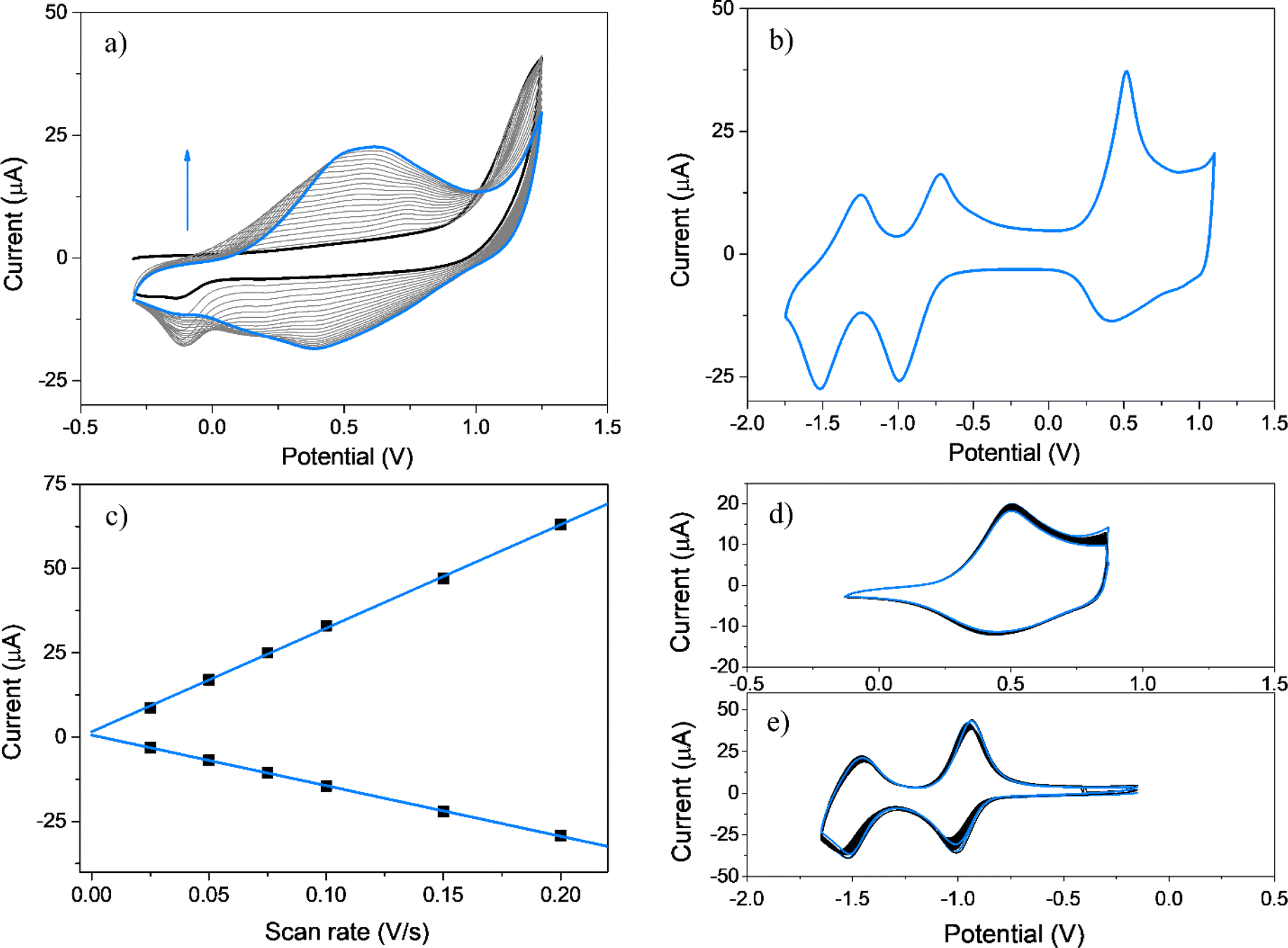 | ||
| Fig. 2 (a) First (black), 2nd–19th (grey) and 20th (cyan) cyclic voltammograms of EDOT-TPPF16. Scan rate: 0.100 V s−1. The measurements were done in DCE containing TBAPF6 using a CG working electrode. (b) Electrochemical response of the electropolymerized film PEDOT-TPPF16 at 0.100 V s−1. (c) Relationship between the peak currents vs. different scan rates (0.025, 0.050, 0.075, 0.100, 0.150, 0.200 V s−1). Data were taken at the potentials of the oxidation and reduction peaks of (b). (d and e) One hundred CV cycles of the film for: (d) oxidation and (e) reduction processes. Scan rate 0.100 V s−1, cyan lines: first and 100th cycle. (b–e) In ACN containing only support electrolyte (TBAP). | ||
| Compound | Wavelength (nm) | Potentialsd | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Soret | QI | QII | QIII | QIV | Oxidation | Reduction | |||
| I | II | I | II | ||||||
| a Peak potential.b Half wave potential.c Solution in ACN.d Potential vs. SCE.e DPV potentials. | |||||||||
| TPPF20 | 406c | 634c | 580c | 534c | 503c | 1.38e | 1.63e | −0.89e | −1.31e |
| PEDOT-TPPF16 | 415 | 643 | 589 | 546 | 509 | 0.60a | — | −1.00a | −1.55a |
| 0.50b | −0.85b | −1.4b | |||||||
| EDOT-TPPF16 | 410c | 635c | 583c | 538c | 504c | 1.25a | — | −1.00a | −1.45a |
| −0.94b | −1.37b | ||||||||
3.4. Characterization of the ambipolar polymeric film. Spectroscopic properties
Spectroscopic and spectroelectrochemical experiments were conducted to verify the formation of the films, investigate their electro-optical properties, and thoroughly analyze the reversibility linked to the redox processes. Specifically, these experiments were carried out by depositing PEDOT-TPPF16 films onto semitransparent ITO electrodes. This experimental setup allowed a detailed exploration of the characteristics and behaviors of the films, offering valuable insights into their electrochemical and optical responses. The semitransparent nature of the ITO electrodes facilitated the examination of the changes in the electronic spectra of the organic polymer in response to electrochemical stimuli. The formation of PEDOT-TPPF16 film on ITO electrode was performed by applying repetitive CV anodic scans for EDOT-TPPF16 in DCE-TBAPF6 (Fig. 3a). It can be seen that during anodic cycling, increases in oxidation–reduction currents are detected, indicating the formation and deposition of the film over the oxide semiconductor. This is a non-trivial result, as the nature of the electrode is changed. The voltammetric response of the electrode (Fig. 3b) is similar to that obtained on GC electrode (Fig. 2b), which presents two redox systems at negative potential values and a broad oxidation. In this case, the peak currents for the redox systems are also linear with the scan rate, confirming the formation of a film over the ITO electrode.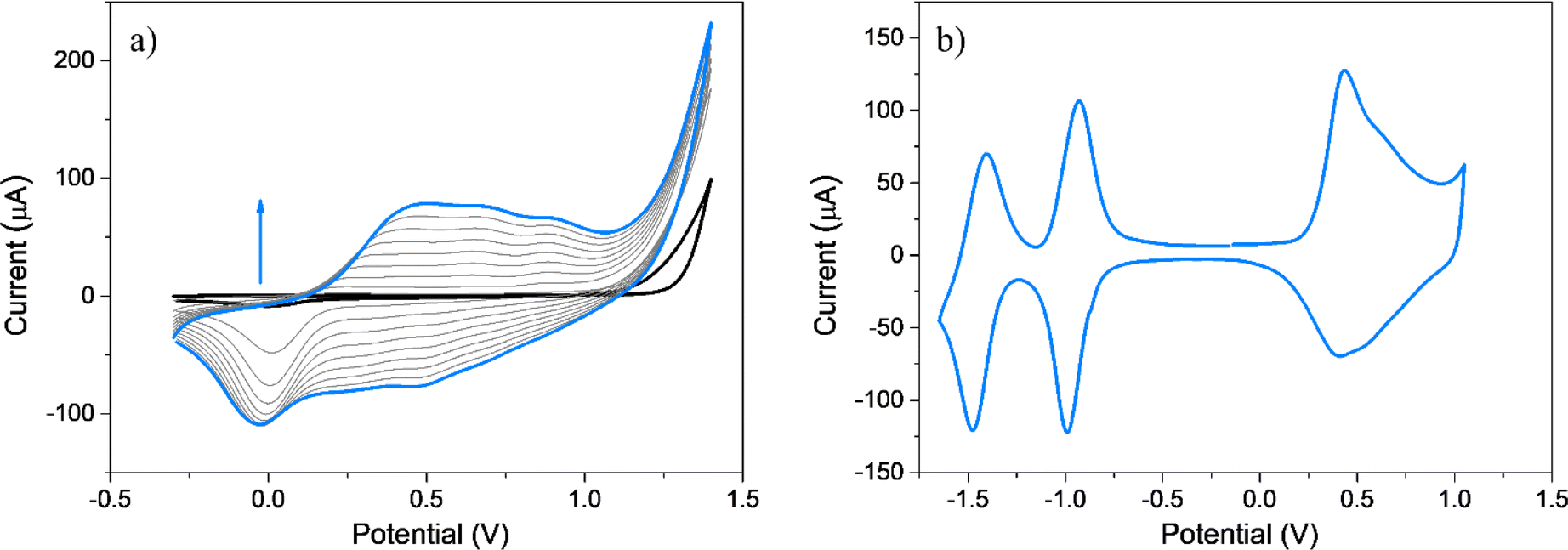 | ||
| Fig. 3 (a) First (black), 2nd–9th (grey) and 10th (cyan) cyclic voltammograms of EDOT-TPPF16. The measurements were done in DCE containing TBAPF6 (0.1 M) using an ITO working electrode. (b) Electrochemical response of the electropolymerized film PEDOT-TPPF16 in ACN containing only TBAP (0.1 M). Scan rate: 0.100 V s−1. | ||
Photophysical properties of EDOT-TPPF16 monomer and PEDOT-TPPF16 electrodeposited polymer were studied and compared in diluted solution and thin solid films, respectively. Fig. 4 displays UV-visible absorption spectra of EDOT-TPPF16 in ACN solution and PEDOT-TPPF16 electrode obtained by placing it in the spectrometer cell holder, both at room temperature. The main absorption characteristics are presented in Table 1. The monomer EDOT-TPPF16 presents the characteristic spectrum of symmetrical free base porphyrin, with a strong absorption in the blue region that belongs to the Soret band (λmax = 410 nm). Additionally, the four characteristic Q bands of the substituted porphyrin are observed in the red region (between 500 and 650 nm).61,79 The sharp electronic transition for the Soret band indicates that the monomer is not aggregated in that solvent. Furthermore, it is worth mentioning that the absorption spectrum of EDOT-TPPF16 closely resembles that of TPPF20 (see Fig. 4). The Soret band of EDOT-TPPF16 exhibits a slight bathochromic shift of ∼4 nm, which is a consequence of the auxochromic effect generated by the oxy substituents.65,66 These outcomes disclose that the external substitution of the PFP groups does not significantly affect the porphyrin-based chromophore. Similarly, the fluorescence emission spectra of TPPF20 and EDOT-TPPF16 in ACN presented a close resemblance (Fig. S9†). Both compounds exhibited the characteristic red fluorescence emission of the pentafluorophenyl-substituted porphyrins, with two emission bands centered around 635 and 700 nm.61,79
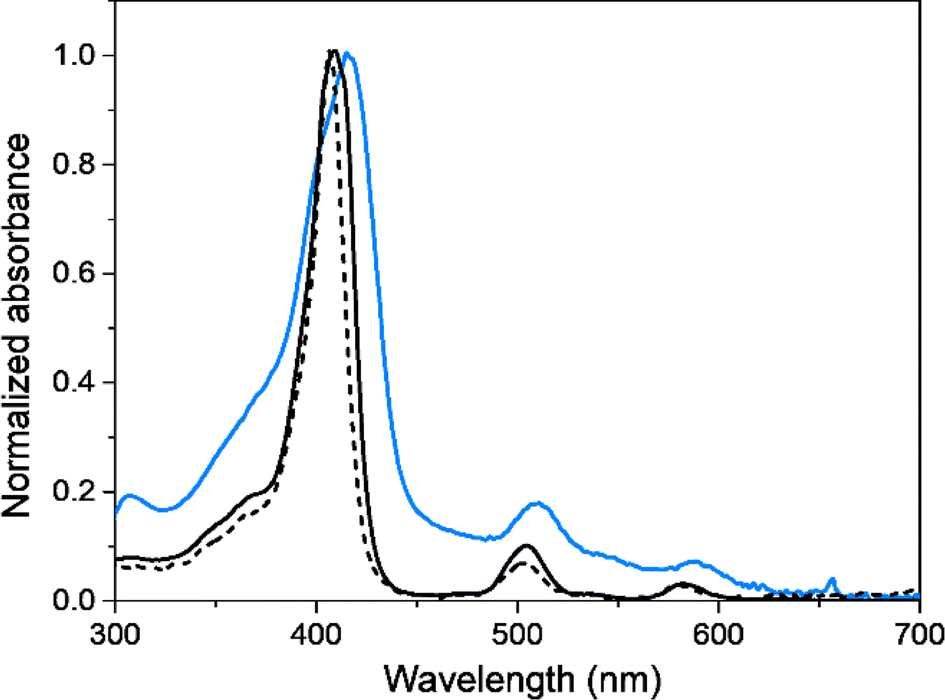 | ||
| Fig. 4 Absorption spectra of TPPF20 (black dashed line), EDOT-TPPF16 monomer (black continuous line) in ACN and PEDOT-TPPF16 electrodeposited polymer on ITO (cyan continuous line). | ||
Regarding the PEDOT-TPPF16 polymer, the UV-visible absorption spectrum exhibits similar spectroscopic features to the monomer EDOT-TPPF16, revealing that the optical properties of the porphyrin-based chromophore are conserved in the polymeric chain. As it can be observed, the electronic transitions corresponding to the tetrapyrrolic macrocycle are clearly defined. The electronic transitions of the porphyrin in the polymer resemble those of the monomer in solution, with well-defined Soret and Q bands. This result not only validates the presence of tetrapyrrolic macrocycle in the film but also demonstrates that the molecular structure of the porphyrin core is maintained intact throughout the electrodeposition process. It is worth noting that there is a broadening and a slight red-shift of the bands of approximately 5 nm compared to the monomer EDOT-TPPF16 in solution, indicating a weak interaction of the porphyrin cores in the polymeric chain. This is not trivial data since the solid state represents a highly aggregated system.
XRD experiment conducted for PEDOT-TPPF16 reflects an amorphous nature of the obtained material (Fig. S10†). This result agrees with PEDOT-based polymeric materials in literature, where a broad diffraction halo is reported at ca. 25°.80–82 This could be indicative of interchain planar ring stacking of the polymer. Additionally, low peaks can be found due to the presence of the electrolyte, but they do not correspond to the PEDOT-TPPF16 structure. In order to get more information about the polymerization mechanism and the chemical structure of the polymer, FT-IR spectra of EDOT-TPPF16 monomer and PEDOT-TPPF16 film were obtained (Fig. S11†). It can be seen that EDOT-TPPF16 monomer presents two bands at around 1184 and 891 cm−1, associated with ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H in-plane and out-of-plane deformation vibrations of the EDOT units, while these bands are absent in the spectra of PEDOT-TPPF16 film. The disappearance of these two peaks indicates an efficient coupling of the EDOT units through the α positions, which supports the proposed polymerization mechanism.83
C–H in-plane and out-of-plane deformation vibrations of the EDOT units, while these bands are absent in the spectra of PEDOT-TPPF16 film. The disappearance of these two peaks indicates an efficient coupling of the EDOT units through the α positions, which supports the proposed polymerization mechanism.83
3.5. Spectroelectrochemical characterization
As already mentioned, spectroelectrochemical studies allow us to obtain information about the chemical structure of the polymer and the redox species involved during the oxidation and reduction processes. The spectra at the different applied potentials were plotted as ΔAbs (subtracting the absorption spectrum at 0.00 V) and are shown in Fig. 5a and b. When the film is oxidized, at applied potentials between 0.00 and 0.40 V, no changes are observed, while at more positive values, the bleaching of the Soret band and two new positive bands at around 440 and 630 nm are detected. The observed spectral changes are predominantly originated from changes in the porphyrin nucleus present in the polymer. Moreover, the observation of clear isosbestic points evidently shows that the formed species are generated by oxidation of the tetrapyrrolic ring, meanwhile the full reversibility of the obtained spectra after film reduction is in agreement with the stable electrochemical response observed after hundreds of successive CV cycles. The new absorption band developed during oxidation agrees with the presence of porphyrin radical cation in the polymeric film (Fig. 5a).84,85 On the other hand, a similar spectroelectrochemical response is observed in the negative potential range (Fig. 5b). Before the film is reduced (0.00 and −0.80 V), there are no changes in the absorption values, but at more negative applied potentials, two new positive bands appear around 440 and 570 nm, accompanied by the bleaching of the Soret band. The absorption bands developed during the reduction processes are comparable to those reported and assigned to the radical anion of the porphyrin macrocycle.86 Therefore, considering the electrochemical, spectral, and spectroelectrochemical data, it is possible to confirm the unaltered presence of the porphyrin macrocycle as photoactive and electroactive centers embedded in the polymeric matrix. The porphyrins are covalently bonded to each other by EDOT dimers, trimers, or longer chains.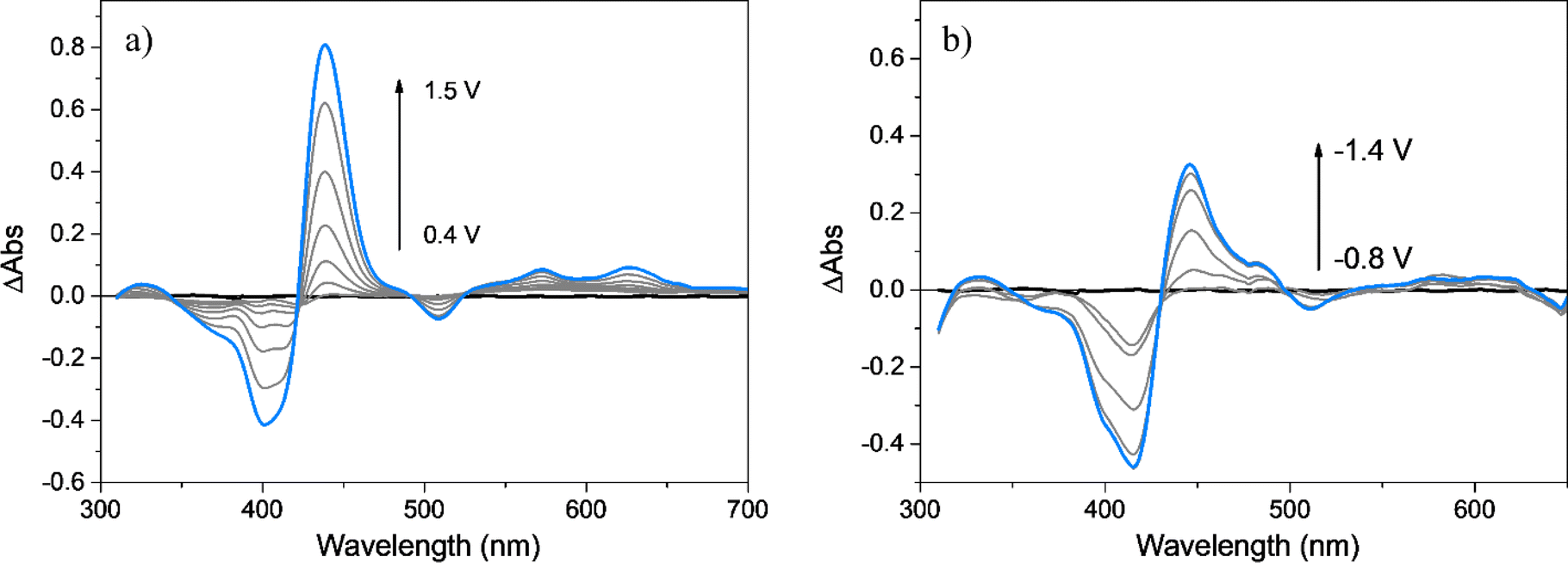 | ||
| Fig. 5 Difference absorption spectra, ΔAbs, of: PEDOT-TPPF16 film deposited over ITO electrode obtained at different applied potentials: (a) 0.4 (black line), 0.9 to 1.4 (gray lines) and 1.5 V (cyan line); (b) −0.8 (black line), −0.9 to −1.3 (gray lines) and −1.4 V (cyan line). All measurements were conducted in ACN (0.10 M TBAP) at 20 mV s−1. | ||
3.6. Surface analysis of the polymeric films
SEM images were acquired to examine the morphological characteristics of the PEDOT-TPPF16 films. Fig. 6a and b displays images of the bare ITO electrode and the PEDOT-TPPF16 film, respectively. The porphyrin film uniformly covers the entire ITO surface, presenting a nearly smooth surface formed by the agglomeration of fused spherical particles measuring about 10–20 nm (as also observed in AFM images, Fig. 6c and d) that follows the roughness pattern of ITO. The small spherical particles interconnect to form a network, creating a three-dimensional structure with void spaces. These void spaces within the film can create extensive contact areas between the liquid electrolyte and the film surface, establishing pathways for effective ion diffusion and migration.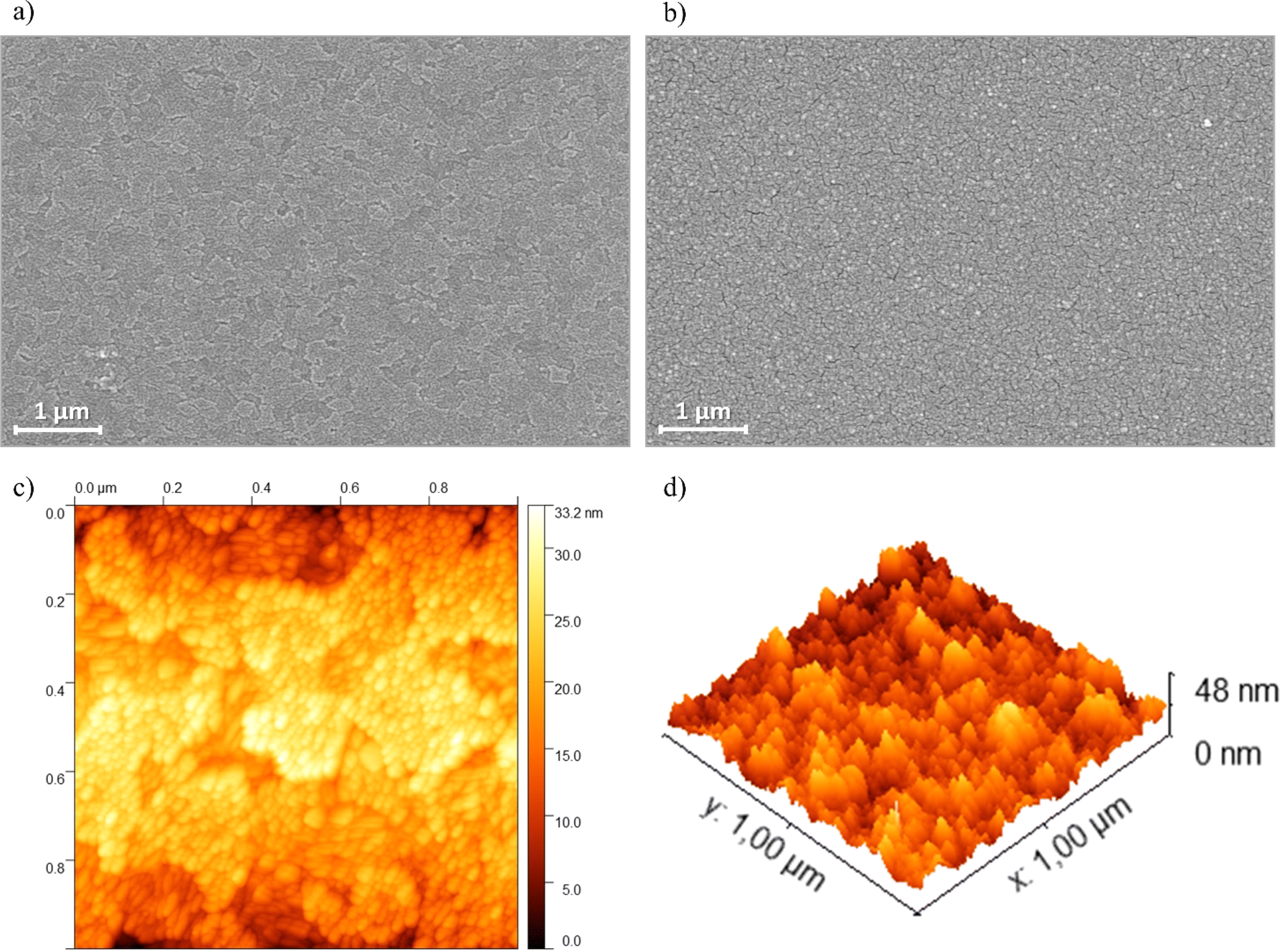 | ||
| Fig. 6 SEM images of (a) bare ITO and (b) PEDOT-TPPF16 film. Scale bars are 1 μm. (c) AFM images of ITO electrode surface (scan size 1 × 1 μm) registered after electrodeposition of porphyrin film. (d) Three-dimensional representation of the electrode surfaces measured by AFM. Films were grown cycling in the same conditions of the exhibited in Fig. 3. | ||
The surface morphologies of the resulting polymer films were further analyzed using AFM. Fig. 6c and d depict typical acoustic AC mode AFM height images of the electrodeposited film on ITO. Consistent with previous reports on electrochemically generated porphyrin films from tetradentate monomers, globular-like microstructures are evident in the analyzed films.55 This observed microstructure appears to result from the nucleation and subsequent growth processes, leading to a homogeneous decoration of the ITO surface. In agreement with this observation, the calculated root mean square roughness (Sq) of PEDOT-TPPF16 film is a little higher (1.9 ± 0.4 nm) than that of bare ITO (1.2 ± 0.2 nm). Fig. 6d illustrates that the polymer surface is highly homogeneous, completely covering the electrode.
3.7. Galvanostatic charge–discharge tests of the film
Owing to the film's reversible oxidation and reduction processes, with high cyclic stability, it can be anticipated that the polymer pseudo-capacitive behaviour could render it suitable as an active component in the development of supercapacitor devices.87 To examine the effectiveness of the polymer as an active material for supercapacitors, we conducted GCD experiments at varied current densities in a three-electrode configuration (ACN 0.10 M, TBAP). The oxidation onset and cutoff potentials were within the range of 0.0–1.0 V (Fig. 7a), while the reduction onset and cutoff potentials were within the range of 0.0 to −1.0 V (Fig. 7b), after the initial reduction peak, as observed in the CV experiments. During the oxidation processes, the voltage undergoes a rapid vertical ascent within the 0 to 0.4 V range. The energy stored in this interval, attributed to the electronic double-layer capacitance, is considered negligible.88,89 On the other hand, within the potential range of 0.4–1.0 V, the responses display a quasi-triangular charge–discharge behavior at various current densities, arising from the pseudocapacitance associated with the reversible redox processes observed in the polymeric material.55,58,90 The charge and discharge curves exhibit nearly symmetrical patterns, indicative of a rapid and reversible electron exchange.91 Furthermore, with increments in the current density, both charge and discharge times experience notable reductions. However, it is noteworthy that even at elevated current regimes of up to 50 A g−1, the film exhibits a specific capacitance retention of approximately 75%, as illustrated in Fig. 7c. Likewise, in the reduction process, charge–discharge curves exhibited similar behavior (Fig. 7b and c), with quasi-triangular performances, but displaying fast potential changes at applied potential close to the redox peak, in agreement with the observed in CV experiments. PEDOT-TPPF16 exhibits maximum specific capacitances of 140 F g−1 and 330 F g−1 (calculated from the discharge curves) for the oxidation and reduction processes, respectively, at a current density of 10 A g−1. These values are inferior to those reported for supercapacitors based on hydrogen-bonded metal porphyrin frameworks,92 but similar to the values reported for electrogenerated porphyrin polymers.26,55,87,92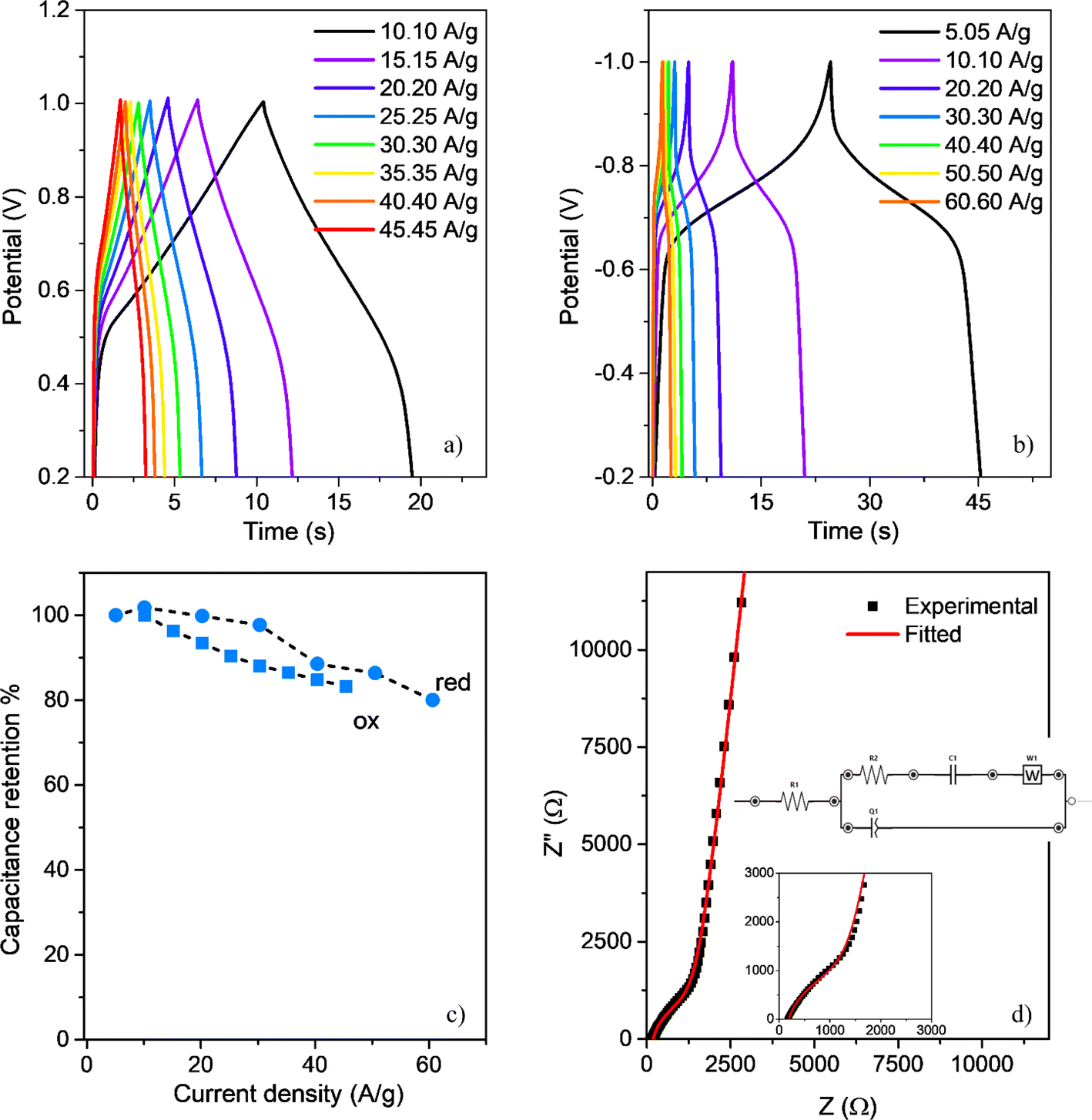 | ||
| Fig. 7 (a) Oxidation and (b) reduction galvanostatic charge/discharge cycles of PEDOT-TPPF16 at different current densities. (c) Capacitance retention % of PEDOT-TPPF16 (oxidation in blue squares and reduction in blue circles) (d) Nyquist plot of PEDOT-TPPF16 at 1.0 V. PEDOT-TPPF16 film was deposited on an GC electrode. All measurements were conducted in ACN containing 0.10 M TBAP. | ||
The observed asymmetry in GCD during the n-doping process (Fig. 7b) could be originated in the leakage process resulting from oxygen reduction reaction. n-doped polymers are less common due to their low stability in the doped state and their tendency to be de-doped by transferring electrons to ambient oxygen.93
Additionally, to further explore into the polymeric material electronic behavior analysis, we conducted EIS experiments in a three-electrode configuration (ACN 0.10 M, TBAP). Fig. 7d exhibits the Nyquist plot that we obtained for a thin film electrode generated in the same condition as the one depicted in Fig. 2a. The gathered data was fitted through the Randles equivalent circuit, illustrated in the inset of Fig. 7d, using the EIS Spectrum Analyzer software. The equivalent circuit includes an ohmic resistance (RES), an interfacial charge transfer resistance (RCT), and two constant phase elements: CDL, which represents an electric double-layer capacitance, and CPS, which represents the pseudocapacitance of the electrode. The diffusion-controlled process into the electrode was simulated with a Warburg element (Zw). The plot shows two regions. At high frequency, an equivalent series resistance (RES) is observed as a semicircle intercepting the real axis at 160 Ω. This represents the ionic and electronic resistance of the electrolyte and electrode, as well as the contacts. At low frequency, the Nyquist plot second region shows the electrode capacitive behavior through a nearly vertical line. The fitted data provided a gravimetric capacitance of 120 F g−1, similar to the value obtained from charge–discharge experiments.
4. Conclusions
This work presents the synthesis of a new fluorinated porphyrin, EDOT-TPPF16, and the subsequent electrochemical synthesis of polymeric films, PEDOT-TPPF16. The monomer design combines the robust perfluorinated porphyrin, known for its exceptional photo and chemical stability, with the versatile and widely used electropolymerizable EDOT group. The electrochemical polymerization of EDOT-TPPF16 generated electroactive polymeric films that showed promising pseudocapacitive behavior in energy storage devices. Various techniques were used to characterize the films. These presented gravimetric capacitances of 140 F g−1 and 330 F g−1 for the cathodic and anodic processes, respectively, at a current density of 10 A g−1. To the best of our knowledge, this is the first time that a perfluorinated porphyrin substituted with EDOT is electropolymerized, representing an important advancement in the field of electroactive polymeric materials.Author contributions
Conceptualization, E. B. P., E. J. G. L., C. A. S., J. C. L., L. P. M., R. E. D., Y. B. P., E. N. D., L. A. O., M. A. G. and D. A. H.; methodology, E. B. P., E. J. G. L., C. A. S., J. C. L., L. P. M., R. E. D., Y. B. P., E. N. D., L. A. O., M. A. G. and D. A. H.; validation, E. B. P., E. J. G. L., C. A. S., J. C. L., L. P. M., R. E. D., Y. B. P.; investigation, E. B. P., E. J. G. L., C. A. S., J. C. L., L. P. M., R. E. D., Y. B. P.; data curation, E. B. P., E. J. G. L., C. A. S., J. C. L., L. P. M., R. E. D., Y. B. P., E. N. D., L. A. O., M. A. G. and D. A. H.; writing—original draft preparation, C. A. S., L. P. M., L. A. O., M. A. G. and D. A. H.; writing—review and editing, C. A. S., L. P. M., R. E. D., S. B. A., L. A. O., M. A. G. and D. A. H.; visualization, E. B. P., E. J. G. L., C. A. S., J. C. L., L. P. M., R. E. D., Y. B. P., E. N. D., L. A. O., M. A. G. and D. A. H.; supervision, C. A. S., L. A. O., M. A. G., and D. A. H.; project administration, C. A. S., L. A. O., M. A. G., and D. A. H.; funding acquisition, C. A. S., L. A. O., M. A. G., and D. A. H. All authors have read and agreed to the published version of the manuscript. Details of the experimental part: E. B. P., J. C. L., and C. A. S. conducted the electrochemistry and impedance measurements, and charge–discharge experiments. E. J. G. L., R. E. D., Y. B. P. and D. A. H. were responsible for the synthesis of the compounds and structure elucidation. L. P. M. performed the SEM and AFM studies. S. B. A. performed XDR and ATR-FTIR experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors are grateful to Secretaría de Ciencia y Técnica, Universidad Nacional de Río Cuarto (Secyt-UNRC), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, PIBAA 2022–2023 28720210101120CO, PIBAA 2022–2023 28720210101058CO, PIP 2021–23 PIP 11220200101208CO) and Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT, PICT 02391/19) of Argentina for financial support. C. A. S., L. P. M., S. B. A., E. N. D., L. A. O., M. A. G., and D. A. H. are scientific members of CONICET. E. B. P., E. J. G. L., J. C. L., Y. B. P. and R. E. D. thank CONICET for doctoral fellowship. The authors greatly acknowledge the technical and imaging assistance of staff from the Laboratorio de Nanoscopía y Nanofotónica (LANN) – CONICET – Universidad Nacional de Córdoba (UNC), Córdoba, Argentina.References
- J. Min Park, J. H. Lee and W.-D. Jang, Coord. Chem. Rev., 2020, 407, 213157 CrossRef.
- R. Costa e Silva, L. Oliveira da Silva, A. de Andrade Bartolomeu, T. J. Brocksom and K. T. de Oliveira, Beilstein J. Org. Chem., 2020, 16, 917–955 CrossRef PubMed.
- M. M. Al Mogren, N. M. Ahmed and A. A. Hasanein, J. Saudi Chem. Soc., 2020, 24, 303–320 CrossRef CAS.
- Y. Matsuo, K. Ogumi, I. Jeon, H. Wang and T. Nakagawa, RSC Adv., 2020, 10, 32678–32689 RSC.
- Z.-L. Qi, Y.-H. Cheng, Z. Xu and M.-L. Chen, Int. J. Mol. Sci., 2020, 21, 5839 CrossRef CAS PubMed.
- C. C. Negut, R.-I. Stefan - van Staden and J. F. van Staden, ECS J. Solid State Sci. Technol., 2020, 9, 051005 CrossRef CAS.
- J. Chen, Y. Zhu and S. Kaskel, Angew. Chem., Int. Ed., 2021, 60, 5010–5035 CrossRef CAS PubMed.
- N. Rabiee, M. T. Yaraki, S. M. Garakani, S. M. Garakani, S. Ahmadi, A. Lajevardi, M. Bagherzadeh, M. Rabiee, L. Tayebi, M. Tahriri and M. R. Hamblin, Biomater, 2020, 232, 119707 CrossRef CAS PubMed.
- J. Tian, B. Huang, M. H. Nawaz and W. Zhang, Coord. Chem. Rev., 2020, 420, 213410 CrossRef CAS.
- A. Akbar, S. Khan, T. Chatterjee and M. Ghosh, J. Photochem. Photobiol., B, 2023, 248, 112796 CrossRef CAS PubMed.
- C. Comuzzi, M. Marino, D. Poletti, M. Boaro and P. Strazzolini, J. Photochem. Photobiol., A, 2022, 430, 113967 CrossRef CAS.
- D. Das and S. Kurungot, Energy Technol., 2020, 8, 2000061 CrossRef CAS.
- M. Xie, J. Liu, L. Dai, H. Peng and Y. Xie, RSC Adv., 2023, 13, 24699–24730 RSC.
- W. Chen, P. Chen, G. Zhang, G. Xing, Y. Feng, Y.-W. Yang and L. Chen, Chem. Soc. Rev., 2021, 50, 11684–11714 RSC.
- Z. Li and Y.-W. Yang, Polym. Chem., 2021, 12, 4613–4620 RSC.
- W. Ji, T.-X. Wang, X. Ding, S. Lei and B.-H. Han, Coord. Chem. Rev., 2021, 439, 213875 CrossRef CAS.
- Z. Li and Y.-W. Yang, Adv. Mater., 2022, 34, 2107401 CrossRef CAS PubMed.
- X. Li, C. Tang, L. Zhang, M. Song, Y. Zhang and S. Wang, Biomimetics, 2023, 8, 171 CrossRef CAS PubMed.
- L. Shu, J. Yu, Y. Cui, Y. Ma, Y. Li, B. Gao and H.-g. Wang, Int. J. Hydrogen Energy, 2022, 47, 10902–10910 CrossRef CAS.
- Y. Zhang, L. Cheng, L. Zhang, D. Yang, C. Du, L. Wan, J. Chen and M. Xie, J. Energy Storage, 2021, 34, 102018 CrossRef.
- Y. Wang, X. Cui, P. Zhang, Y. Wang and W. Lu, Environ. Technol. Innovation, 2023, 29, 102972 CrossRef CAS.
- Q. Liu, Q. Sun, J. Shen, H. Li, Y. Zhang, W. Chen, S. Yu, X. Li and Y. Chen, Coord. Chem. Rev., 2023, 482, 215078 CrossRef CAS.
- W.-C. Wang, Y.-W. Lin, S.-H. Peng, C.-T. Chuang, C.-C. Chang and C.-S. Hsu, Org. Electron., 2020, 86, 105899 CrossRef CAS.
- Z. Liu, Z. Huang, Y. Chen, T. Xu, H. Yu, X. Guo, L. Yan, M. Zhang, W.-Y. Wong and X. Wang, Macromol. Chem. Phys., 2020, 221, 1900446 CrossRef CAS.
- J. Tian and W. Zhang, Prog. Polym. Sci., 2019, 95, 65–117 CrossRef CAS.
- J. E. Durantini, R. Rubio, C. Solis, L. Macor, G. M. Morales, M. I. Mangione, D. A. Heredia, E. N. Durantini, L. Otero and M. Gervaldo, Sustainable Energy Fuels, 2020, 4, 6125–6140 RSC.
- Z. Chen, J. Wang, S. Zhang, Y. Zhang, J. Zhang, R. Li and T. Peng, ACS Appl. Energy Mater., 2019, 2, 5665–5676 CrossRef CAS.
- Y. Zhang, L. Cheng, L. Zhang, D. Yang, C. Du, L. Wan, J. Chen and M. Xie, J. Energy Storage, 2021, 34, 102018 CrossRef.
- R. Guilard, K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, New York, 2003 Search PubMed.
- R. Luo, M. Chen, F. Zhou, J. Zhan, Q. Deng, Y. Yu, Y. Zhang, W. Xu and Y. Fang, J. Mater. Chem. A, 2021, 9, 25731–25749 RSC.
- M. Gervaldo, M. Funes, J. Durantini, L. Fernandez, F. Fungo and L. Otero, Electrochim. Acta, 2010, 55, 1948–1957 CrossRef CAS.
- J. Durantini, L. Otero, M. Funes, E. N. Durantini, F. Fungo and M. Gervaldo, Electrochim. Acta, 2011, 56, 4126–4134 CrossRef CAS.
- H. Ma, Y. Chen, X. Li and B. Li, Adv. Funct. Mater., 2021, 31, 2101861 CrossRef CAS.
- B. Stöckle, D. Y. W. Ng, C. Meier, T. Paust, F. Bischoff, T. Diemant, R. J. Behm, K.-E. Gottschalk, U. Ziener and T. Weil, Macromol. Symp., 2014, 346, 73–81 CrossRef.
- D. A. Heredia, E. J. Gonzalez Lopez, E. N. Durantini, J. Durantini, T. Dittrich, J. Rappich, L. Macor, C. Solis, G. M. Morales, M. Gervaldo and L. Otero, Electrochim. Acta, 2019, 311, 178–191 CrossRef CAS.
- D. Heredia, L. Fernandez, L. Otero, M. Ichikawa, C.-Y. Lin, Y.-L. Liao, S.-A. Wang, K.-T. Wong and F. Fungo, J. Phys. Chem. C, 2011, 115, 21907–21914 CrossRef CAS.
- T. Patois, B. Lakard, S. Monney, X. Roizard and P. Fievet, Synth. Met., 2011, 161, 2498–2505 CrossRef CAS.
- K. Cysewska, J. Karczewski and P. Jasiński, Electrochim. Acta, 2015, 176, 156–161 CrossRef CAS.
- R. K. Gupta, Organic Electrodes: Fundamental to Advanced Emerging Applications, Springer Nature, 2022 Search PubMed.
- M. A. Soldatov, V. V. Butova, D. Pashkov, M. A. Butakova, P. V. Medvedev, A. V. Chernov and A. V. Soldatov, Nanomaterials, 2021, 11, 619 CrossRef CAS PubMed.
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS PubMed.
- S. H. Mir, L. A. Nagahara, T. Thundat, P. Mokarian-Tabari, H. Furukawa and A. Khosla, J. Electrochem. Soc., 2018, 165, B3137 CrossRef CAS.
- X. Zhang, Z. Xiao, X. Liu, P. Mei and Y. Yang, Renewable Sustainable Energy Rev., 2021, 147, 111247 CrossRef CAS.
- E. Reynoso, A. M. Durantini, C. A. Solis, L. P. Macor, L. A. Otero, M. A. Gervaldo, E. N. Durantini and D. A. Heredia, RSC Adv., 2021, 11, 23519–23532 RSC.
- K. Sun, S. Zhang, P. Li, Y. Xia, X. Zhang, D. Du, F. H. Isikgor and J. Ouyang, J. Mater. Sci.: Mater. Electron., 2015, 26, 4438–4462 CrossRef CAS.
- S. Nie, Z. Li, Y. Yao and Y. Jin, Front. Chem., 2021, 9, 803509 CrossRef CAS PubMed.
- I. V. Shershnev, A. S. Kopylov, A. V. Cherkasova and A. B. Solovieva, Russ. J. Phys. Chem. B, 2022, 16, 1277–1284 CrossRef CAS.
- A. S. Kopylov, N. A. Aksenova, M. A. Savko, I. V. Shershnev, T. S. Zarkhina, A. V. Krivandin, O. V. Shatalova, A. V. Cherkasova, P. S. Timashev and A. B. Solovieva, Russ. J. Phys. Chem., 2022, 96, 444–449 CrossRef CAS.
- D. Tao, L. Feng, Y. Chao, C. Liang, X. Song, H. Wang, K. Yang and Z. Liu, Adv. Funct. Mater., 2018, 28, 1804901 CrossRef.
- Z. Yang, D. Tao, W. Zhong, Z. Liu, L. Feng and M. Chen, Biomaterials, 2022, 280, 121250 CrossRef CAS PubMed.
- N. K. Zaitsev, P. V. Melnikov, V. A. Alferov, A. V. Kopytin and K. E. German, Procedia Eng., 2016, 168, 309–312 CrossRef CAS.
- M. Manathanath, M. Xie, C. Arunkumar, Z. Wang, J. Zhao and S. Sujatha, Dyes Pigm., 2019, 165, 117–127 CrossRef CAS.
- Q. Cao, Q. Yin, Q. Chen, Z.-B. Dong and B.-H. Han, Chem.–Eur. J., 2017, 23, 9831–9837 CrossRef CAS PubMed.
- C. Solis, E. Baigorria, M. E. Milanesio, G. Morales, E. N. Durantini, L. Otero and M. Gervaldo, Electrochim. Acta, 2016, 213, 594–605 CrossRef CAS.
- R. Rubio, M. B. Suarez, M. E. Pérez, D. A. Heredia, G. M. Morales, E. N. Durantini, L. Otero, M. Gervaldo and J. E. Durantini, Electrochim. Acta, 2023, 458, 142552 CrossRef CAS.
- H. Zhang, Y. Zhang, C. Gu and Y. Ma, Adv. Energy Mater., 2015, 5, 1402175 CrossRef.
- E. J. Gonzalez Lopez, M. Renfige Rodriguez, S. C. Santamarina, L. Macor, L. A. Otero, M. A. Gervaldo, A. M. Durantini, E. N. Durantini, J. E. Durantini and D. A. Heredia, ACS Appl. Polym. Mater., 2023, 5, 943–956 CrossRef CAS.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401 CrossRef.
- C. J. Raj, M. Rajesh, R. Manikandan, S. Park, J. H. Park, K. H. Yu and B. C. Kim, Ionics, 2018, 24, 2335–2342 CrossRef CAS.
- C. J. Raj, B. C. Kim, W.-J. Cho, W.-g. Lee, S.-D. Jung, Y. H. Kim, S. Y. Park and K. H. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 13405–13414 CrossRef CAS PubMed.
- D. A. Heredia, S. R. Martínez, A. M. Durantini, M. E. Pérez, M. I. Mangione, J. E. Durantini, M. A. Gervaldo, L. A. Otero and E. N. Durantini, ACS Appl. Mater. Interfaces, 2019, 11, 27574–27587 CrossRef CAS PubMed.
- J. I. T. Costa, A. C. Tomé, M. G. P. M. S. Neves, J. A. S. Cavaleiro and J. Porphyr, Phthalocyanines, 2011, 15, 1116–1133 CrossRef CAS.
- N. V. S. D. K. Bhupathiraju, W. Rizvi, J. D. Batteas and C. M. Drain, Org. Biomol. Chem., 2016, 14, 389–408 RSC.
- M. C. Malacarne, S. Banfi, A. S. Alberton, E. Caruso and J. Porphyr, Phthalocyanines, 2019, 23, 1047–1056 CrossRef CAS.
- D. A. Heredia, J. E. Durantini, D. D. Ferreyra, E. Reynoso, E. J. Gonzalez Lopez, A. M. Durantini, M. E. Milanesio and E. N. Durantini, J. Photochem. Photobiol., B, 2021, 225, 112321 CrossRef CAS PubMed.
- E. J. Gonzalez Lopez, S. R. Martínez, V. Aiassa, S. C. Santamarina, R. E. Domínguez, E. N. Durantini and D. A. Heredia, Pharmaceutics, 2023, 15, 392 CrossRef CAS PubMed.
- R. Lyubimenko, D. Busko, B. S. Richards, A. I. Schäfer and A. Turshatov, ACS Appl. Mater. Interfaces, 2019, 11, 31763–31776 CrossRef CAS PubMed.
- W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem.–Eur. J., 2013, 19, 5654–5664 CrossRef CAS PubMed.
- K. L. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, New York, 1999, vol. 8 Search PubMed.
- C. Brückner and N. Hewage, Oxidation and Reduction of Porphyrins in Fundamentals of Porphyrin Chemistry, ed. P. J. Brothers and M. O. Senge, 2022, pp. 303–347, DOI:10.1002/9781119129301.ch6.
- Y. Cui, L. Zeng, Y. Fang, J. Zhu, C. H. Devillers, D. Lucas, N. Desbois, C. P. Gros and K. M. Kadish, ChemElectroChem, 2016, 3, 228–241 CrossRef CAS.
- K. M. Kadish and M. M. Morrison, J. Am. Chem. Soc., 1976, 98, 3326–3328 CrossRef CAS PubMed.
- H. Randriamahazaka, V. Noël and C. Chevrot, J. Electroanal. Chem., 1999, 472, 103–111 CrossRef.
- Q. Pei, G. Zuccarello, M. Ahlskog and O. Inganäs, Polymer, 1994, 35, 1347–1351 CrossRef CAS.
- J. Roncali, P. Blanchard and P. Frere, J. Mater. Chem., 2005, 15, 1589–1610 RSC.
- L. Groenendaal, G. Zotti, P.-H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855–879 CrossRef CAS.
- A. Blacha-Grzechnik, M. Krzywiecki, R. Motyka and M. Czichy, J. Phys. Chem. C, 2019, 123, 25915–25924 CrossRef CAS.
- F. Zaar, S. Olsson, R. Emanuelsson, M. Strømme and M. Sjödin, Electrochim. Acta, 2022, 424, 140616 CrossRef CAS.
- S. C. Santamarina, D. A. Heredia, A. M. Durantini and E. N. Durantini, Polymers, 2022, 14, 4936 CrossRef CAS PubMed.
- S. V. Selvaganesh, J. Mathiyarasu, K. L. N. Phani and V. Yegnaraman, Nanoscale Res. Lett., 2007, 2, 546–549 CrossRef CAS.
- Q. Zhao, R. Jamal, L. Zhang, M. Wang and T. Abdiryim, Nanoscale Res. Lett., 2014, 9, 557 CrossRef PubMed.
- B. Gupta, M. Mehta, A. Melvin, R. Kamalakannan, S. Dash, M. Kamruddin and A. K. Tyagi, Mater. Chem. Phys., 2014, 147, 867–877 CrossRef CAS.
- Q. Xie, Y. Xu, Z. Wang, C. Xu, P. Zou, Z. Lin, C. Xu, C. Yang, F. Kang and C.-P. Wong, PLoS One, 2016, 11, e0166529 CrossRef PubMed.
- C. Paliteiro and A. Sobral, Electrochim. Acta, 2005, 50, 2445–2451 CrossRef CAS.
- R. A. Krüger, A. S. Terpstra and T. C. Sutherland, CJC University of Calgary 50th Anniversary Virtual Issue, 2016, vol. 1, pp. 214–220 Search PubMed.
- Y. Fang, Y. G. Gorbunova, P. Chen, X. Jiang, M. Manowong, A. A. Sinelshchikova, Y. Y. Enakieva, A. G. Martynov, A. Y. Tsivadze, A. Bessmertnykh-Lemeune, C. Stern, R. Guilard and K. M. Kadish, Inorg. Chem., 2015, 54, 3501–3512 CrossRef CAS PubMed.
- M. A. Gervaldo, Y. M. R. Rodriguez, R. A. Rubio, L. P. Macor, C. A. Solis, J. E. Durantini and L. A. Otero, in Organic Electrodes: Fundamental to Advanced Emerging Applications, ed. R. K. Gupta, Springer International Publishing, Cham, 2022, pp. 379–397, DOI:10.1007/978-3-030-98021-4_20.
- C. Lei, F. Markoulidis, Z. Ashitaka and C. Lekakou, Electrochim. Acta, 2013, 92, 183–187 CrossRef CAS.
- S.-M. Chen, R. Ramachandran, V. Mani and R. Saraswathi, Int. J. Electrochem. Sci., 2014, 9, 4072–4085 CrossRef.
- X. Wu, X. Feng, J. Yuan, X. Yang, H. Shu, C. Yang, Z. liu, J. Peng, E. Liu, S. Tan and P. Gao, Energy Storage Mater., 2022, 46, 252–258 CrossRef.
- T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef PubMed.
- W. Zhao, J. Peng, W. Wang, B. Jin, T. Chen, S. Liu, Q. Zhao and W. Huang, Small, 2019, 15, 1901351 CrossRef PubMed.
- S. Debnath, C. J. Boyle, D. Zhou, B. M. Wong, K. R. Kittilstved and D. Venkataraman, RSC Adv., 2018, 8, 14760–14764 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00945b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |