
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergism in carbon nanotubes and carbon-dots: counter electrode of a high-performance dye-sensitized solar cell†
A M Mahmudul Hasana and
Md. Abu Bin Hasan Susan *ab
*ab
aDepartment of Chemistry, University of Dhaka, Dhaka-1000, Bangladesh. E-mail: susan@du.ac.bd
bDhaka University Nanotechnology Center (DUNC), University of Dhaka, Dhaka-1000, Bangladesh
First published on 4th March 2024
Abstract
Dye-sensitized solar cells (DSSCs) play a crucial role in the realm of renewable energy technology by converting solar energy into electrical energy in an efficient and cost-effective way. In the pursuit of improving the photoconversion efficiency (PCE) of DSSCs, this work aims at fabricating a new counter electrode (CE) using a binary composite of heteroatom-doped carbon dots (C-dots) and functionalized multi-walled carbon nanotubes (o-MWCNTs). We demonstrate that this binary composite exhibits superior performance to pristine o-MWCNTs, resulting in a remarkable enhancement in the PCE. The PCE of the o-MWCNT/C-dots composite was measured at an impressive 4.28%, significantly outperforming the pristine o-MWCNT electrode, which yielded an efficiency of 2.24%. The enhanced performance of the o-MWCNT/C-dots composite can be attributed to the synergistic effects of heteroatom-doped C-dots since their binding to the o-MWCNTs by activated oxygenic surface functional groups increases the surface area from 218 to 253 m2 g−1. This enhanced surface area results from the reduction of π–π stacking interactions of the individual tubes and production of a new hollow channel in the structure that provides an ideal scaffold for I2 adsorption and electron transfer. We demonstrate the role of C-dots on MWCNT's property modulation toward higher PCE by density functional theory (DFT) calculation and electrochemical analysis. Electron-excess N and S doped C-dots exhibit strong catalytic activity, allowing for rapid electron transfer processes in the CE-electrolyte surface via the donor acceptor mechanism, whereas electron-deficient B doped C-dots undermine the cell performance by forming a charge recombination trap at the CE surface. The synthesized composite has higher redox reversibility up to 100 CV cycles and chemical stability, studied by the post-performance material characterization. The findings offer a promising avenue for the development of high-performance DSSCs, which will help to promote sustainable and renewable energy technologies.
Introduction
In the face of growing environmental concerns and the pressing need to combat climate change, the world is witnessing a remarkable shift towards sustainable energy sources. Solar energy, in particular, has emerged as a key component in the global transition to a cleaner and greener future.1–3 With the dawn of a new decade, the necessity of solar energy and solar cells has become increasingly apparent, offering unparalleled benefits for both the environment and human society. In terms of affordability, dye-sensitized solar cells (DSSCs) have a distinct advantage over traditional solar cells.4,5 DSSCs offer substantial environmental benefits. Unlike fossil fuel combustion, which releases greenhouse gases and contributes to climate change, DSSCs generate electricity through a clean and emission-free process.6–9 The operational mechanism of DSSCs (Scheme 1) is straightforward. A photosensitizer, typically an organic dye (e.g., N719) is immobilized onto a mesoporous semiconductor material (i.e., TiO2) known as the photoanode. This sensitizer effectively captures a broad range of solar radiation across the spectrum and initiates generation of electrons. Subsequently, these photo-generated electrons rapidly migrate from the excited state of the dye molecules to the conduction band (CB) of the semiconducting material located on the photoanode. The cations of the dye molecules are formed through electron transfer and are then reduced by the I−/I3− redox couple, which acts as an electrolyte. It is crucial to promptly reduce the redox cations to prevent charge recombination with the electrons present in the CB of the semiconductor. The process of reduction occurs in the oxidized redox pair as a result of the influx of electrons from the external circuit, which are received by the counter electrode (CE). The reduction process of I−/I3− is facilitated by an electrocatalyst that is located at the CE.10 Zhang et al.11 described the reactions involved in the reduction mechanism of I3−.| I3− → I2 (sol) + I− (sol) |
| I2 (sol) + 2 (surface active site) → 2I (adsorbed) |
| I (adsorbed) + e− → I− |
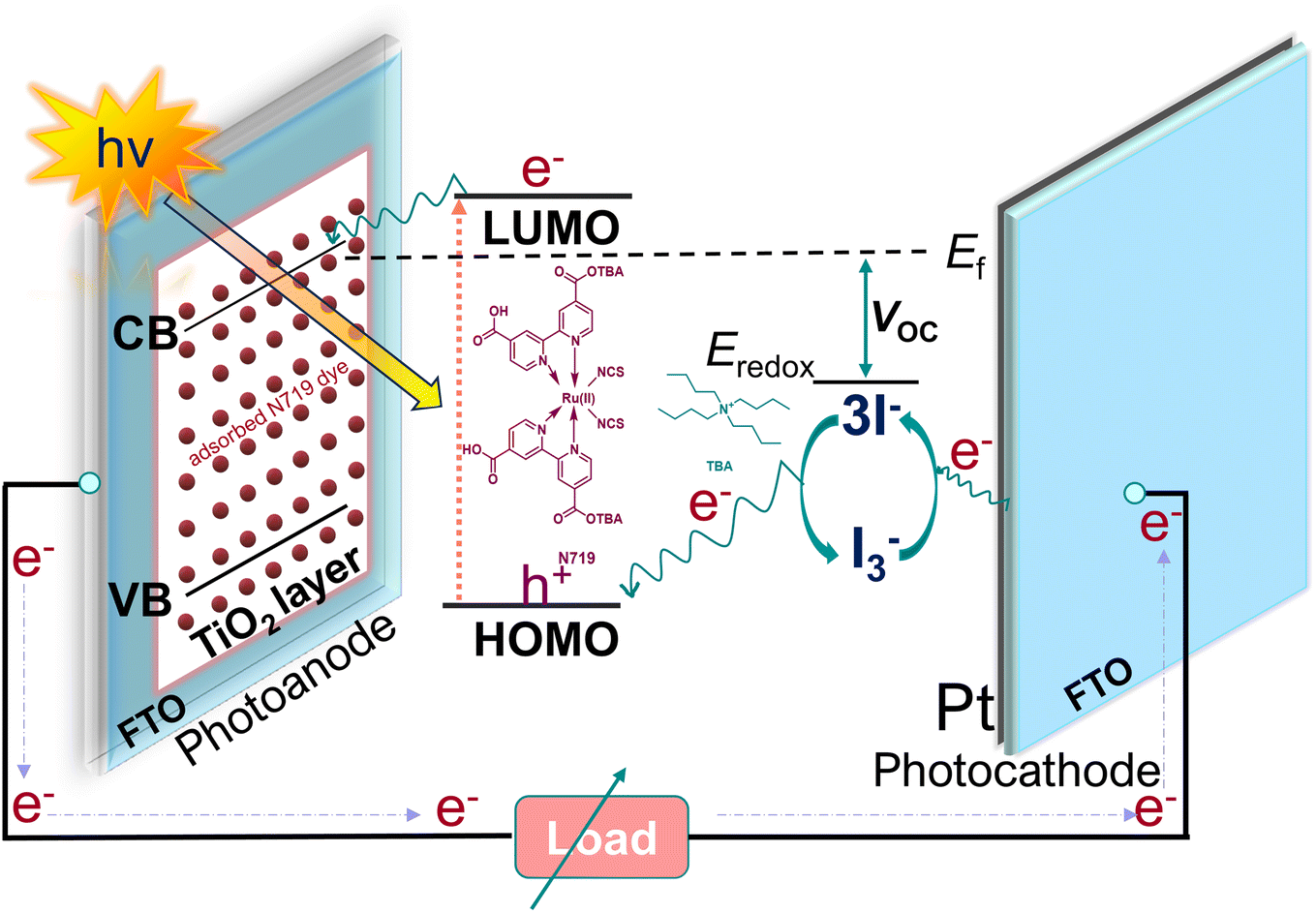 | ||
| Scheme 1 Functioning mechanism of a dye-sensitized solar cell (DSSC). | ||
The CE accelerates the reduction of I3− to I− by providing its surface-active sites. The possession of a significant surface area is crucial for the effective adsorption of mediators in the context of CE.12,13 In addition, it is important for the CE to demonstrate enhanced electrical conductivity in order to facilitate efficient charge mobility. This high mobility is necessary to prevent the recombination of oxidized dye cations with the electrons present in the CB of the TiO2. The selection of an appropriate electrocatalyst plays a significant role in augmenting the essential open circuit voltage (VOC) of DSSCs.14 The VOC is a significant metric for evaluating the performance of a DSSC. According to energy band diagram, the VOC is determined by the disparity between the energy level associated with the reduction potential of the redox mediator and the quasi-Fermi level of TiO2 on the photoanode when exposed to light (Scheme 1). The enhanced electrocatalytic performance observed in the CE can be attributed to the decreased likelihood of cations in the electrolyte to undergo diffusion onto the semiconductor surface and subsequently recombine.15,16 The decrease in photogenerated electron-electrolyte cation recombination leads to a higher carrier concentration in the CB of TiO2, resulting in an increase in the quasi-Fermi level and ultimately the VOC of the cell.
Due to its outstanding electrical conductivity and catalytic properties, platinum (Pt) has been widely recognized as a highly effective material for the CE in DSSCs.17 Nevertheless, in view of the growing emphasis on environmentally and economically viable energy options, researchers have progressively explored alternative options to Pt. Carbon-based materials have garnered significant interest as a feasible substitute owing to their notable attributes such as elevated electrical conductivity, thermal and chemical durability, as well as improved efficiency and cost-effectiveness. Using a graphite-carbon black mixture, Kay and Grätzel were the first to successfully integrate carbon-based material in DSSCs, achieving a photovoltaic efficiency of 6.7%.18 Subsequent to this study, several more carbon-based materials, including carbon black,19,20 activated carbon,21,22 porous carbon,23 carbon nanotubes,24–26 carbon nanofibers,27 graphite,28 and graphene,29–31 have been employed as electrocatalytic substances for the CE in DSSCs. Porous carbons have several advantageous characteristics that make them a promising material for electrocatalysis. The DSSC system is benefitted from the intrinsic porous structure of the material, which is characterized by a high surface area and an interconnected pore network.32 The unique structure creates an optimal platform for electrochemical processes. The presence of porosity facilitates effective diffusion of electrolytes and transfer of electrons, hence minimizing charge transfer resistance (RCT) and improving the overall performance of the cell.33–35
This study presents a novel binary composite consisting of functionalized multi-walled carbon nanotubes (o-MWCNT) and carbon dots (C-dots), which serves as a cost-effective CE for DSSCs. MWCNTs exhibit enhanced electrical conductivity due to delocalized π electron through the sp2 hybridized hollow carbon structure with variable chirality, improved thermal stability, and possess a higher surface area (>200 m2 g−1).36,37 The sp2 hybridized smooth graphitic plane creates faster electron transport channels. However, because of the van der Waals force and π–π stacking interaction between the individual tubes, MWCNT has limited dispersion and coalesces the tube with surface defect.38,39 This aggregation limits the available surface-active site for I2 adsorption and charge transfer reaction. In this study, we aimed at modifying the electrocatalytic activity and surface property of MWCNT by the chemical surface oxidation with concentrated H2SO4 and HNO3. Several studies have shown that surface functionalization and foreign molecule binding can improve both the surface area and conductivity, hence the electrocatalytic activity of MWCNT.40–43
The functionalization process enhances the surface area of MWCNTs, hence promoting the mobility of charge carriers and providing additional binding sites for C-dots via noncovalent interaction. Sometimes harsh oxidation conditions damage the conducting plane of MWCNT during surface activation and compromised the conductivity.44 To compensate for this conductivity loss in this study we loaded the C-dots in MWCNT that create a well penetrated network architecture featuring certain surface functional groups.45 The binding of these two species via surface functional group (Fig. 1B) lowers the system energy and it becomes thermodynamically favorable.36 Furthermore, the distinct characteristics of C-dots, resulting from their nanoscale dimensions,46–48 higher surface area and remarkable quantum confinement effects49,50 provide low charge transfer resistance in I−/I3− solution. For an efficient CE material, the synergism between MWCNT and C-dots is essential. C-dots require a conductive support with a high surface area to prevent self-aggregation and enhance the overall surface area of the composite. MWCNT is conductive with limited dispersion while C-dots are generally nonconductive. Binding of these two species together synergistically enhances the dispersion of MWCNT and fast flowing electron density. In addition, the increased specific surface area of C-dots improves electrocatalytic activity (ECA) and makes charge transport within the DSSC more effective. Because of the precise tunability of electrical properties by heteroatom doping, they can also better align with the dye-sensitizer and make electron injection and transit easier.51 This study aimed at investigating the synergism of the MWCNT and C-dots and its influence on the performance of DSSC by different heteroatom doped C-dots loading into o-MWCNT 3D network. The o-MWCNT/C-dots binary composite exhibits a reduction in charge transfer resistance (RCT) and an increase in charge recombination resistance, as determined using electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV). The o-MWCNT/C-dots binary composite show enhanced ECA compared to o-MWCNT. This is the first attempt to use an environmentally friendlier approach to synthesize surface charged C-dots using a natural precursor and its synergism to the MWCNT for higher photoconversion efficiency (PCE) in DSSC via both the computational and experimental approach.
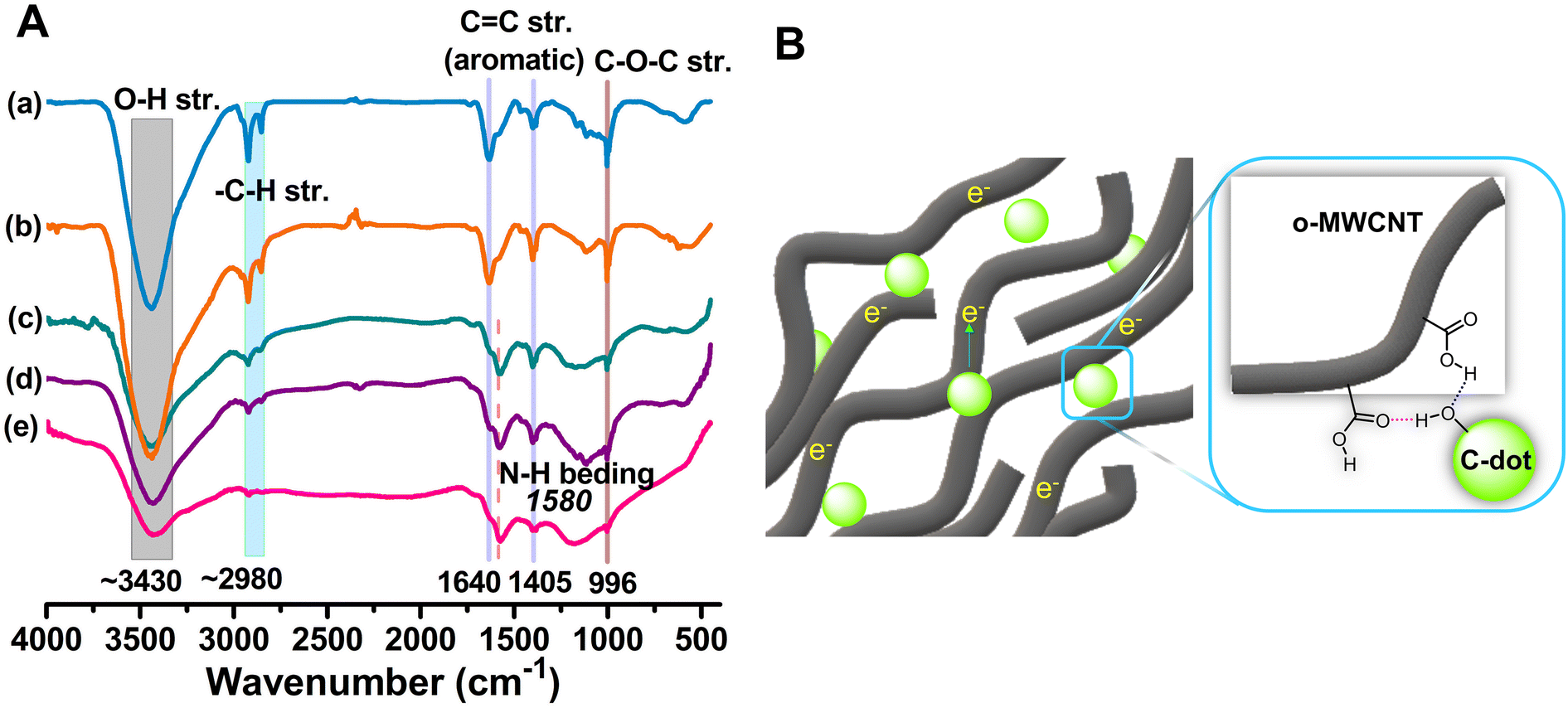 | ||
| Fig. 1 (A) FTIR spectra of the o-MWCNT and o-MWCNT/C-dots (10 wt%) binary composites [(a) o-MWCNT (b) o-MWCNT/NBS-CD (c) o-MWCNT/NB-CD (d) o-MWCNT/NS-CD and (e) o-MWCNT/N-CD]. (B) Possible binding scheme of C-dots to the o-MWCNT. | ||
Experimental
Synthesis of materials
C-dots were synthesized from watermelon juice precursor by hydrothermal method52 (Fig. S1†). N-doped C-dots (N-CD) were synthesized in situ by heating 60 mL of freshly squeezed watermelon juice at 180 °C for 3 h in a 100 mL of Teflon-equipped stainless-steel autoclave. Under elevated temperature and pressure conditions, the organic molecules present in watermelon juice undergo fragmentation, resulting in the formation of C-dots. An additional doping agent was added into the autoclave in each case to synthesize heteroatom doped C-dots. Sulfamic acid (NH2SO3H) and H3BO3 were used for S and B sources (Fig. S1†). MWCNTs were chemically oxidized to mitigate their agglomeration properties. 4.00 g commercial MWCNT was first dispersed into 100 mL DI water via ultra-sonification for 3 h. Then a mixture of concentrated H2SO4 (98%) and HNO3 (67%) at a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was added to the dispersion at 0 °C with constant stirring and again ultra-sonicated for 2 h. The resulting black slurry was then settled down and it was filtered in a double rings filter paper via a Buckner funnel. The obtained black product was then washed with DI water until the pH of the washed water was about 7. Then the product was dried in a vacuum oven at 80 °C overnight. Binary composite was synthesized by sono-mechanical method (Fig. S2†). 5 g of o-MWCNT was dispersed into 50 mL dimethylformamide (DMF) via ultrasonication for 1 h. A specific weight percentage of C-dots was added to the dispersion. The dispersion was then once more sonicated for 2 h and vigorously stirred for 24 h at room temperature. The resulting black slurry was then heated at 140 °C in a vacuum oven for complete removal of DMF and to produce a solid dry o-MWCNT/C-dots binary composite. The composites synthesized in this process are (a) o-MWCNT/N-CD (5 wt%), (b) o-MWCNT/N-CD (10 wt%), (c) o-MWCNT/N-CD (15 wt%), (d) o-MWCNT/N-CD (50 wt%), (e) o-MWCNT/NB-CD (10 wt%), (f) o-MWCNT/NS-CD (10 wt%), and (g) o-MWCNT/NBS-CD (10 wt%).
:1 was added to the dispersion at 0 °C with constant stirring and again ultra-sonicated for 2 h. The resulting black slurry was then settled down and it was filtered in a double rings filter paper via a Buckner funnel. The obtained black product was then washed with DI water until the pH of the washed water was about 7. Then the product was dried in a vacuum oven at 80 °C overnight. Binary composite was synthesized by sono-mechanical method (Fig. S2†). 5 g of o-MWCNT was dispersed into 50 mL dimethylformamide (DMF) via ultrasonication for 1 h. A specific weight percentage of C-dots was added to the dispersion. The dispersion was then once more sonicated for 2 h and vigorously stirred for 24 h at room temperature. The resulting black slurry was then heated at 140 °C in a vacuum oven for complete removal of DMF and to produce a solid dry o-MWCNT/C-dots binary composite. The composites synthesized in this process are (a) o-MWCNT/N-CD (5 wt%), (b) o-MWCNT/N-CD (10 wt%), (c) o-MWCNT/N-CD (15 wt%), (d) o-MWCNT/N-CD (50 wt%), (e) o-MWCNT/NB-CD (10 wt%), (f) o-MWCNT/NS-CD (10 wt%), and (g) o-MWCNT/NBS-CD (10 wt%).
Electrochemical measurements
The electrochemical experiments were performed using a three-electrode cell setup, consisting of Pt disk (diameter: 2 mm) working electrode (WE), Pt wire CE, and an Ag/Ag+ nonaqueous reference electrode (RE). Electrolyte solution for CV and EIS experiments was prepared with 0.5 M LiI, 0.05 M iodine (I2), and 0.1 M LiClO4 in acetonitrile. To prepare the electrode, 95 wt% composite material (i.e., o-MWCNT/C-dots binary composite) was mixed with 5 wt% polyvinylidene fluoride (PVDF) binder in 100 μL ethanol. The mixture was sonicated for 30 min and fabricated onto the polished Pt disk WE surface via drop casting46 method. The fabricated electrode was then dried at room temperature for 24 h and 80 °C for 2 h. CV and EIS were performed in a CHI 760E electrochemical analyzer where the same amount (0.85 mg) of samples were fabricated on the Pt disk WE for each measurement. EIS was recorded in a frequency range of 1 Hz to 1 MHz in the form of Nyquist plot.Preparation of the dye-sensitized TiO2 photoanode
The photoanode was fabricated through the application of a TiO2 layer onto the conductive fluorine doped tin oxide (FTO) glass substrate. FTO glasses (thickness 1.15 mm, 14 Ω cm−1) were cleaned by sonication with soap water, followed by DI water and ethanol, each for 15 min. Afterward, a compact blocking layer of TiO2 was deposited onto the dried FTO substrate by immersing it into 40 mM aqueous titanium butoxide, Ti(OBu)4 solution for 30 min at 80 °C and then cleaned with DI water and ethanol. TiO2 paste was prepared by sonicating 0.5 g of TiO2 nanoparticles powder in 10 mL of ethanol for 3 h. After vigorously stirring the solution for 30 min, 5 wt% Nafion perfluorinated resin binder solution was added. Using a doctor blade technique,47 the produced TiO2 paste was applied to the FTO over the blocking layer, covering a surface area of 1 cm2 and a thickness of 0.3 mm. After drying the films for 15 min at 120 °C, the films were annealed at 450 °C for 15 min in a muffle furnace. The photoanode was immersed in a 0.45 mM N719 dye solution in acetonitrile for 16 h to sensitize the TiO2 coat. The electrode was then washed with DI and dried under N2 for 5 min.Preparation of the o-MWCNT/C-dots CE
20 mg of the synthesized o-MWCNT/C-dots composite materials were dispersed in 4 mL ethanol. Then 100 μL 5 wt% Nafion perfluorinated resin binder solution was added to the dispersion and stirred vigorously for 30 min. Then the mixture was transferred to a mortar and made into a sticky shampoo-like paste. A 6–8 μm thick layer was coated on the previously washed and dried FTO glass substrate in 1 cm2 area via the doctor blade technique. The electrode was then dried at 80 °C for 30 min and then annealed at 150 °C for 15 min.Preparation of the Pt thin film CE
The thermal decomposition method was employed to deposit the Pt catalyst on the FTO. A clean FTO glass was heated to 470 °C for 15 min. A drop of 0.9 mM H2PtCl6. 6H2O solution in ethanol was drop casted on the FTO substrates and annealed at 470 °C for 15 min.Assembly of the DSSC
Dye-sensitized TiO2 photoanode and o-MWCNT/C-dots fabricated photocathode were sealed together by binder clips with a Surlyn spacer around the TiO2 active area (0.25 cm2). The liquid redox electrolyte (I−/I3−) was injected into assembled cells and prepared for the photovoltaic performance analysis. The I−/I3− redox electrolyte consisted of 0.5 M LiI, 0.05 M I2, and 0.5 M 4-tert-butylpyridine in acetonitrile. I–V experiments were performed using ZAHNER, Xpot, 26356, Germany, under simulated sunlight irradiation (AM 1.5, 100 mW cm−2).Computational details
The molecular structures of C-dots were drawn using open-source Avogadro software. The molecular geometry optimization and frequency calculation of the compound was carried out by employing the density functional theory (DFT)53 at B3LYP level and 6-311G basis set with the help of Gaussian 16 software package. The optimized structure of the compounds has been used to calculate the electrostatic potential, highest molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) energy by Gaussian 16 software package.54,55Results and discussion
Characterization of materials
The formation of a rigid o-MWCNT/C-dots binary composite can be depicted by the FTIR spectrum (Fig. 1A) of the composite. FTIR spectra in Fig. S3A† confirms that chemical oxidation of MWCNT56 results in the formation of various hydrophilic surface functional groups, such as carboxylic acid (–COOH), hydroxyl (–OH), and carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). In Fig. 1A, the O–H stretching vibration and the presence of the –COOH groups are confirmed by the intense broad band at 3430 cm−1. The O–H and N–H vibration bands in the composite have overlapped. The intensity of the band is noticeably lower in the composite, suggesting that C-dots have binding interaction with the 3D network of o-MWCNT via their surface functional groups, forming a compact rigid structural unit (Fig. 1B). The stretching vibration of the C–H bond is represented by the doublet vibration band at 2980 and 2830 cm−1. As the binary composites contain N-doped C-dots, a band at 1580 cm−1 results from the N–H bending mode of vibration. The reduced peak intensity for the composite affirms the formation of composite without disturbing the backbone structure and property of o-MWCNT.
O). In Fig. 1A, the O–H stretching vibration and the presence of the –COOH groups are confirmed by the intense broad band at 3430 cm−1. The O–H and N–H vibration bands in the composite have overlapped. The intensity of the band is noticeably lower in the composite, suggesting that C-dots have binding interaction with the 3D network of o-MWCNT via their surface functional groups, forming a compact rigid structural unit (Fig. 1B). The stretching vibration of the C–H bond is represented by the doublet vibration band at 2980 and 2830 cm−1. As the binary composites contain N-doped C-dots, a band at 1580 cm−1 results from the N–H bending mode of vibration. The reduced peak intensity for the composite affirms the formation of composite without disturbing the backbone structure and property of o-MWCNT.
The size of the synthesized C-dots was measured by dynamic light scattering (DLS) experiment. DLS pattern (Fig. 2A) demonstrates that synthesized C-dots have a particle size of 2–10 nm and a high degree of dispersion, with no observable agglomeration. This can be attributed to the repulsive forces exerted by negatively charged surface groups. In a DLS experiment, smaller particles exhibit a greater extent of Brownian motion, and it is defined by the translational diffusion coefficient (D), which depends on the size of the particle and its surface structure as well as the concentration and type of the solvated ions.57 The value of the D can be converted into the hydrodynamic particle size by the Stokes–Einstein equation: DH = kBT/3πηD; where, DH = hydrodynamic diameter, kB = Boltzmann constant, T = absolute temperature, η = viscosity and D = diffusion coefficient. In Fig. 2A, N-CD shows the shortest hydrodynamic size, measuring 5.0 nm. Both NB-CD and NS-CD exhibit a comparable DH of 5.6 nm, but NBS-CD demonstrates a slightly larger size of 6.5 nm. The incorporation of external heteroatom doping has led to an alteration in the dimensions of the carbon dots, even when subjected to the same hydrothermal treatment conditions across all samples. FESEM and EDS analyses with elemental mapping of C-dots (Fig. 2B and S4†) show the formation of targeted heteroatom doped quantum size molecule that is also supported by experimental results from DLS. Therefore, the DLS and FESEM analyses confirm the successful hydrothermal synthesis of C-dots from biomolecule precursor.
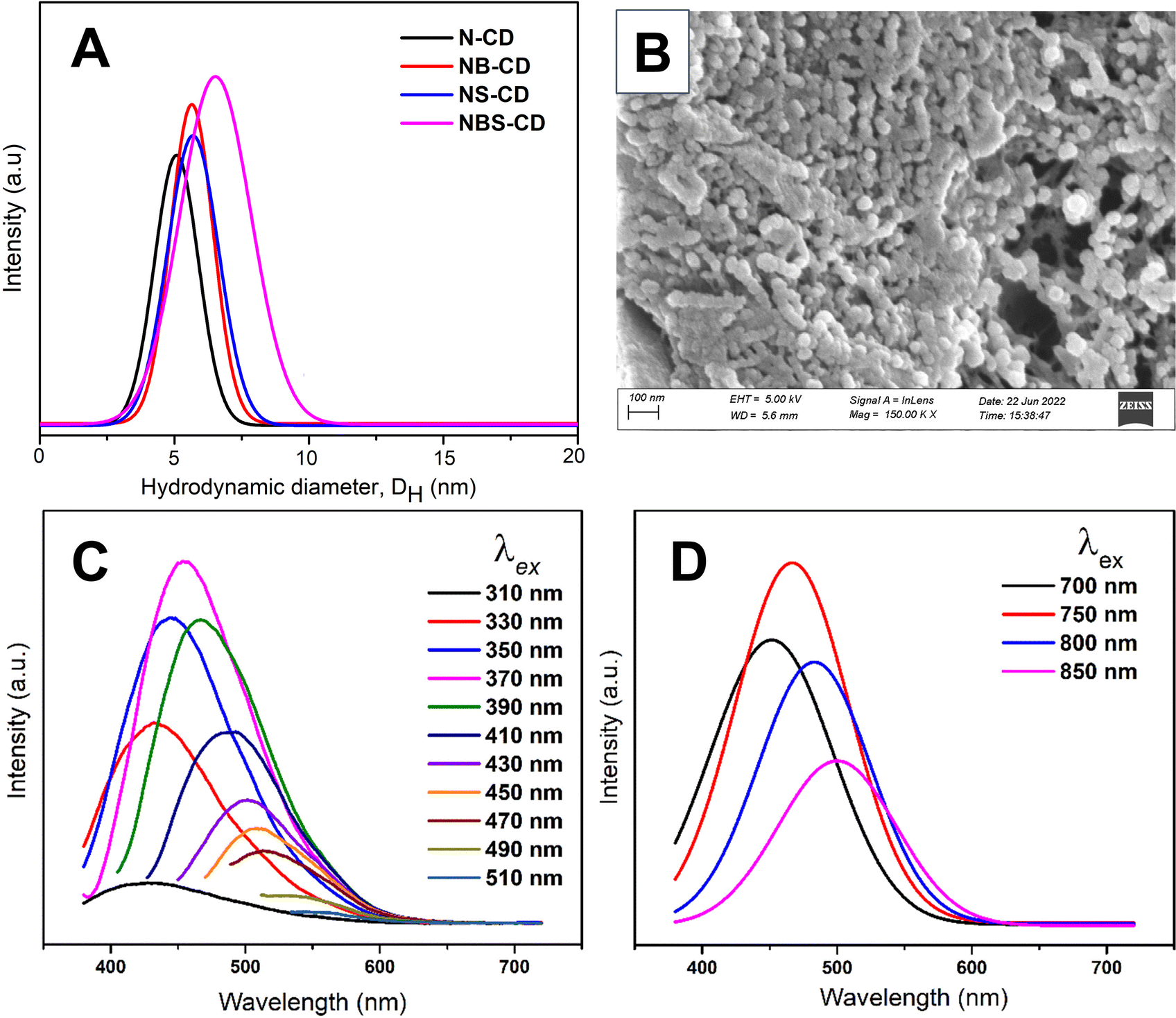 | ||
| Fig. 2 (A) DLS pattern of heteroatom doped C-dots, (B) FESEM images of N-CD, and (C and D) photoluminescence spectra of N-CD. | ||
Synthesized heteroatom doped C-dots are photoluminescent. Because of the surface functional group C-dots are readily soluble in H2O forming a brown colored solution in visible light and have a highly vibrant green luminescence when exposed to ultraviolet light (UV) at a wavelength of 265 nm (Fig. S1†). C-dot has energy up and down conversion property. It can absorb UV, turn this into NIR light and vice versa. As excitation wavelength (λex) increases from 310 to 370 nm, the photoluminescence (PL) intensity of N-CD increases (Fig. 2C) gradually and the emission peak position has a redshift until the maximized peak intensity at λex = 370 nm. When further increased to λex > 370 nm, the PL intensity gradually reduces with a red shift. Similarly, the up-converted PL emission peaks are shifted from 450 to 500 nm by tuning the excitation wavelength from 700 to 850 nm (Fig. 2D). This excitation and emission relationship of C-dots is mostly ascribed to the interaction between the fluorophores and their surrounding environment. This interaction is particularly influenced by the presence of excitation energy traps associated with oxygenic groups (–COOH, –OH, C–O–C, etc.) of N-CD. Moreover, emission wavelength (λem) is highly dependent on the solvent environment. The excited fluorophore can interact with solvent molecules, changing its energy levels and emission characteristics. A similar PL pattern is observed for all the synthesized heteroatom doped C-dots (Fig. S5†). This unique photoluminescence property of the synthesized C-dots provides additional support for its successful synthesis. The steady state photoluminescence study of the o-MWCNT/N-CD (5%) composite also shows a similar kind of emission behavior (Fig. S6A†). o-MWCNT doesn't have any PL property whereas the composite shows a broad and higher wavelength emission peak shifting from the pristine C-dots. The PL emission of the composite structure can be attributed to the photogenerated electron transfer between C-dots and MWCNTs. When the composite is excited by lower wavelength high energy photon, transition of electrons from C-dots to MWCNTs takes place. Time resolved photoluminescence spectra (Fig. S6B†) ensure this charge transfer process. The average PL lifetime of C-dot reduces from 6.08 ns to 4.20 ns after incorporating it in MWCNT network. This faster decay process of C-dots results from the photogenerated electron transfer to the MWCNT.87 This experimental finding also suggests the presence of higher electron density in the composite under solar irradiation while use in the DSSC.
FESEM images of o-MWCNT/N-CD binary composites with varying C-dots content are illustrated in Fig. 3. The position of C-dots within the o-MWCNT network is indicated by a red circle. The outcome demonstrates that C-dots are evenly dispersed across the o-MWCNT 3D network resulting in a more exfoliated structure than pristine MWCNT (Fig. S7†). The inclusion of C-dots causes the o-MWCNT network to become more porous and conductive, as evidenced by the glossy surface of the material (Fig. 3A).58 C-dots prevent o-MWCNT aggregation via binding to the surface functional group of the tube. C-dots were readily incorporated into the network because of their smaller size, which prevented the individual tubes from clumping together and formed a rigid composite system via a noncovalent supramolecular interaction of the surface groups of the individual material. However, the composite structure aggregates when C-dots loading exceed the optimal amount (15 wt%), likely due to the fact that an excessive number of nuclei formed by C-dots can congregate among themselves, hence decreasing the total surface area and conductivity as indicated by a comparatively dark surface (Fig. 3C and D). When the network was overloaded with C-dots, the active region of the o-MWCNT backbone structure was impeded and it breaks the backbone structure of o-MWCNT and reduces their thermal stability (Fig. S8†).
 | ||
| Fig. 3 FESEM image of (A) o-MWCNT/N-CD (5 wt%), (B) o-MWCNT/N-CD (10 wt%), (C) o-MWCNT/N-CD (15 wt%), and (D) o-MWCNT/N-CD (50 wt%). (E) XRD pattern and (F) N2 sorption isotherm of MWCNT, o-MWCNT, and o-MWCNT/N-CD (5 wt%) binary composite. | ||
The chemical functionalization and property modification of MWCNTs via incorporation of C-dots was also confirmed by the X-ray diffraction (XRD) analysis. The XRD pattern (Fig. 3E) of the pristine MWCNTs shows peaks at 2θ angles of 25.81° and 43.21°, that correspond to the (002) and (100) crystallographic planes of the carbon atoms, respectively.59 The same reflection peaks are also observed in o-MWCNT with a slight peak shifting at lower 2θ. This peak shifting proves the increment of the individual tube spacing according to Bragg's equation (2dsinθ = nλ) and more porous structure resulting from the oxygenic sites formation in the tube via chemical oxidation that supports the previous report.60,61 The aggregation of MWCNTs resulting from van der Waals and π–π interactions has been significantly reduced after acid treatment. The first broad peak at 002 plane of o-MWCNT/N-CD (5 wt%) indicates the effective binding of the C-dots in o-MWCNT via supramolecular H-bond formation without collapsing the original backbone structure of MWCNT. N2 physisorption analysis of the materials strongly supports the XRD findings.
Fig. 3F shows the change of the surface area of MWCNT upon chemical oxidation and incorporation of 5 wt% N-CD by N2 isotherm analysis. The adsorption/desorption isotherms of all samples are type-I and type-IV in the relative pressure range of 0.1–0.9, implying the development of microporous to mesoporous carbonaceous materials consistent with the work of Moralı et al.62 Pristine MWCNT poses 218 m2 g−1 BET surface area (SBET) which increases with chemical oxidation and 5 wt% C-dots loading in it. Furthermore, comparatively large hysteresis in o-MWCNT and o-MWCNT/N-CD (5 wt%) can be attributed to occur either due to the formation of new pore channels or probable pore swelling resulting from the synergistic interaction of o-MWCNT and C-dots. The superior surface area and probable porous channel formation accelerate the surface adsorption of I2 as well as the charge transfer kinetics of the I3− reduction.11
DFT calculation of heteroatom doped C-dots
For understanding the effect of heteroatom doping on the electronic properties of C-dots a DFT calculation has been carried out with B3LYP exchange correlation functional and 6-311G basis set. For this study the heteroatom doped C-dots have been designed based on the doping concentration result obtained from EDS analysis (Fig. S4†). It is acceptable to regard C-dots as tiny pieces of quasi-zero-dimensional (0 D) graphene sheets.63,64 There are just single atomic layers in ideal C-dots. In this work, a total of 85 atoms and 14 hexagonal rings have been used to mimic a single layer of C-dots. To ensure the accuracy of the computation, the bond lengths of graphene sheets and the modeled C-dots are compared. The bond length of C-dots is 1.43 Å, which is consistent with previous observations.65,66The heteroatom doped C-dots have distinct electronic structures and HOMO–LUMO electronic distributions (Fig. 4). This reflects that the electron density from the dopant heteroatom has significant influence on the overall band structure and electronic property of the C-dots. N and S are electron donors, which can be assigned from the HOMO electronic distribution of N-CD, NS-CD, and NBS-CD whereas the B is an electron deficient dopant that is reflected on the LUMO structure of the NB-CD and NBS-CD. N contributes greater electron density to the HOMO structure of NS-CD than S does, making N a more effective donor than S. When NB-CD forms a composite with o-MWCNT, it creates some charge recombination site and hinder the electron transfer process from CE to the I3− for dye regeneration in the cell. The presence of NB-CD is likely to have a negative impact on cell performance, while the inclusion of composites such as N-CD and NS-CD is expected to improve cell performance.
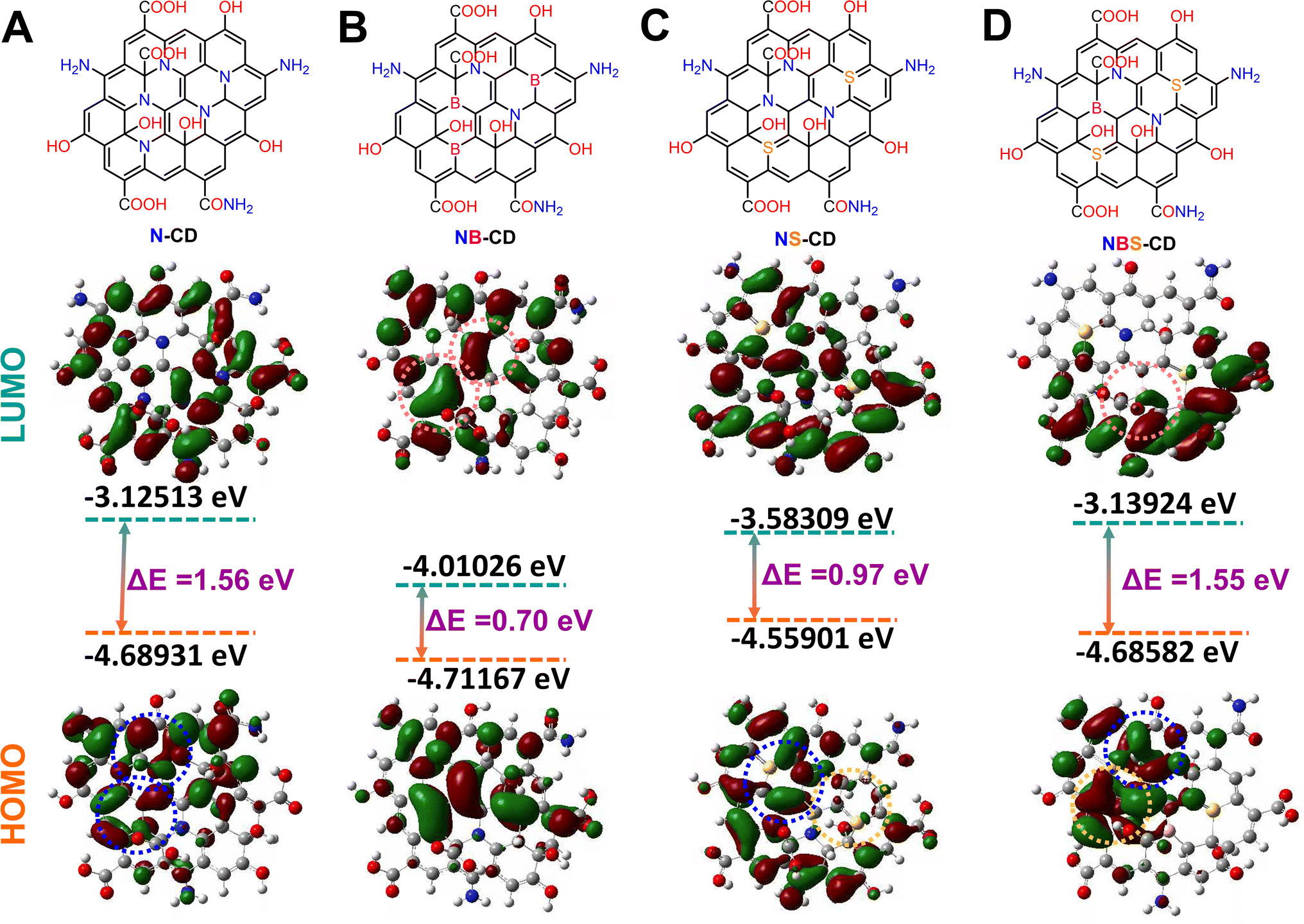 | ||
| Fig. 4 Model structure and HOMO–LUMO energy distribution of (A) N-CD, (B) NB-CD, (C) NS-CD, and (D) NBS-CD obtained from DFT calculation. | ||
Electrochemical analysis
The ECA of heteroatom doped C-dots were examined by CV and EIS analysis. ECA of the CE can be assessed by three parameters such as cathodic peak potential (EPC), cathodic current density (JPC), and the peak-to-peak separation potential (Epp) between the reduction and oxidation peaks of the I3−/I− redox couple. A good electrocatalytic reaction is faster at low overpotential and produces large amount of cathodic current. In this way, ECA is inversely interrelated to the magnitude of the Epp and directly related to the JPC.67 Fig. 5A shows that N-CD, NB-CD, NS-CD, and NBS-CD have cathodic peak potentials of 0.08, 0.08, 0.09, and 0.12 V respectively. For N-CD and NB-CD the reduction reaction occurs at low overpotential showing the good ECA toward the I3− reduction. The Epp value for all N-CD, NB-CD, and NS-CD is similar (0.64 V) whereas the Epp value for NBS-CD is 0.57 V, lower than the other C-dots. The lower the value of Epp, the higher the rate constant of the electrochemical redox reaction is. A high rate constant indicates fast charge transfer through the electrode surface and slower charge recombination process. On the other hand, N-CD has the largest cathodic current density (−17.48 mA cm−2) to indicate the faster reaction and superior ECA toward I3− reduction.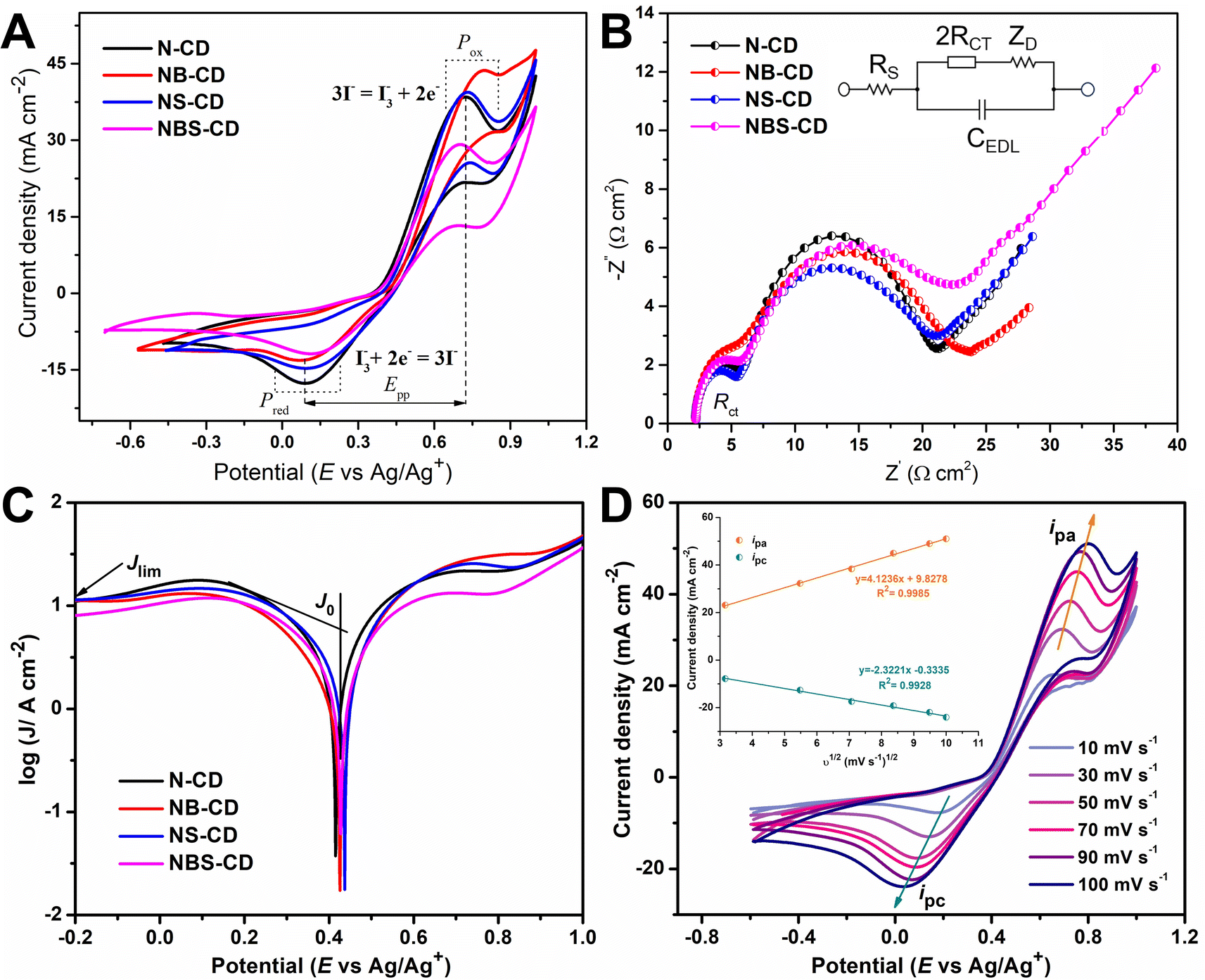 | ||
| Fig. 5 (A) Cyclic voltammogram, (B) Nyquist plot (C), Tafel polarization curve of heteroatom doped C-dots, and (D) cyclic voltammogram and Randles–Sevcik plot of N-CD at different scan rates. | ||
The reduction of internal impedance through nano-structural design is also important in the realm of applications of DSSCs. The impedance analysis has been shown by Nyquist plot in Fig. 5B where real and imaginary impendances are drawn in x and y-axis respectively. The typical Nyquist plot (Fig. 5B) shows a well-defined semicircle with one small curve. The high-frequency region (around 100 kHz) intercept on the real axis (Z′′) characterizes the solution resistance (RS) of the CE. The first semicircle at the medium frequency region is associated with the charge transfer resistance (RCT) and the corresponding constant phase element (CPE) of electrolyte and CE, while the other small semicircle at the low-frequency region determines the Nernst diffusion impedance (ZW or ZD) of an electrolyte. In this respect, this work mainly emphasizes the RS and RCT values of counter electrodes, as the value of Nernst diffusion impedance is insignificant.
Fig. 5B shows the Nyquist plot of heteroatom doped C-dots in an applied frequency of 1 Hz to 1 MHz. N-CD and NS-CD CE have the lowest RCT of 3.2 Ω, signifying low RCT as compared to their counterparts. This is in accordance with the high catalytic activity of I3− reduction in the CV results (Fig. 5A). Although NBS-CD has the lowest Epp value in the CV, the lowest value of JPC and the highest value of RCT make its performance worse than N-CD. Among the four-heteroatom doped C-dots sample, N-CD shows the best electrocatalytic activity toward the I3− reduction that can be attributed to the lowest value of RCT in the Nyquist plot (Fig. 5B) and the highest value of JPC obtained from CV analysis. In a broad context, electron-rich N and S doped C-dots exhibit superior electrocatalytic performance compared to electron-deficient B doped C-dots.
The Tafel polarization technique was employed to analyze the charge transfer kinetics at the CE/electrolyte interface using the same symmetric cell used for EIS. The Tafel polarization curve illustrates the correlation between the logarithmic current density (logJ) and voltage (V). In Tafel plot, the middle zone or Tafel zone is characterized by a steep slope that dictates the ECA of the electrode. The exchange current density (J0) and limiting diffusion current density (Jlim) can be determined from the polarization profile (Fig. 5C), providing additional evidence for the electrochemical performance of the electrode. Greater values of J0 and Jlim indicate superior performance of the material as an electrocatalyst. In Fig. 5C, N-CD and NS-CD have larger J0 value than NB-CD and NBS-CD indicating higher ECA of electron donating heteroatom doped C-dots. J0 from Tafel plot can be correlated with RCT of EIS by eqn (1) where R is the ideal gas constant, T is the temperature, n is the number of electrons involved in the reduction reaction of I3− to I− and F is the Faraday constant.
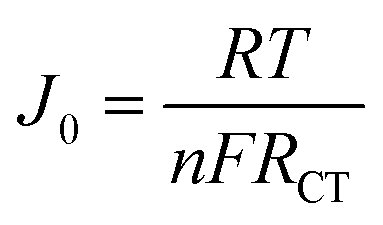 | (1) |
Based on the given relationship, a larger J0 value indicates a lesser contribution of RCT to the electrode that is also reflected in the EIS and Tafel plot of C-dots. The Randles–Sevcik plot of N-CD (Fig. 5D) in the context of cyclic voltammetry has been used to analyze the electrochemical behavior of a redox couple at the electrode–electrolyte interface. In the Randles–Sevcik plot, the system follows the ideal behavior described by the equation Ip = nFACov1/2, such as a reversible redox process, negligible uncompensated resistance, and uniform electrode properties. This linear ideal behavior is also observed for the o-MWCNT/N-CD binary composite material (Fig. S11†).
Fig. 6A shows that o-MWCNT/C-dots have higher ECA than o-MWCNT. A larger enclosed redox reaction area of a CV represents a higher electrochemically active electrode surface. The CVs of the o-MWNCT/N-CD composite in all compositions show a higher enclosed area than the pristine o-MWCNT. So, the incorporation of N-CD in the o-MWCNT modifies the 3D network structure of o-MWCNT with higher surface area and provides enhanced electrocatalytic activity. However, o-MWCNT/C-dots (50 wt%) show deteriorated ECA than o-MWCNT. These findings are in line with what was observed through the FESEM analysis. The ECA determining parameters from CV and EIS analysis of a CE in DSSC (JPC, EPP, RS, and RCT) values are listed in Table 1.
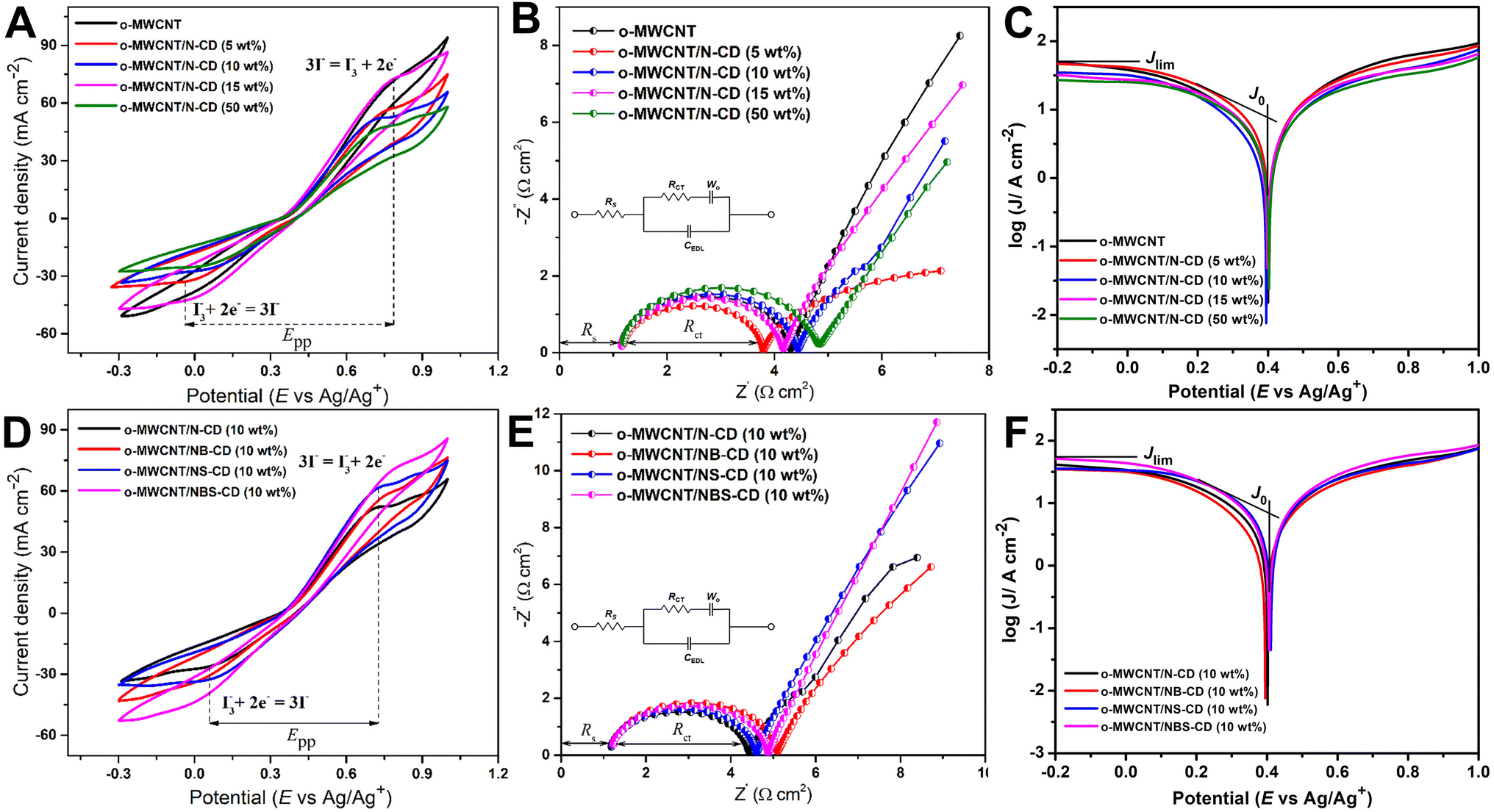 | ||
| Fig. 6 (A and D) Cyclic voltammogram, (B and E) Nyquist plot, and (C and F) Tafel polarization plot of heteroatom doped C-dots and o-MWCNT/C-dots binary composite at different compositions in an electrolyte containing 0.5 M LiI, 0.05 M I2, and 0.1 M LiClO4 in acetonitrile. Scan rate is 50 mV s−1 for CV and frequency is 1 to 106 Hz for Nyquist plot. | ||
| Samples | JPC (mA cm−2) | EPP (V) | RS (Ω cm2) | RCT (Ω cm2) |
|---|---|---|---|---|
| o-MWCNT | −38.11 | 0.79 | 1.14 | 3.16 |
| o-MWCNT/N-CD (5 wt%) | −32.86 | 0.77 | 1.14 | 2.65 |
| o-MWCNT/N-CD (10 wt%) | −26.71 | 0.67 | 1.14 | 3.33 |
| o-MWCNT/N-CD (15 wt%) | −41.06 | 0.77 | 1.14 | 3.01 |
| o-MWCNT/N-CD (50 wt%) | −23.49 | 0.69 | 1.14 | 3.72 |
| o-MWCNT/N-CD (10 wt%) | −26.71 | 0.67 | 1.14 | 3.33 |
| o-MWCNT/NB-CD (10 wt%) | −31.90 | 0.73 | 1.14 | 3.96 |
| o-MWCNT/NS-CD (10 wt%) | −32.43 | 0.67 | 1.14 | 3.46 |
| o-MWCNT/NBS-CD (10 wt%) | −42.10 | 0.76 | 1.14 | 3.75 |
50 wt% C-dots loading shows less electrocatalytic properties than o-MWCNT due to the breakdown of the backbone structure of o-MWCNT and reduction of the surface area and conductivity of the composite. In Nyquist plot (Fig. 6B), the semicircle diameter of o-MWCNT/N-CD (5 wt%) CE is lower than the other CEs, and increases with increased C-dots content up to 50%, followed by slight decrease for 15% loading. This low RCT value suggests that N-CD improves the charge transfer rate of o-MWCNT for the cathodic reaction. The slope for the impedance profile in the low-frequency region related to reactant diffusion behavior is higher for o-MWCNT than that of binary composite, demonstrating that o-MWCNT is favorable to the reactant diffusion process. The low RCT of composite material indicates the creation of new charge movement channel upon C-dots loading. This channel basically arises from the high surface area, porosity, and conductivity of the material that make the charge transfer dynamics faster through the electrode electrolyte interface.68–70 The Tafel polarization curve (Fig. 6C) demonstrates that the o-MWCNT/N-CD (5 wt%) exhibits larger Jlim and J0 values compared to o-MWCNT, indicating a higher ECA of the material.
There is a significant change in impedance and CV data while in the binary composite N-CD is co-doped with other heteroatoms (Fig. 6D & F). The highest value of Jpc (−42.10 mA cm−2) is obtained for NBS-CD (10 wt%), while the lowest value of Epp (0.67 V) is obtained for the N-CD and NS-CD. NB-CD and NBS-CD loaded binary composite has higher value of Epp and RCT than the other composites while that values are smaller for N-CD and NS-CD. Thus, composite with electron deficient B doped C-dots undermine the I3−/I− redox reaction rate while composite with electron donating N and S co-doped C-dots make the process effective and faster. The findings obtained from the Tafel plot (Fig. 6D) corroborate the observations made in CV and EIS experiments. Specifically, the composites doped with electron-donating heteroatoms (N, S) exhibit superior values of Jlim and J0 compared to the electron-deficient B-doped composite.
Photovoltaic performance analysis
The photovoltaic efficiency of the fabricated DSSCs (see ESI† for details) was measured under a simulated irradiation of AM 1.5 illumination (1 sun, 100 mW cm−2). Photocathode was made by a 6–9 μm thick layer of composite materials on the FTO by doctor blade method. The measurement of surface roughness and thickness by atomic force microscopy (AFM) shows the formation of a homogeneous coating with a 110 nm average roughness without any significant surface defect (Fig. S15†). The performance of this prototype was then compared to that of a DSSC constructed with a Pt thin film photocathode. The conversion efficiency of DSSC is influenced by various elements, including reflectance, thermodynamic efficiency, conduction efficiency, charge carrier separation, and collection efficiency. These performance determining parameters are directly related to the fill factor (FF), short circuit current (ISC), and open-circuit voltage (VOC) of a cell that can be attained by the J–V curve analysis of a solar cell.71When the voltage across a solar cell is zero, or when the solar cell is short-circuited, the current flowing through it is known as the ISC. The highest voltage that a solar cell can produce at zero current is known as the VOC. The amount of forward bias on the solar cell resulting from the bias of the junction with the light-generated current is reflected in the VOC.72–74 FF is a parameter that, in conjunction with VOC and ISC, determines the maximum power of a solar cell. The FF is defined as the ratio of the maximum power (Pmax= Imp × Vmp) from the solar cell to the product of VOC and ISC so that:
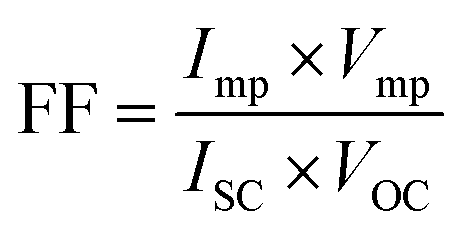 | (2) |
Efficiency is a widely employed metric for evaluating and contrasting the performance of DSSCs. Efficiency can be formally defined as the quantitative measure of the ratio between the energy output generated by a solar cell and the energy input received from the sun. The efficiency (η) of a solar cell is quantified as the proportion of incident power that is transformed into electrical energy. It is formally defined as

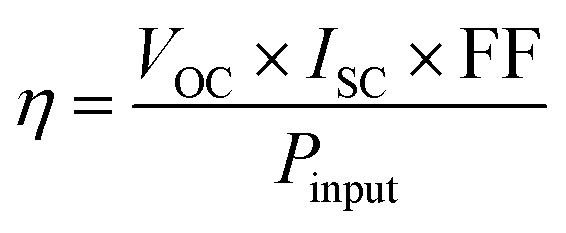 | (3) |
For this work the input power (Pinput) for efficiency calculations is 1 kW m−2 or 100 mW cm−2.
The J–V analysis (Fig. 7A) of binary composite fabricated DSSCs show that the PCE of C-dots loaded o-MWCNT has higher PCE than pristine o-MWCNT. The highest PCE at 5 wt% N-CD loading is 4.28% with the VOC of 0.71 V, JSC of 17.24 mA cm−2, and FF of 35%, respectively, which is higher than that of o-MWCNT/N-CD (10 wt%) CE (η = 3.98%) by 7%. Table 2 shows that C-dots content sputtered on the o-MWCNT surface enhances the PCE and other performance indication parameters (e.g., VOC, JSC, FF) of the cell. The threshold of C-dots content is 5 wt%. The PCE of o-MWCNT/N-CD (5 wt%) fabricated DSSC is 91% and 28% higher than the pristine o-MWCNT and Pt thin film fabricated DSSC, respectively. Higher loading could cover the individual conductive tube and create more surface defect resulting the deflection of carrier mobility and reduced PCE. But 5 wt% doesn't impact its conductive behavior rather it enhances the surface property of the o-MWCNT and increases the PCE of the cell. When the N-CD loading amount exceeds 50 wt% the efficiency of the cell reduces with a lower value of VOC, JSC, and FF. Nevertheless, excessive N-CD loading could promote the charge recombination at photoelectrode due to the slower reduction rate, resulting in decreased PCE.
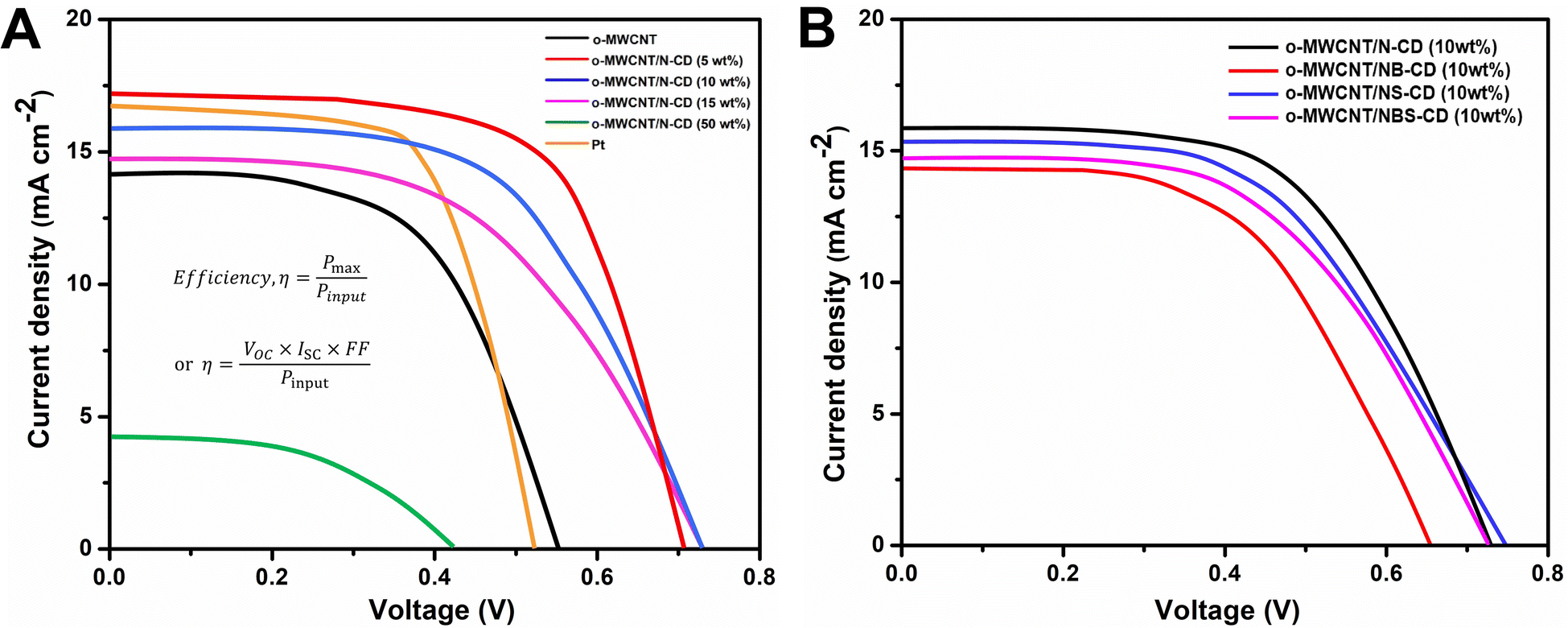 | ||
| Fig. 7 Photovoltaic performance analysis of DSSCs fabricated with (A) o-MWCNT, Pt thin film and o-MWCNT/N-CD (@different wt%) and (B) o-MWCNT/10 wt% C-dots (@N, S, and B co-doped) binary composites. | ||
| Sample | JSC (mA cm−2) | VOC (V) | FF (%) | PCE, η (%) |
|---|---|---|---|---|
| o-MWCNT | 14.08 | 0.55 | 29 | 2.24 |
| o-MWCNT/N-CD (5 wt%) | 17.24 | 0.71 | 35 | 4.28 |
| o-MWCNT/N-CD (10 wt%) | 16.07 | 0.73 | 34 | 3.98 |
| o-MWCNT/N-CD (15 wt%) | 14.96 | 0.73 | 28 | 3.05 |
| o-MWCNT/N-CD (50 wt%) | 4.19 | 0.43 | 25 | 0.45 |
| Pt | 16.49 | 0.48 | 41 | 3.17 |
| o-MWCNT/NB-CD (10 wt%) | 14.31 | 0.65 | 29 | 3.06 |
| o-MWCNT/NS-CD (10 wt%) | 15.37 | 0.74 | 33 | 3.49 |
| o-MWCNT/NBS-CD (10 wt%) | 14.74 | 0.73 | 31 | 3.33 |
Presence of electron donating and accepting heteroatom doped C-dots in o-MWCNT in the context of PCE has also been investigated. Fig. 7B depicts the J–V curve of the DSSC fabricated with 10 wt% heteroatom doped C-dots loaded o-MWCNT. Among all the combinations, o-MWCNT/N-CD shows the best performance because of the electron donating property of N that makes the composite more conductive, provides high surface area, and fasten the charge transfer process in the CE electrolyte interface that is also proven from CV, EIS, and DFT analysis of the material. A proper amount of N-CD loading in the o-MWCNT network can significantly improve the performance of o-MWCNT/N-CD binary composite CE based DSSCs and stand up as a promising candidate to replace Pt in DSSCs.
Cyclability testing and post cycling material characterization
The longevity of the DSSC with synthesized o-MWCNT/N-CD (5 wt%) CE material was tested for 100 CV cycles at 50 mV s−1 throughout a potential window from −0.30 V to 1.00 V (Fig. 8A). The presence of overlapping CV peaks and consistent anodic and cathodic peak currents indicate that o-MWCNT/N-CD (5 wt%) possesses reversible redox activity, exceptional chemical stability, prolonged life cyclability, and firm adhesion to the electrode surface. After running 100 cycles the tested material was recollected, vigorously washed with MeOH and acetone to remove any adsorbed dye or I2 and dried under vacuum. At the same time 50 mg o-MWCNT/N-CD (5 wt%) was dipped into a 0.5 M LiI, and 0.05 M I2 solution in acetonitrile for 7 days. Material was collected by filtration, washed with acetone and dried under vacuum. The chemical stability of the collected dried material was characterized by FTIR and XRD analyses.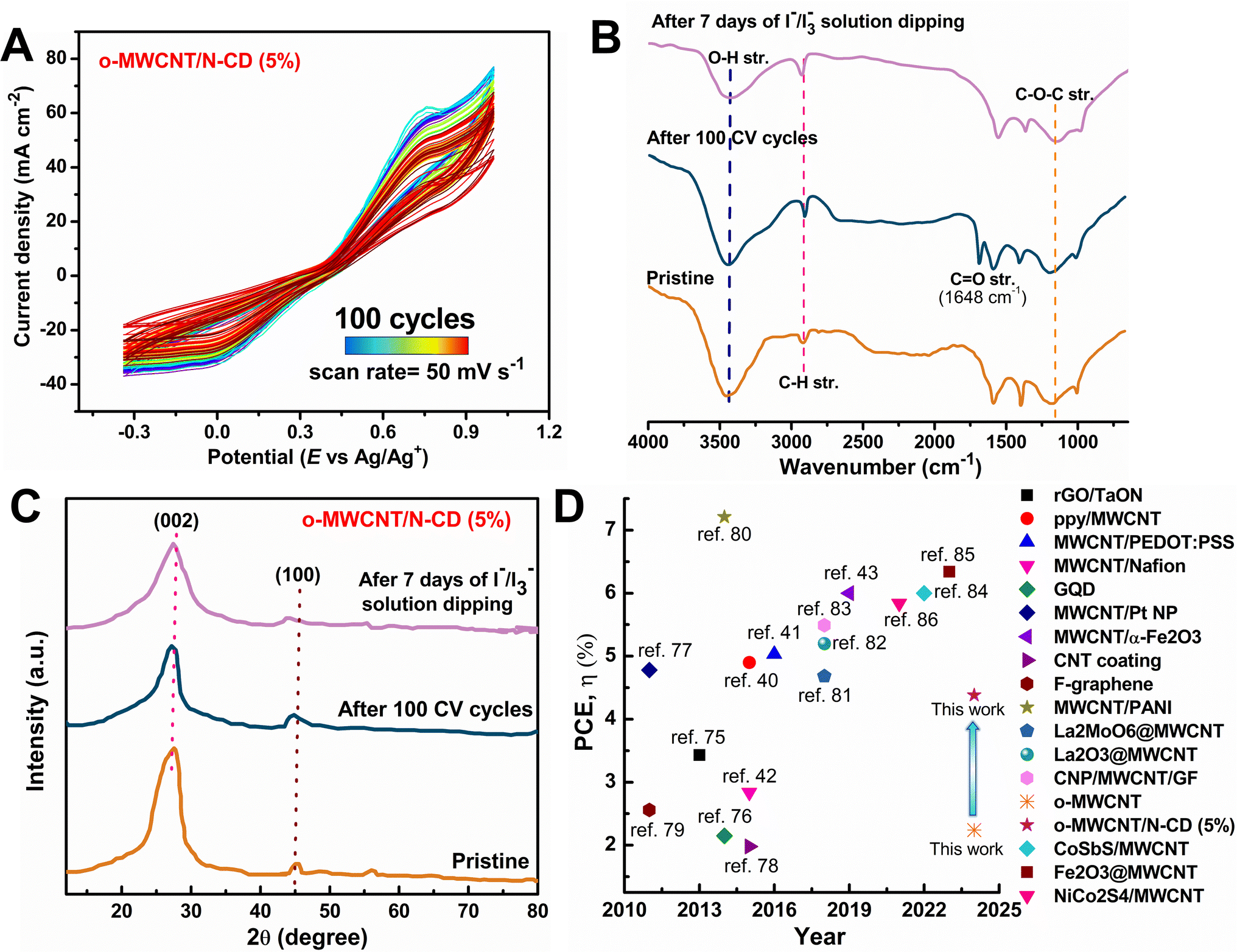 | ||
| Fig. 8 (A) Stability testing of o-MWCNT/N-CD (5 wt%) by 100 CV cycles, (B) FTIR spectra, and (C) XRD pattern of the post-performance analyzed material. (D) Literature PCE data comparison plot for a similar kind of CE material. | ||
FTIR spectra (Fig. 8B) of the o-MWCNT/N-CD (5 wt%) after 100 cycling and I3−/I− solution dipping are similar to the pristine material. The CO stretching band at 1648 cm−1 originates from the surface adsorbed acetone. XRD pattern of the treated material (Fig. 8C) shows similar characteristics. Adsorbed I2 may be the cause minor peak broadening and decreased intensity of the treated material, which in turn may have led to a slight decrease in the crystallinity of the material. The findings of stability tests of the material demonstrate outstanding chemical stability without any significant degradation, extended life cycle, and strong adherence to the electrode surface.
This work shows a complete study of a new CE design for DSSCs via synergism of MWCNT and C-dots, successful characterization of materials, evaluation of electrochemical performance, and characterization of post-performance material stability. Our developed materials overperformed different CEs based on carbonaceous and polymeric materials reported in the literature40–43,74–86 (Fig. 8D and Table S1†). The synthesis of heteroatom doped C-dots and o-MWCNT/C-dots binary composite is totally reproducible with the reported synthesis route. We also tested the electrochemical performance of o-MWCNT/N-CD (5 wt%) with 10 trials via CV and EIS experiment. The variation of the performance evaluation parameter JPC, EPP, and RCT are shown in Fig. S16.† The variation of all the parameters within 10 trials are set between small standard deviations that can be considered as experimental and or human error. This validates the high degree of reproducibility of the reported work.
Conclusions
In conclusion, this study presents a promising avenue for enhancing the efficiency and sustainability of DSSCs through the utilization of a novel carbon-based binary composite CE. The successful synthesis of C-dots from readily available natural precursors signifies a promising breakthrough in environmentally friendly materials science. Furthermore, the introduction of heteroatoms into the C-dots, while avoiding toxic chemicals, demonstrated the feasibility of tailoring the electrocatalytic performance of the composite. The key finding of this study is the superior electrocatalytic performance of the o-MWCNT/C-dots binary composite compared to its MWCNT precursor achieved through the synergistic effect of o-MWCNT/C-dots with a favorable supramolecular binding. This enhancement can be attributed to the increased porosity of the composite material, effectively mitigating the agglomeration behavior of MWCNTs caused by π–π stacking and van der Waals interactions among individual tubes. Notably, N-doped carbon dots exhibited the highest catalytic activity among the heteroatom-doped counterparts. Electron donating property of N and S make C-dots a better electrocatalyst than electron acceptor B doped C-dots. This study also suggests that an optimal level of individual material mixing is crucial to design a composite material. Out of all the synthesized binary composites, o-MWCNT/N-CD (5 wt%) has the highest PCE that is 91% higher than pristine o-MWCNT since it satisfies every need for being a CE, including increased surface area, quicker charge kinetics, reduced charge recombination, and a surface defect site. Finally, this research facilitates the enhancement of solar energy usage and the reduction of the carbon footprint linked to conventional energy sources.Author contributions
Mahmudul Hasan: writing – original draft preparation, data curation, formal analysis, methodology, reviewing and editing, and Md. Abu Bin Hasan Susan: conceptualization, writing – reviewing and editing, supervision, funding acquisition, resources.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
AMMH acknowledges research fellowships from the Semiconductor Technology Research Centre at the University of Dhaka, Bangladesh and the National Science and Technology (NST) Fellowship, Bangladesh. The authors also express gratitude for the financial support received from the University Grants Commission of Bangladesh and Ministry of Science and Technology, Bangladesh, to support their research endeavors. The authors are grateful to Prof. Austin M. Evans, Department of Chemistry, University of Florida and his funding source, Department of Defense for a Multidisciplinary University Research Initiative Award (W911NF2310260) for providing the AFM assistance. The authors also express their gratitude to the Centre for Advanced Research in Sciences of the University, of Dhaka, Bangladesh for permitting the use of photoluminescence spectrophotometer. The authors also acknowledge analytical services from the Genome Centre of Jashore University of Science and Technology, Bangladesh for FESEM and EDX and Renewable Energy Research division of BCSIR for their assistance in conducting the evaluation of the performance of DSSCs.References
- B. Goss Levi, J. Renewable Sustainable Energy, 2013, 5, 032801 CrossRef.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed.
- I. K. Popoola, M. A. Gondal, J. M. AlGhamdi and T. F. Qahtan, Sci. Rep., 2018, 8, 12864 CrossRef PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- G. J. Meyer, ACS Nano, 2010, 4, 4337–4343 CrossRef CAS PubMed.
- S. M. McCullough, J. M. Evans, T. Moot, A. D. Taggart, L. Troian-Gautier and J. F. Cahoon, ACS Appl. Energy Mater., 2020, 3, 1496–1505 CrossRef CAS.
- G. P. Smestad, Sol. Energy Mater. Sol. Cells, 2003, 76, 1–2 CrossRef CAS.
- D. Kuang, P. Wang, S. Ito, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 7732–7733 CrossRef CAS PubMed.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218–12219 CrossRef CAS PubMed.
- F. Yang, X. Tian, K. Zhang, X. Zhang and L. Liu, ECS J. Solid State Sci. Technol., 2018, 7, P311 CrossRef CAS.
- B. Zhang, D. Wang, Y. Hou, S. Yang, X. H. Yang, J. H. Zhong, J. Liu, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, Sci. Rep., 2013, 3, 1836 CrossRef PubMed.
- J. Han, H. Kim, D. Y. Kim, S. M. Jo and S.-Y. Jang, ACS Nano, 2010, 4, 3503–3509 CrossRef CAS PubMed.
- M. R. Al-bahrani, X. Xu, W. Ahmad, X. Ren, J. Su, Z. Cheng and Y. Gao, Mater. Res. Bull., 2014, 59, 272–277 CrossRef CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- T. K. Das, P. Ilaiyaraja and C. Sudakar, ACS Appl. Energy Mater., 2018, 1, 765–774 CrossRef CAS.
- X. Liu, Z. Mao, J. Liu, F. Meng, X. Shi, X. Xue and B. Zhao, ACS Appl. Energy Mater., 2022, 5, 8391–8399 CrossRef CAS.
- S. S. Jeon, C. Kim, J. Ko and S. S. Im, J. Phys. Chem. C, 2011, 115, 22035–22039 CrossRef CAS.
- A. Kay and M. Grätzel, Sol. Energy Mater. Sol. Cells, 1996, 44, 99–117 CrossRef CAS.
- T. N. Murakami, S. Ito, Q. Wang, M. K. Nazeeruddin, T. Bessho, I. Cesar, P. Liska, R. Humphry-Baker, P. Comte, P. Péchy and M. Grätzel, J. Electrochem. Soc., 2006, 153, A2255 CrossRef CAS.
- J. Ou, J. Xiang, J. Liu and L. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 14862–14870 CrossRef CAS PubMed.
- K. Imoto, K. Takahashi, T. Yamaguchi, T. Komura, J.-i. Nakamura and K. Murata, Sol. Energy Mater. Sol. Cells, 2003, 79, 459–469 CrossRef CAS.
- A. A. Arbab, K. C. Sun, I. A. Sahito, M. B. Qadir, Y. S. Choi and S. H. Jeong, ACS Appl. Mater. Interfaces, 2016, 8, 7471–7482 CrossRef CAS PubMed.
- S.-Q. Fan, B. Fang, J. H. Kim, B. Jeong, C. Kim, J.-S. Yu and J. Ko, Langmuir, 2010, 26, 13644–13649 CrossRef CAS PubMed.
- Y.-A. Leu, M.-H. Yeh, L.-Y. Lin, T.-J. Li, L.-Y. Chang, S.-Y. Shen, Y.-S. Li, G.-L. Chen, W.-H. Chiang, J.-J. Lin and K.-C. Ho, ACS Sustain. Chem. Eng., 2017, 5, 537–546 CrossRef CAS.
- M. Chen, J. Liu, X.-Q. Wang, L.-L. Shao, Z.-Y. Yuan, X. Qian, Y.-N. Wang, A.-X. Ding and Y.-S. Tian, ACS Appl. Energy Mater., 2021, 4, 13952–13962 CrossRef CAS.
- M.-H. Yeh, L.-Y. Lin, C.-L. Sun, Y.-A. Leu, J.-T. Tsai, C.-Y. Yeh, R. Vittal and K.-C. Ho, J. Phys. Chem. C, 2014, 118, 16626–16634 CrossRef CAS.
- M. Younas, T. N. Baroud, M. A. Gondal, M. A. Dastageer and E. P. Giannelis, J. Power Sources, 2020, 468, 228359 CrossRef CAS.
- G. Veerappan, K. Bojan and S.-W. Rhee, ACS Appl. Mater. Interfaces, 2011, 3, 857–862 CrossRef CAS PubMed.
- S. S. Nemala, P. Kartikay, K. S. Aneja, P. Bhargava, H. L. M. Bohm, S. Bohm and S. Mallick, ACS Appl. Energy Mater., 2018, 1, 2512–2519 CrossRef CAS.
- J. D. Roy-Mayhew, D. J. Bozym, C. Punckt and I. A. Aksay, ACS Nano, 2010, 4, 6203–6211 CrossRef CAS PubMed.
- R. Bajpai, S. Roy, P. Kumar, P. Bajpai, N. Kulshrestha, J. Rafiee, N. Koratkar and D. S. Misra, ACS Appl. Mater. Interfaces, 2011, 3, 3884–3889 CrossRef CAS PubMed.
- D.-Y. Kang, Y. Lee, C.-Y. Cho and J. H. Moon, Langmuir, 2012, 28, 7033–7038 CrossRef CAS PubMed.
- A. M. M. Hasan, M. A. Hasan, A. Reza, M. M. Islam and M. A. B. H. Susan, Mater. Today Commun., 2021, 29, 102732 CrossRef CAS.
- H. S. Jung and J.-K. Lee, J. Phys. Chem. Lett., 2013, 4, 1682–1693 CrossRef CAS PubMed.
- F. T. Krauss, I. Pantenburg and B. Roling, Adv. Mater. Interfaces, 2022, 9, 2101891 CrossRef CAS.
- F. Ding, P. Larsson, J. A. Larsson, R. Ahuja, H. Duan, A. Rosén and K. Bolton, Nano Lett., 2007, 8, 463–468 CrossRef PubMed.
- T. H. V. Kumar, J. Rajendran, R. D. Nagarajan, G. Jeevanandam, A. N. Reshetilov and A. K. Sundramoorthy, ACS Omega, 2021, 6, 5146–5157 CrossRef CAS PubMed.
- T. H. V. Kumar, J. Rajendran, R. Atchudan, S. Arya, M. Govindasamy, M. A. Habila and A. K. Sundramoorthy, Environ. Res., 2023, 238, 117193 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, ChemSusChem, 2011, 4, 913–925 CrossRef CAS PubMed.
- P. Gemeiner, J. Kuliček, M. Mikula, M. Hatala, Ľ. Švorc, L. Hlavatá, M. Mičušík and M. Omastová, Synth. Met., 2015, 210, 323–331 CrossRef CAS.
- A. Ali, S. M. Shah, S. Bozar, M. Kazici, B. Keskin, M. Kaleli, S. Akyürekli and S. Günes, Nanotechnology, 2016, 27, 384003 CrossRef PubMed.
- K. Prabakaran, S. Mohanty and S. K. Nayak, RSC Adv., 2015, 5, 40491–40504 RSC.
- F. Mousavi and A. A. Taherpour, Electrochim. Acta, 2019, 318, 617–624 CrossRef CAS.
- K. Li, Y. Xiao, Y. Zhao, Y. Xia, J. Ding, Q. He, J. Ling and G. Li, Inorg. Chem. Commun., 2022, 145, 110025 CrossRef CAS.
- D. Das, I. V. Sabaraya, T. Zhu, T. Sabo-Attwood and N. B. Saleh, Environ. Sci. Technol., 2018, 52, 8233–8241 CrossRef CAS PubMed.
- Z. Ye, G. Li, J. Lei, M. Liu, Y. Jin and B. Li, ACS Appl. Bio Mater., 2020, 3, 7095–7102 CrossRef CAS PubMed.
- K. A. S. Fernando, S. Sahu, Y. Liu, W. K. Lewis, E. A. Guliants, A. Jafariyan, P. Wang, C. E. Bunker and Y.-P. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 8363–8376 CrossRef CAS PubMed.
- W. Xie, R. Gomes, T. Aubert, S. Bisschop, Y. Zhu, Z. Hens, E. Brainis and D. Van Thourhout, Nano Lett., 2015, 15, 7481–7487 CrossRef CAS PubMed.
- B. Omogo, F. Gao, P. Bajwa, M. Kaneko and C. D. Heyes, ACS Nano, 2016, 10, 4072–4082 CrossRef CAS PubMed.
- P. B. Sorokin, P. V. Avramov, L. A. Chernozatonskii, D. G. Fedorov and S. G. Ovchinnikov, J. Phys. Chem. A, 2008, 112, 9955–9964 CrossRef CAS PubMed.
- R. Kawano, H. Matsui, C. Matsuyama, A. Sato, M. A. B. H. Susan, N. Tanabe and M. Watanabe, J. Photochem. Photobiol., A, 2004, 164, 87–92 CrossRef CAS.
- A. Prasannan and T. Imae, Ind. Eng. Chem. Res., 2013, 52, 15673–15678 CrossRef CAS.
- W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 100, 12974–12980 CrossRef CAS.
- H. B. Schlegel, J. M. Millam, S. S. Iyengar, G. A. Voth, A. D. Daniels, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 2001, 114, 9758–9763 CrossRef CAS.
- H. P. Hratchian and M. J. Frisch, J. Chem. Phys., 2011, 134, 204103 CrossRef PubMed.
- J. A. Steckel, K. D. Jordan and P. Avouris, J. Phys. Chem. A, 2002, 106, 2572–2579 CrossRef CAS.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, E. P. Gray, C. P. Higgins and J. F. Ranville, Environ. Sci. Technol., 2012, 46, 12272–12280 CrossRef CAS PubMed.
- M. R. Hajaligol, P. A. Martoglio Smith, J. B. Wooten and V. Baliga, Energy Fuels, 2000, 14, 1083–1093 CrossRef.
- M. Farbod, S. K. Tadavani and A. Kiasat, Colloids Surf., A, 2011, 384, 685–690 CrossRef CAS.
- S. Sagar, N. Iqbal and A. Maqsood, J. Phys.: Conf. Ser., 2013, 439, 012024 CrossRef CAS.
- X. He, X. Xu, G. Bo and Y. Yan, RSC Adv., 2020, 10, 2180–2190 RSC.
- U. Moralı, H. Demiral and S. Şensöz, J. Cleaner Prod., 2018, 189, 602–611 CrossRef.
- A. S. Hassanien, R. A. Shedeed and N. K. Allam, J. Phys. Chem. C, 2016, 120, 21678–21684 CrossRef CAS.
- M. Aslan and H. Eskalen, Fullerenes, Nanotubes Carbon Nanostruct., 2021, 29, 1026–1033 CrossRef CAS.
- V. J. Surya, K. Iyakutti, H. Mizuseki and Y. Kawazoe, IEEE Trans. Nanotechnol., 2012, 11, 534–541 Search PubMed.
- M. Pumera and C. H. A. Wong, Chem. Soc. Rev., 2013, 42, 5987–5995 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- W. J. Lee, E. Ramasamy, D. Y. Lee and J. S. Song, ACS Appl. Mater. Interfaces, 2009, 1, 1145–1149 CrossRef CAS PubMed.
- R. Stalder, D. Xie, A. Islam, L. Han, J. R. Reynolds and K. S. Schanze, ACS Appl. Mater. Interfaces, 2014, 6, 8715–8722 CrossRef CAS PubMed.
- B. He, Q. Tang, M. Wang, H. Chen and S. Yuan, ACS Appl. Mater. Interfaces, 2014, 6, 8230–8236 CrossRef CAS PubMed.
- I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486–489 CrossRef CAS PubMed.
- K. M. Kim, Masud, J.-M. Ji and H. K. Kim, ACS Appl. Energy Mater., 2020, 4, 1302–1312 CrossRef.
- L. Kavan, P. Liska, S. M. Zakeeruddin and M. Grätzel, ACS Appl. Mater. Interfaces, 2014, 6, 22343–22350 CrossRef CAS PubMed.
- R. Ramanathan, M. Zinigrad, D. Kasinathan and R. K. Poobalan, ACS Appl. Energy Mater., 2022, 5, 11506–11516 CrossRef CAS.
- Y. Li, H. Wang, Q. Feng, G. Zhou and Z.-S. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8217–8224 CrossRef CAS PubMed.
- W. Wei, K. Sun and Y. H. Hu, J. Mater. Chem. A, 2014, 2, 16842–16846 RSC.
- H.-Y. Wang, F.-M. Wang, Y.-Y. Wang, C.-C. Wan, B.-J. Hwang, R. Santhanam and J. Rick, J. Phys. Chem. C, 2011, 115, 8439–8446 CrossRef CAS.
- S. Widodo, G. Wiranto and M. N. Hidayat, Energy Procedia, 2015, 68, 37–44 CrossRef CAS.
- S. Das, P. Sudhagar, V. Verma, D. Song, E. Ito, S. Y. Lee, Y. S. Kang and W. Choi, Adv. Funct. Mater., 2011, 21, 3729–3736 CrossRef CAS.
- H. Niu, S. Qin, X. Mao, S. Zhang, R. Wang, L. Wan, J. Xu and S. Miao, Electrochim. Acta, 2014, 121, 285–293 CrossRef CAS.
- K. Wu, J. Zhao, Y. Xiong, B. Ruan and M. Wu, J. Rare Earths, 2018, 36, 1278–1283 CrossRef CAS.
- K. Wu, J. Zhao, Y. Xiong, B. Ruan and M. Wu, Ionics, 2018, 24, 4055–4061 CrossRef CAS.
- X. Luo, Y. Zhang and S. H. Kim, Nanoscale Res. Lett., 2018, 13, 274 CrossRef PubMed.
- I. J. Peter, S. Vijaya, S. Anandan, S. Karazhanov and P. Nithiananthi, J. Electrochem. Soc., 2022, 169, 056518 CrossRef CAS.
- K. Wu, F. Nie, H. Zhao, X. Qi, P. Li and M. Wu, Int. J. Hydrogen Energy, 2023, 48, 33571–33579 CrossRef CAS.
- V. Gayathri, I. John Peter, K. Ramachandran, S. Karazhanov and C. Raja Mohan, Energy Fuels, 2021, 35, 13360–13369 CrossRef CAS.
- S. Chandra, S. Pradhan, S. Mitra, P. Patra, A. Bhattacharya, P. Pramanik and A. Goswami, Nanoscale, 2014, 6, 3647–3655 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00601a |
| This journal is © The Royal Society of Chemistry 2024 |