
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer evaluations of iodoquinazoline substituted with allyl and/or benzyl as dual inhibitors of EGFRWT and EGFRT790M: design, synthesis, ADMET and molecular docking†
Ahmed K. B. Aljohani a,
Khaled El-Adl*bc,
Basmah Almohaywid,
Omar M. Alatawie,
Marwa Alsulaimanyf,
Ahmed El-morsyg,
Sara A. Almadanih,
Hussam Y. Alharbii,
Majed S. Aljohanii,
Felemban Athary Abdulhaleem Mj,
Hanan E. M. Osmanjk and
Samy Mohamadyl
a,
Khaled El-Adl*bc,
Basmah Almohaywid,
Omar M. Alatawie,
Marwa Alsulaimanyf,
Ahmed El-morsyg,
Sara A. Almadanih,
Hussam Y. Alharbii,
Majed S. Aljohanii,
Felemban Athary Abdulhaleem Mj,
Hanan E. M. Osmanjk and
Samy Mohamadyl
aPharmacognosy and Pharmaceutical Chemistry Department, College of Pharmacy, Taibah University, Al-Madinah Al-Munawarah 41477, Saudi Arabia. E-mail: eladlkhaled74@azhar.edu.eg; khaled.eladl@hu.edu.eg; eladlkhaled74@yahoo.com
bChemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
cPharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City, 11884, Cairo, Egypt
dDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Khalid University, Abha 61421, Saudi Arabia
eDepartment of Chemistry, Faculty of Science, University of Tabuk, Tabuk 47512, Saudi Arabia
fDepartment of Pharmacognosy & Pharmaceutical Chemistry, College of Pharmacy, Taibah University, Medina 42353, Saudi Arabia
gPharmaceutical Chemistry Department, College of Pharmacy, The Islamic University, Najaf, Iraq
hDepartment of Pharmacology and Toxicology, College of Pharmacy, Taibah University, Medina 42353, Saudi Arabia
iDepartment of Chemistry, Faculty of Science, Taibah University, Yanbu, Saudi Arabia
jBiology Department, Faculty of Science, Umm Al-Qura University, Makkah 21955, Saudi Arabia
kBotany and Microbiology Department, Al-Azhar University, Cairo 11651, Egypt
lPharmaceutical Chemistry Department, Faculty of Pharmacy, The British University in Egypt, Cairo, Egypt
First published on 6th March 2024
Abstract
Fifteen new iodoquinazoline derivatives, 5a,b to 18, are reported in this study and their anticancer evaluation as dual inhibitors of EGFRWT and EGFRT790M. The new derivatives were designed according to the target of structural requirements of receptors. Cytotoxicity of our compounds was evaluated against MCF-7, A549, HCT116 and HepG2 cell lines using MTT assay. Compounds 18, 17 and 14b showed the highest anticancer effects with IC50 = 5.25, 6.46, 5.68 and 5.24 μM, 5.55, 6.85, 5.40 and 5.11 μM and 5.86, 7.03, 6.15 and 5.77 μM against HepG2, MCF-7, HCT116 and A549 cell lines, respectively. The eight highly effective compounds 10, 13, 14a, 14b, 15, 16, 17 and 18 were inspected against VERO normal cell lines to evaluate their cytotoxicity. Our conclusion was that compounds 10, 13, 14a, 14b, 15, 16, 17 and 18 possessed low toxicity against VERO normal cells with IC50 increasing from 43.44 to 52.11 μM. All compounds were additionally assessed for their EGFRWT and EGFRT790M inhibitory activities. Additionally, their ability to bind with EGFRWT and EGFR receptors was confirmed by molecular docking. Compound 17 exhibited the same inhibitory activity as erlotinib. Compounds 10, 13, 14b, 16 and 18 excellently inhibited VEGFR-2 activity with IC50 ranging from 0.17 to 0.50 μM. Moreover, compounds 18, 17, 14b and 16 remarkably inhibited EGFRT790M activity with IC50 = 0.25, 0.30, 0.36 and 0.40 μM respectively. As planned, compounds 18, 17 and 14b showed excellent dual EGFRWT/EGFRT790M inhibitory activities. Finally, our compounds 18, 17 and 14b displayed good in silico ADMET calculated profiles.
1. Introduction
The globally most fatal type of all cancers is lung cancer. Every year 7 million people die due to cancer. Lung cancer is estimated to account for 25% of these deaths.1 Lung cancer is categorized into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC).2 NSCLC is the fatal type of lung cancer with approximately 1.5 million recorded cases with below 20% survival rate. Targeted therapy intended at signaling pathways and cancer-particular molecules showed fewer side effects than chemotherapy.3 In anticancer therapy, one of the most important targets is PTKs (protein tyrosine kinases), as they modify the signaling pathway of growth factors.4PTKs play an important role in controlling the cancer physiological cycle and are found on cell membranes of living organisms.5 EGFR is one of the PTKs that was documented to be the most vital target for the managing of growth, proliferation, and differentiation of cancers.6 Most of the NSCLC therapeutic agents/EGFR inhibitors have a shared structure of quinazoline (Fig. 1).1 In the first year of treatment with these drugs, more than 50% of the patients developed resistance by T790M mutation.7 As such, many second-generation EGFR inhibitors, such as afatinib and dacomitinib (Fig. 1), were designed and developed to manage the resistance caused by the T790M mutation.1 The reported objective was not attained due to serious side effects produced by second-generation EGFR inhibitors, such as diarrhea and skin rashes. Many third-generation NSCLC therapeutic agents/EGFR inhibitors, such as AZD9291 and WZ4002 (Fig. 1), were designed and synthesized to manage the developed resistance caused by EGFRT790M/L858R mutations.8 This study targets the computational design of new EGFRWT/EGFRT790M inhibitors with enhanced inhibitory effects, as well as a docking study and prediction of pharmacokinetic properties of these new EGFR inhibitors.
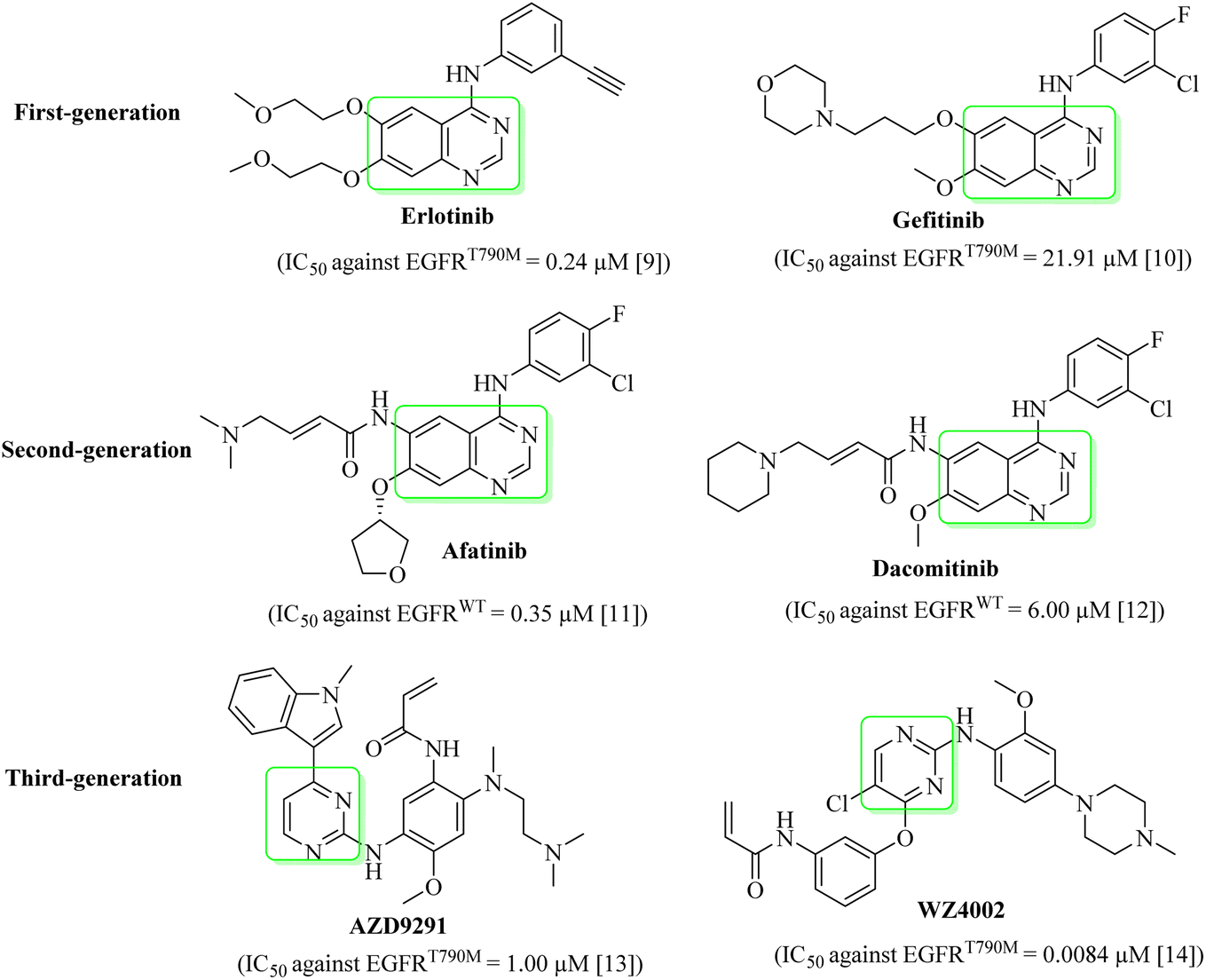 | ||
| Fig. 1 Quinazoline- and pyrimidine-based EGFR inhibitors.9–14 | ||
The current research is an extension to our earlier studies15–30 to achieve anticancer agents. This study showed the effectiveness of hybridization between iodoquinazolinone and different moieties to produce anticancer agents targeting EGFRWT and EGFRT790M. We obtained a new series of iodoquinazolinone joined to different moieties through acetamide linker as dual EGFRWT/EGFRT790M inhibitors for cancer treatment against A549, HCT116, MCF-7 and HepG2. In silico molecular docking study was performed for our derivatives to discover their binding modes in EGFR active sites and/or predict their ADMET properties.
1.1. Rationale and structure-based design
In this research, we used iodo-substitution of quinazoline derivatives as anticancer agents due to iodine high atomic number, ease of attachment to organic compounds, and the specificity of its uptake by the human body, and radioactive isotopes of iodine can also be used to treat thyroid cancer. Moreover, there are many approved drugs containing iodo substitutions, such as 2-iodo-4′-methoxychalcone, amiodarone and iodochlorhydroxyquinoline (clioquinol).31 Our compounds were constructed according to the structural requirements of inhibitors of EGFR. A new series of iodoquinazolines was synthesized through bioisosteric variations of erlotinib at four points: (1) a flat hetero-aromatic system that occupies the adenine binding pocket, (2) a terminal hydrophobic head (occupying hydrophobic region I) which was replaced by allyl and/or benzyl moieties, (3) an NH group (as HBD and/or HBA) which was replaced in our derivatives with a carbonyl group as the HB acceptor, and (4) a hydrophobic tail (occupying hydrophobic region II) which was replaced with different side chain moieties (Fig. 2 and 3).32–37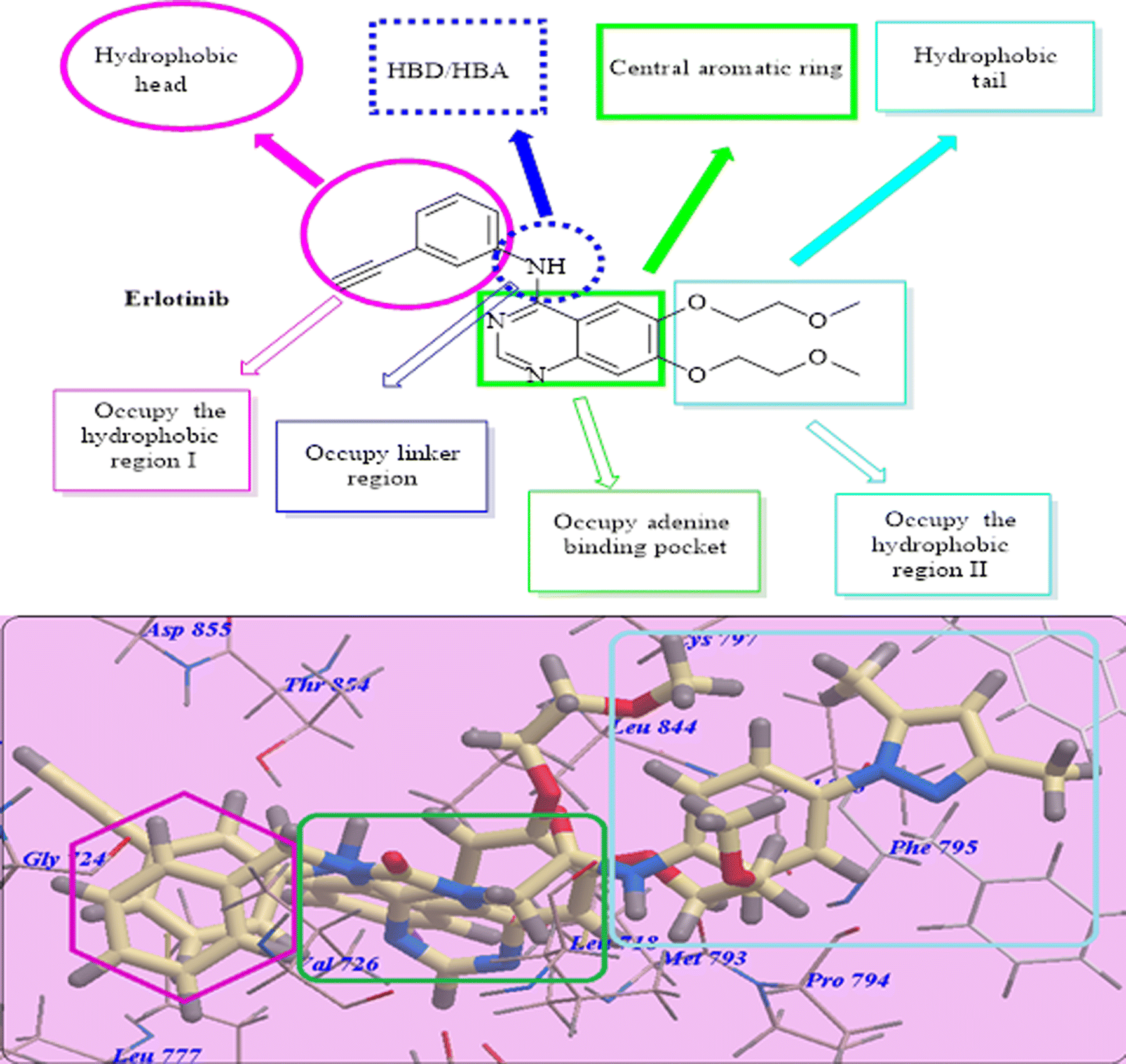 | ||
| Fig. 2 Structural requirements for erlotinib as a reported EGFR-TK inhibitor. | ||
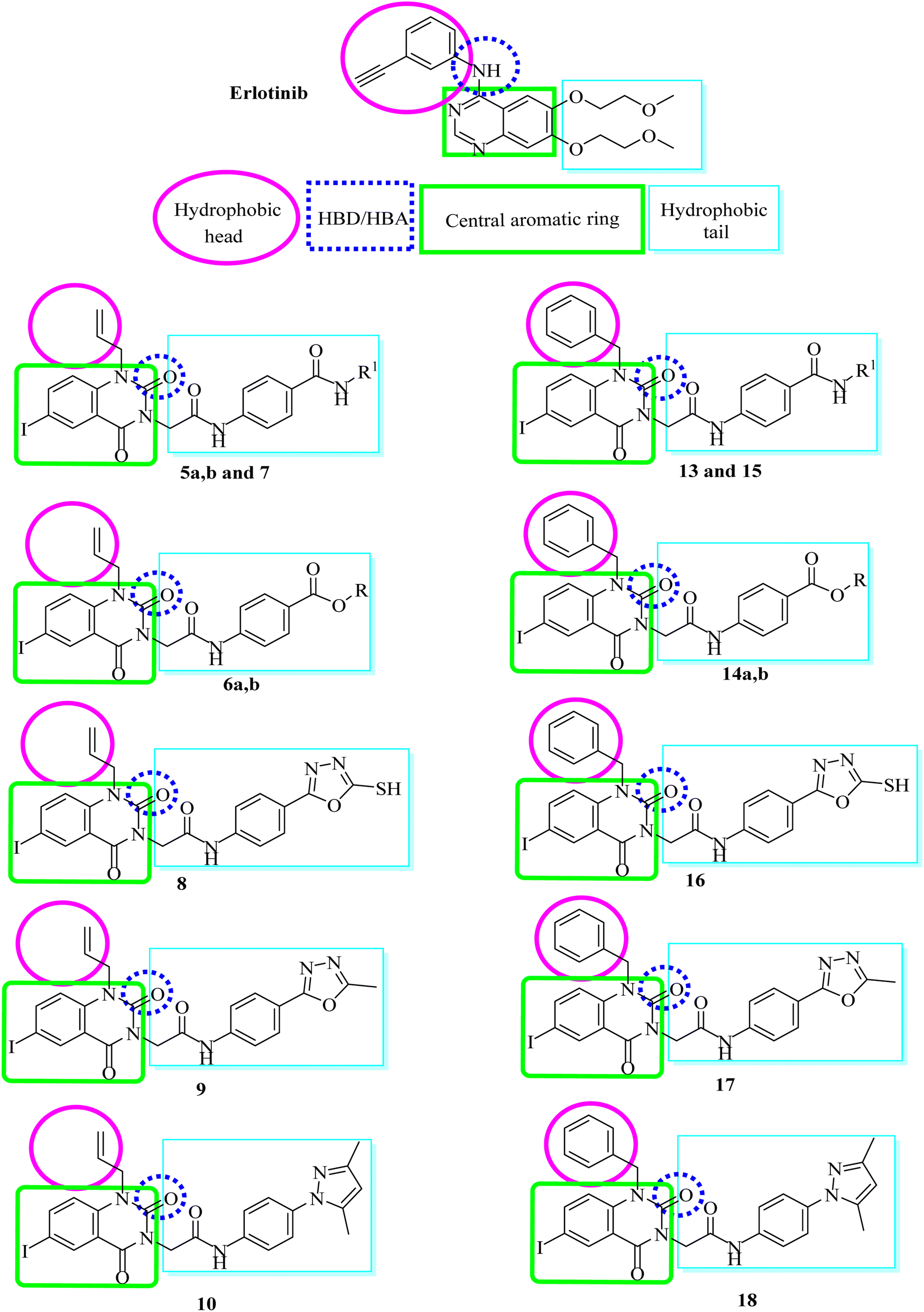 | ||
| Fig. 3 Basic structural features of EGFR inhibitors. | ||
2. Results and discussion
2.1. Chemistry
The synthetic pathways for derivatives 5a,b to 18 are illustrated in Schemes 1 and 2. This started with the reaction of chloroacetyl chloride with suitable amine Ia–d to give corresponding chloroacetamide IIa–d.38,39 The chloroactamide IIa underwent reaction with appropriate methyl and/or ethylamine according to the mixed anhydride procedure39 to give derivatives IIIa,b correspondingly. Reaction of anthranilic acid (1) with iodine afforded 5-iodoanthranilic acid (2) which underwent reaction with allyl bromide to give the corresponding N-allyl derivative (3).40 Fusion of 5-iodo-N-allylanthranilic acid (3) with urea afforded 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4).40 In situ reaction of 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4) with the appropriate chloroacetamide IIIa,b afforded corresponding benzamide derivatives 5a,b. 1H-NMR spectrum of derivative 5b as an example revealed characteristic bands for the new N-allyl group at 5.10–5.18 for CH2CH and CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH and at 5.88–5.92 for CHCH2. Moreover, it showed signals for CH3 and CH2 at 1.21–125 and 3.82–3.88, respectively. 13C-NMR is also consistent with the formed structure. Moreover, reaction of 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4) with the appropriate chloroacetamide IIb,c afforded corresponding ester derivatives 6a,b. The 13C-NMR and 1H-NMR spectra of compounds 6a,b were consistent with formed structures. 1H-NMR spectrum of derivative 6b as a model displayed characteristic bands for the ethyl group at 1.35–1.38 (triplet signal for CH2CH3) and 4.32–4.35 (quartet signal for CH2CH3) in addition to characteristic singlet signals for CHCH2 and NH at 5.98 and 10.64, respectively. Reaction of ester 6b with hydrazine hydrate afforded the corresponding hydrazide derivative 7, which showed the disappearance of characteristic bands for ethyl ester and presence of NH2, NH and hydrazide carbonyl bands, in its IR spectrum. Cyclization of acid hydrazide 7 with carbon disulfide, acetic anhydride and/or acetyl acetone afforded the corresponding oxadiazole-5-thiol 8, 5-methyloxadiazole 9 and/or dimethylpyrazole derivative 10, respectively (Scheme 1). IR and 1H-NMR spectra for compounds 8, 9 and 10 exhibited the disappearance of hydrazide characteristic bands. In addition, 1H-NMR spectrum for compound 10 presents two singlet signals at 1.99 and 2.19 for two CH3 moieties of the pyrazole group. Moreover, 13C-NMR is consistent with the formed structure.
CH and at 5.88–5.92 for CHCH2. Moreover, it showed signals for CH3 and CH2 at 1.21–125 and 3.82–3.88, respectively. 13C-NMR is also consistent with the formed structure. Moreover, reaction of 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4) with the appropriate chloroacetamide IIb,c afforded corresponding ester derivatives 6a,b. The 13C-NMR and 1H-NMR spectra of compounds 6a,b were consistent with formed structures. 1H-NMR spectrum of derivative 6b as a model displayed characteristic bands for the ethyl group at 1.35–1.38 (triplet signal for CH2CH3) and 4.32–4.35 (quartet signal for CH2CH3) in addition to characteristic singlet signals for CHCH2 and NH at 5.98 and 10.64, respectively. Reaction of ester 6b with hydrazine hydrate afforded the corresponding hydrazide derivative 7, which showed the disappearance of characteristic bands for ethyl ester and presence of NH2, NH and hydrazide carbonyl bands, in its IR spectrum. Cyclization of acid hydrazide 7 with carbon disulfide, acetic anhydride and/or acetyl acetone afforded the corresponding oxadiazole-5-thiol 8, 5-methyloxadiazole 9 and/or dimethylpyrazole derivative 10, respectively (Scheme 1). IR and 1H-NMR spectra for compounds 8, 9 and 10 exhibited the disappearance of hydrazide characteristic bands. In addition, 1H-NMR spectrum for compound 10 presents two singlet signals at 1.99 and 2.19 for two CH3 moieties of the pyrazole group. Moreover, 13C-NMR is consistent with the formed structure.
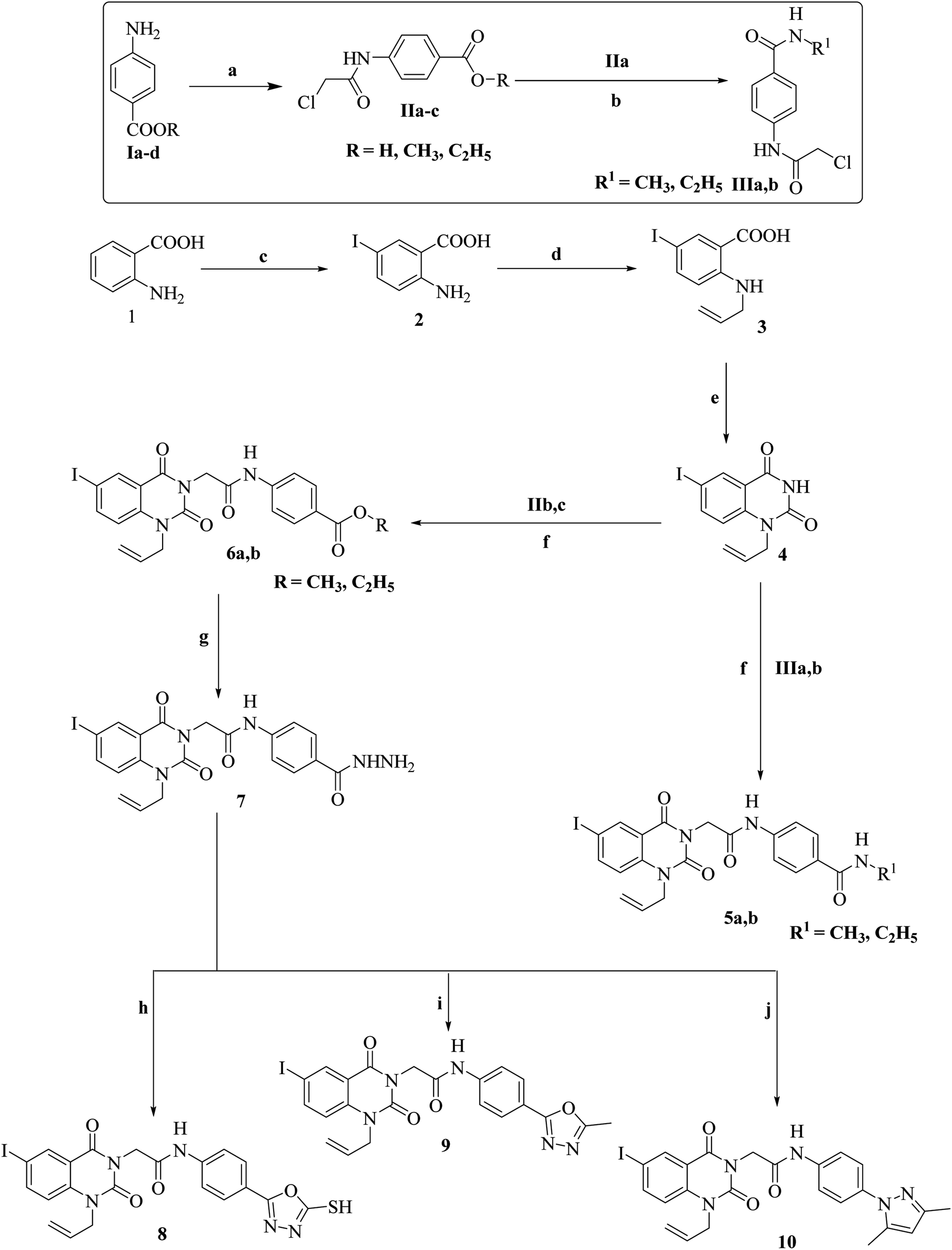 | ||
| Scheme 1 Synthetic route for compounds 5a,b–10. Reaction condition and yield: (a) CICH2COCl/DMF/K2CO3, 0–5 °C, 1 h, 85%; (b) ECF/TEA/DMF/NH2-R1, 0–5 °C, 1 h, 80–82%; (c) I2/KOH/H2O/stirring RT, overnight, 87%; (d) allyl bromide/K2CO3/ethanol/reflux, 6 h, 84%; (e) NH2CONH2/fusion/1 h, 85%; (f) acetone/ K2CO3/reflex, 12 h, 72–76%; (g) NH2NH2/ethanol/reflux 6 h, 80%; (h) CS2/KOH/H2O/ethanol/reflux/24 h, 76%; (i) AC2O/reflux/3 h, 78%; (j) acetylacetone/dioxane/TEA/reflux, 4 h, 72%. | ||
 | ||
| Scheme 2 Synthetic route for compounds 13a,b–18. Reaction condition and yield: (a) benzyl bromide/K2CO3/ethanol/reflux, 6 h, 82%; (b) NH2CONH2/fusion/1 h, 85%; (c) acetone/K2CO3/reflex, 12 h, 70–75%; (d) NH2NH2/ethanol/reflux 6 h, 80%; (e) CS2/KOH/H2O/ethanol/reflux/24 h, 76%; (f) AC2O/reflux/3 h, 78%; (g) acetylacetone/dioxane/TEA/reflux, 4 h, 72%. | ||
Conversely, reaction of 5-iodoanthranilic acid (2) with benzyl bromide afforded the corresponding N-benzyl derivative (11).40 Fusion of 5-iodo-N-benzylanthranilic acid (11) with urea afforded 6-iodo-1-benzylquinazoline-2,4(1H,3H)-dione (12).40 In situ reaction of 6-iodo-1-benzylquinazoline-2,4(1H,3H)-dione (12) with chloroacetamide IIIa afforded the corresponding benzamide derivative 13. 1H-NMR spectrum of derivative 13 revealed characteristic bands for the new N-benzyl group at 5.43 for NCH2. Additionally, it showed a signal for NHCH3 at 3.06. Moreover, reaction of 6-iodo-1-benzylquinazoline-2,4(1H,3H)-dione (12) with the appropriate chloroacetamide IIb,c afforded corresponding ester derivatives 14a,b. 13C-NMR and 1H-NMR spectra of compounds 14a,b were consistent with the formed structures. 1H-NMR spectrum of derivative 14b as a model displayed characteristic bands for the ethyl group at 1.26–1.29 (triplet signal for CH2CH3) and 4.24–4.27 (quartet signal for CH2CH3) in addition to characteristic singlet signals for CH2CO, NCH2 and NH at 4.84, 5.38 and 10.67, respectively. Reaction of ester 14b with hydrazine hydrate afforded the corresponding hydrazide derivative 15, which showed the disappearance of the characteristic bands for ethyl ester and presence of NH2, NH and hydrazide carbonyl bands, in its IR spectrum. Cyclization of acid hydrazide 14 with carbon disulfide, acetic anhydride and/or acetyl acetone afforded the corresponding oxadiazole-5-thiol 16, 5-methyloxadiazole 17 and/or dimethylpyrazole derivative 18, respectively (Scheme 2). IR and 1H-NMR spectra for compounds 8, 9 and 10 exhibited the disappearance of hydrazide characteristic bands. In addition, 1H-NMR spectrum for compound 18 presents two singlet signals at 2.17 and 2.53 for two CH3 moieties of the pyrazole group. Additionally, 13C-NMR is consistent with the formed structure.
2.2. Docking studies
For molecular docking investigations, Molsoft software was used. The PDB IDs for EGFR-TK wild-type and mutated one, EGFRWT (PDB: 4HJO)41 and EGFRT790M (PDB ID 3W2O),38 were used in each experiment, respectively.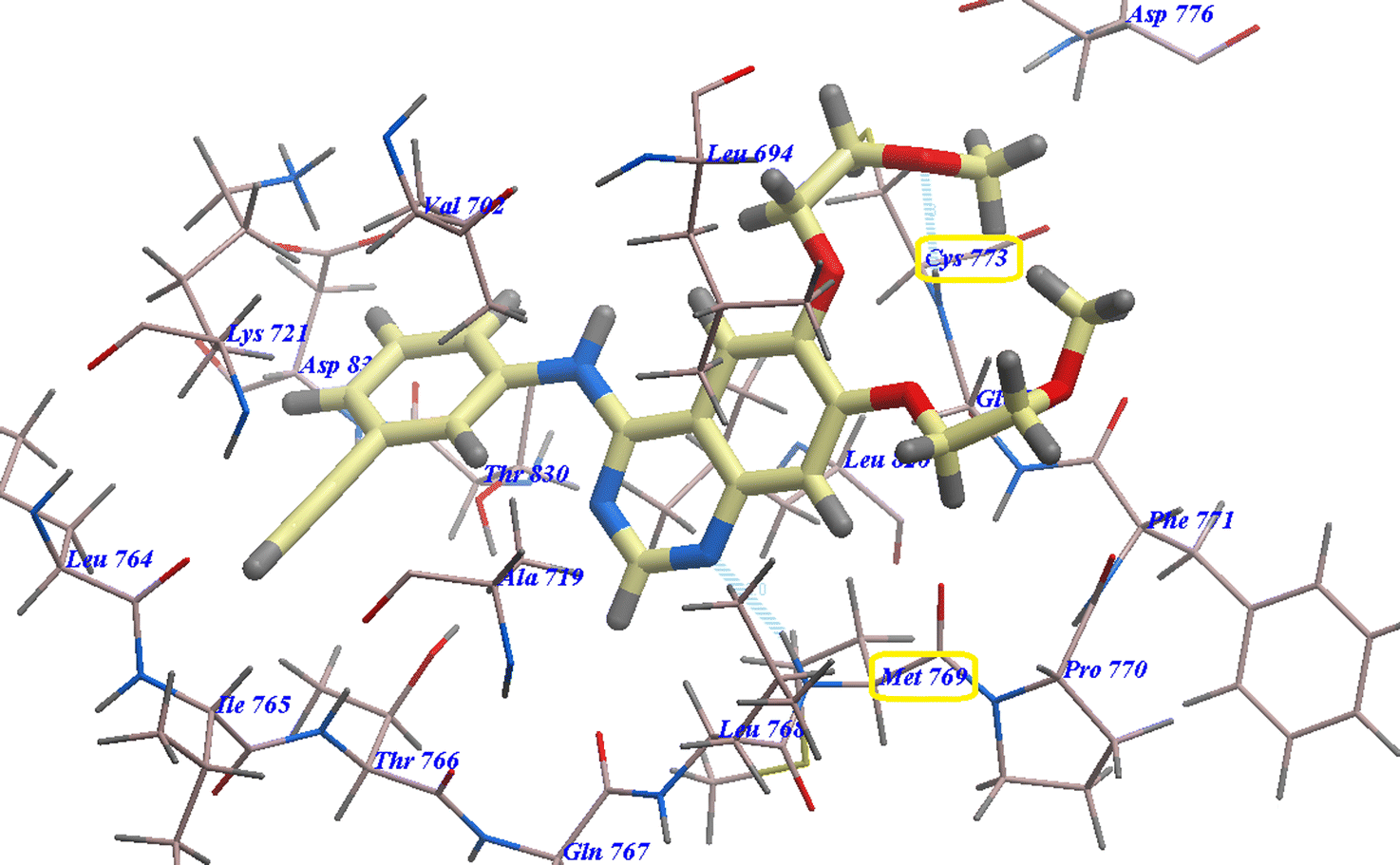 | ||
| Fig. 4 Erlotinib expected binding mode with 4HJO. | ||
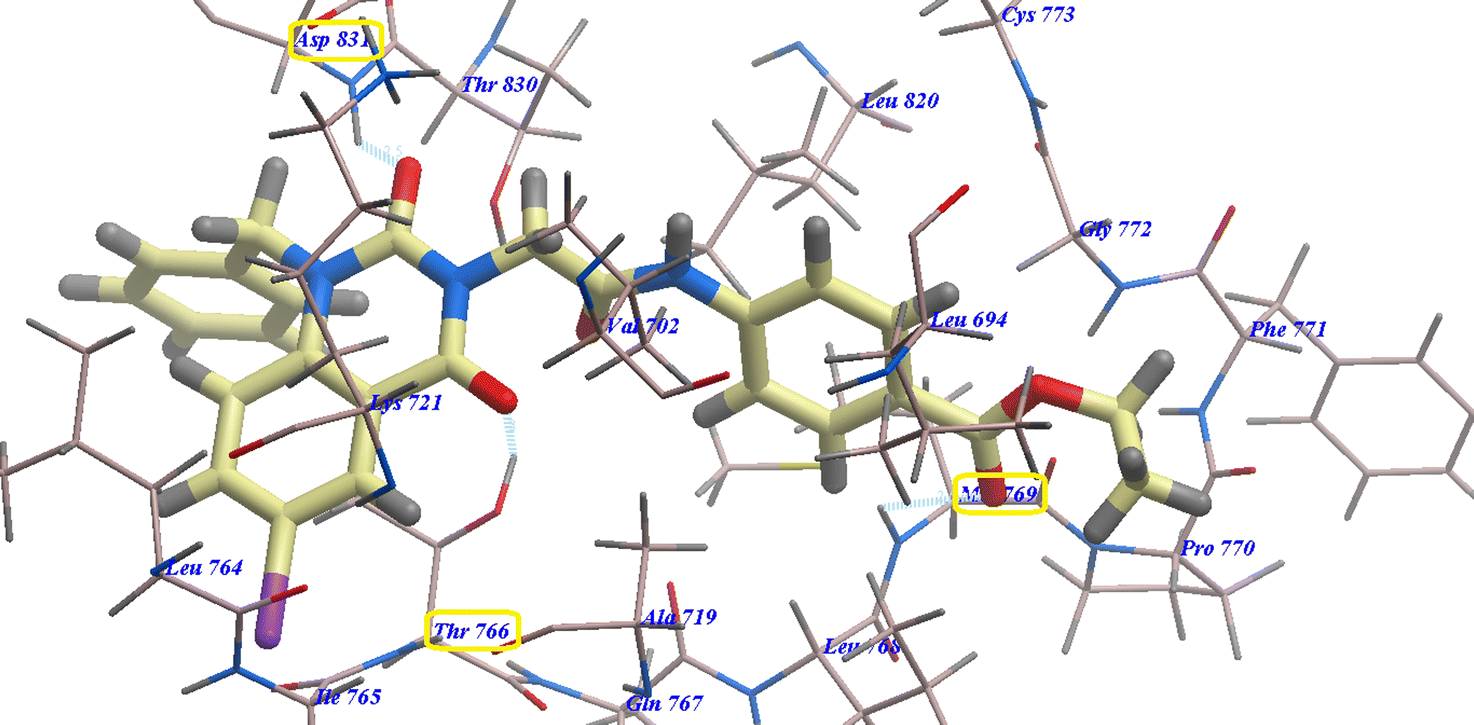
Compound 18 presented undistinguishable binding features from those of erlotinib. It displayed a binding energy of −109.97 kcal mol−1 and three H-bonds with Asp831 (2.47 Å), Thr766 (2.96 Å) and Met769 (2.95 Å) (Fig. 5).
 | ||
| Fig. 5 18 expected binding mode with 4HJO. | ||
Compound 17 presented nearly undistinguishable docking from that of erlotinib and 18. It exhibited a binding energy of −108.87 kcal mol−1 (Table 1) and four H-bonds with Asp831 (2.48 Å), Thr766 (2.96 Å), Met769 (1.70 Å) and Gly772 (2.96 Å) (Fig. 6). Moreover, compound 14b exhibited a binding energy of −108.27 kcal mol−1 and formed three H-bonds with Asp831 (2.46 Å), Thr766 (2.66 Å) and Met769 (2.54 Å) (Fig. 7).
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 5a | −89.65 | 13 | −98.86 |
| 5b | −95.87 | 14a | −98.84 |
| 6a | −91.11 | 14b | −108.27 |
| 6b | −99.13 | 15 | −97.98 |
| 7 | −88.36 | 16 | −108.12 |
| 8 | −97.98 | 17 | −108.87 |
| 9 | −99.12 | 18 | −109.97 |
| 10 | −102.09 | Erlotinib | −92.64 |
 | ||
| Fig. 6 17 expected binding mode with 4HJO. | ||
 | ||
| Fig. 7 14b expected binding mode with 4HJO. | ||
 | ||
| Fig. 8 Docking results for erlotinib with 3W2O. | ||
Compound 17 exhibited a nearly undistinguishable pattern from that of erlotinib. It demonstrated a binding energy of −95.80 kcal mol−1 (Table 2) and five H-bonds with Thr854 (2.95 Å), Val726 (2.96 Å), Cys797 (2.96 Å), Met793 (2.60 Å) and Phe795 (2.95 Å) (Fig. 9).
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 5a | −78.53 | 13 | −93.30 |
| 5b | −82.40 | 14a | −86.89 |
| 6a | −82.32 | 14b | −94.74 |
| 6b | −84.74 | 15 | −82.06 |
| 7 | −70.72 | 16 | −91.70 |
| 8 | −84.27 | 17 | −95.80 |
| 9 | −86.18 | 18 | −95.07 |
| 10 | −85.42 | Erlotinib | −82.77 |
 | ||
| Fig. 9 Docking of 17 in the active site of 3W2O. | ||
Compounds 18 and 14b presented identical docking patterns to those of erlotinib and 17, with affinities of −95.07 kcal mol−1 (forming 4 H-bonds) (Fig. 10) and −94.74 kcal mol−1 (forming 4 H-bonds) (Fig. 11), respectively.
 | ||
| Fig. 10 Docking of 18 in the active site of 3W2O. | ||
 | ||
| Fig. 11 Docking of 14b in the active site of 3W2O. | ||
2.3. Biological evaluation
| Compound | IC50a (μM) | ||||||
|---|---|---|---|---|---|---|---|
| HepG2 | MCF-7 | HCT116 | A549 | VERO | EGFRWT | EGFRT790M | |
| a IC50 values are the mean ± SD of three separate experiments.b NT: not tested. | |||||||
| 5a | 15.00 ± 1.5 | 13.22 ± 1.0 | 13.44 ± 1.5 | 10.60 ± 1.5 | NTb | 1.32 ± 0.25 | 1.62 ± 0.20 |
| 5b | 12.53 ± 1.0 | 11.26 ± 1.1 | 11.00 ± 1.3 | 9.50 ± 1.5 | NTb | 1.00 ± 0.15 | 1.33 ± 0.10 |
| 6a | 13.03 ± 1.4 | 12.25 ± 1.7 | 12.05 ± 1.5 | 10.40 ± 1.5 | NTb | 1.15 ± 0.25 | 1.54 ± 0.50 |
| 6b | 11.58 ± 1.8 | 10.95 ± 1.6 | 9.79 ± 1.8 | 9.39 ± 1.5 | NTb | 0.95 ± 0.35 | 1.12 ± 0.50 |
| 7 | 16.50 ± 1.6 | 15.20 ± 1.9 | 15.15 ± 1.7 | 12.20 ± 1.5 | NTb | 1.50 ± 0.15 | 1.90 ± 0.50 |
| 8 | 11.15 ± 1.9 | 10.77 ± 1.8 | 9.31 ± 1.8 | 9.00 ± 1.5 | NTb | 0.88 ± 0.35 | 0.96 ± 0.50 |
| 9 | 11.00 ± 1.4 | 10.44 ± 1.7 | 8.62 ± 1.5 | 7.06 ± 1.5 | NTb | 0.80 ± 0.25 | 0.89 ± 0.50 |
| 10 | 7.35 ± 1.0 | 7.85 ± 0.9 | 6.96 ± 1.1 | 6.90 ± 1.5 | 43.44 ± 0.31 | 0.38 ± 0.35 | 0.54 ± 0.50 |
| 13 | 8.00 ± 1.3 | 8.25 ± 1.1 | 7.36 ± 1.1 | 7.10 ± 1.1 | 46.88 ± 0.43 | 0.50 ± 0.15 | 0.61 ± 0.10 |
| 14a | 9.00 ± 1.2 | 8.35 ± 1.5 | 8.50 ± 1.1 | 7.87 ± 1.1 | 50.22 ± 0.31 | 0.60 ± 0.35 | 0.75 ± 0.50 |
| 14b | 5.86 ± 1.0 | 7.03 ± 1.1 | 6.15 ± 1.5 | 5.77 ± 1.3 | 49.14 ± 0.32 | 0.25 ± 0.35 | 0.36 ± 0.10 |
| 15 | 9.50 ± 1.0 | 8.80 ± 1.0 | 8.95 ± 1.0 | 8.18 ± 1.0 | 52.11 ± 0.31 | 0.65 ± 0.15 | 0.80 ± 0.10 |
| 16 | 6.54 ± 1.0 | 7.36 ± 1.0 | 6.67 ± 1.0 | 6.20 ± 1.0 | 43.78 ± 0.42 | 0.33 ± 0.25 | 0.43 ± 0.10 |
| 17 | 5.55 ± 0.7 | 6.85 ± 0.9 | 5.40 ± 1.5 | 5.11 ± 1.1 | 46.11 ± 0.31 | 0.15 ± 0.35 | 0.30 ± 0.50 |
| 18 | 5.25 ± 0.7 | 6.46 ± 0.9 | 5.68 ± 1.5 | 5.24 ± 1.1 | 45.50 ± 0.31 | 0.17 ± 0.35 | 0.25 ± 0.50 |
| Erlotinib | 7.73 ± 0.67 | 13.91 ± 1.3 | 8.20 ± 0.34 | 5.49 ± 0.45 | NTb | 0.15 ± 0.02 | 0.24 ± 0.22 |
Regarding HepG2, compounds 10, 13, 14a, 15 and 16 exhibited very good anticancer effects with IC50 ranging from 6.54 to 9.50 μM. Derivatives 5b, 6a, 6b, 8 and 9 with IC50 ranging from 11.00 to 13.03 μM presented good cytotoxicity. In contrast, derivatives 5a and 7 displayed moderate anticancer effects with IC50 = 15.00 and 16.50 μM, respectively.
Concerning MCF-7, compounds 10, 13, 14a, 15 and 16 exhibited very good anticancer effects with IC50 ranging from 7.36 to 8.80 μM. Derivatives 5a, 5b, 6a, 6b, 8 and 9 with IC50 ranging from 10.44 to 13.44 μM presented good cytotoxicity. Conversely, derivative 7 displayed moderate anticancer effects with IC50 = 15.20 μM.
Derivatives 6b, 8, 9, 10, 13, 14a, 15 and 16 exhibited very good anticancer effects with IC50 ranging from 6.67 to 9.79 μM against HCT116. Derivatives 5a, 5b, and 6a with IC50 = 13.44, 11.00 and 12.05 μM displayed good cytotoxicity. Conversely, derivative 7 revealed moderate anticancer effect with IC50 = 15.15 μM.
Derivatives 5b, 6b, 8, 9, 10, 13, 14a, 15 and 16 exhibited very good anticancer effects with IC50 ranging from 6.20 to 9.50 μM against A549. Derivatives 5a, 6a and 7 with IC50 = 10.60, 10.40 and 12.20 μM showed good cytotoxicity.
2.3.1.1. Selectivity index (SI). To evaluate toxicity induced by our iodoquinazoline derivatives, the eight highly effective derivatives 10, 13, 14a, 14b, 15, 16, 17 and 18 were tested against VERO normal cells. All results revealed that each compound possessed low toxicity against VERO normal cells with IC50 ranging from 43.44 to 52.11 μM. One criterion for an anticancer drug to be good is that it should not affect non-cancer cells. A molecule could be considered as highly selective for cancer cells if it presents SI value ≥5. A molecule with moderate selectivity presents SI value >2, while low selectivity is considered if the SI is lower than 2.43 In this study, derivatives 10, 13, 14a, 14b, 15, 16, 17 and 18 are correspondingly 5.91-, 5.86-, 5.58-, 8.39-, 5.49-, 6.69-, 8.31- and 8.67-fold more toxic against HepG2 than VERO cells. More, derivatives 10, 13, 14a, 14b, 15, 16, 17 and 18 are correspondingly 5.53-, 5.68-, 6.01-, 6.99-, 5.92-, 5.95-, 6.73- and 7.04-fold more toxic in MCF-7 than in VERO cells. Furthermore, structures 10, 13, 14a, 14b, 15, 16, 17 and 18 have respectively 6.24-, 6.37-, 5.91-, 7.99-, 5.82-, 6.56-, 7.69- and 8.43-fold grander lethalness in HCT116 than in VERO cells. Additionally, products 10, 13, 14a, 14b, 15, 16, 17 and 18 have respectively 6.30-, 6.60-, 6.38-, 8.52-, 6.37-, 7.06-, 8.63- and 8.90-fold more toxicity in A549 than in VERO cells. All compounds displayed high selectivity against tested cancer cell lines with SI values >5 which revealed a high selectivity against the tested cancer cell lines.
The molecular modeling and biological data obtained suggested that the high activities of benzyl derivatives may be due to that the benzyl moiety was expected to occupy the hydrophobic region I more than the allyl one. In addition, the carbonyl groups of quinazoline scaffold formed different H-bonds with amino acid residues in the linker and/or adenine binding sites. Moreover, the long hydrophobic side chains attached to the quinazoline scaffold at position-3 occupied the hydrophobic region II. All the mentioned structural elements are necessary for higher activities and/or binding with the active sites of EGFR receptors.
Based on the structures of synthesized derivatives and data in Table 3, we can divide the tested compounds into two groups.
Compounds 5a,b, 6a,b, and 7 with 1-allyl substitutions and amide, ester and hydrazide, respectively, belong to the first group. Moreover, this group contains compounds 13, 14a,b and 15 with 1-benzyl substitutions and amide, ester and hydrazide respectively.
Compound 14b with a more hydrophobic ethyl ester side chain exhibited higher anticancer activities than compound 13 with methyl amide and compound 14a with methyl ester side chains against the four tested cell lines. Compound 15 with a hydrazide side chain showed the lowest activities among this group.
In the same manner, compound 6b with a more hydrophobic ethyl ester side chain exhibited higher anticancer activities than compound 5b with ethyl amide, compound 6a with methyl ester and compound 5a with methyl amide side chains against the four tested cell lines. Compound 7 with a hydrazide side chain showed the lowest activities among this group.
The second group consists of compounds 16, 17 and 18 with 1-benzyl substitutions and heteroaromatic distal hydrophobic side chains. Additionally, compounds 8, 9 and 10 with 1-allyl substitutions and heteroaromatic distal hydrophobic side chains belong to this group.
Compound 18 with a highly lipophilic electron-donating 3,5-dimethylpyrazole moiety showed stronger activity than derivative 17 with 5-methyloxadiazole, and compound 16 with 5-mercapto-oxadiazole moiety against MCF-7 and HepG2. While against A549 and HCT116, the order of activity is 17 > 18 > 16.
Conversely, derivative 10 with a highly lipophilic electron-donating 3,5-dimethylpyrazole moiety showed stronger activity than derivative 9 with 5-methyloxadiazole and compound 8 with 5-mercapto-oxadiazole moiety against the four MCF-7, HepG2, A549 and HCT116 cell lines.
2.4. ADMET; in silico studies profile
In silico prediction of the highly active derivatives 14b, 17 and 18 was calculated for their physicochemical character evaluation and the proposed ADMET profile. It was predicted using pkCSM descriptor algorithm procedures46 and matched to the rule of five described by Lipinski.47 Good absorption properties were expected for the molecules that follow at least three of the following rules: (i) no more than five hydrogen bond donors, (ii) no more than 10 hydrogen bond acceptors, (iii) molecular weight less than 500, (iv) not more than 5 for log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. In the current study, our new compounds 14b, 17 and 18 violate the molecular weight rule, while the standard anticancer agent erlotinib does not violate any rule.
P. In the current study, our new compounds 14b, 17 and 18 violate the molecular weight rule, while the standard anticancer agent erlotinib does not violate any rule.
As a result of obtaining data from Table 4, we can assume that compounds 14b, 17 and 18 have very good GIT absorption in humans (92.177 to 99.768), which indicates that they more easily cross different biological membranes.48 Thus, they may show a significantly high bioavailability through GIT. Concerning CNS penetrability, our prepared compounds can reach the CNS, with CNS permeability values of −2.299 to −1.962, comparable to that of erlotinib (−3.216).
| Parameter | 14b | 17 | 18 | Erlotinib |
|---|---|---|---|---|
| Physicochemical properties | ||||
| Molecular weight | 583.382 | 593.381 | 605.436 | 393.443 |
| LogP |
3.6315 | 3.81322 | 4.25734 | 3.4051 |
| Rotatable bonds | 7 | 6 | 6 | 10 |
| Acceptors | 7 | 8 | 7 | 7 |
| Donors | 1 | 1 | 1 | 1 |
| Surface area | 213.847 | 218.102 | 225.872 | 169.532 |
|
||||
| Absorption | ||||
| Water solubility | −5.318 | −4.679 | −5.204 | −4.736 |
| Human intest. absorption | 92.177 | 92.423 | 99.768 | 94.58 |
| Substrate for P-glycoprotein | Yes | Yes | Yes | No |
| Inhibitor of P-glycoprotein I | Yes | Yes | Yes | Yes |
| Inhibitor of P-glycoprotein II | Yes | Yes | Yes | Yes |
|
||||
| Distribution | ||||
| Permeability through BBB | −1.106 | −1.338 | −1.084 | −0.745 |
| Permeability to CNS | −2.299 | −2.225 | −1.962 | −3.216 |
|
||||
| Metabolism | ||||
| CYP2D6 substrate | No | No | No | No |
| CYP3A4 substrate | Yes | Yes | Yes | Yes |
| Inhibition of CYP1A2 | No | No | No | Yes |
| Inhibition of CYP2C19 | Yes | Yes | Yes | Yes |
| Inhibition of CYP2C9 | Yes | Yes | Yes | Yes |
| Inhibition of CYP2D6 | No | No | No | No |
| Inhibition of CYP3A4 | Yes | Yes | Yes | Yes |
|
||||
| Excretion | ||||
| Clearance | −0.482 | −0.556 | −0.587 | 0.702 |
|
||||
| Toxicity | ||||
| Human max. tolerated dose | 0.37 | 0.701 | 0.559 | 0.839 |
| Acute toxic activity (LD50) | 2.683 | 2.816 | 2.621 | 2.393 |
| Chronic toxic activity (LOAEL) | 1.366 | 1.32 | 0.862 | 1.37 |
| Hepatotoxic effect | Yes | Yes | Yes | Yes |
It is well known that CYP3A4, a major drug-metabolizing enzyme, could be inhibited by erlotinib and by our derivatives 14b, 17 and 18. Elimination was expected depending on the total clearance which is a considerable factor in deciding dose intervals. The data showed that erlotinib exhibited higher clearance rates compared with our new compounds which demonstrated low clearance values. Thus, erlotinib could be eliminated faster, and as a result, supposed to have shorter dosing intervals. Unlike erlotinib, the prepared compounds exhibited low clearance rates, which signifies a long duration of action and extended dosing intervals. Toxicity is the final ADMET profile factor studied. As presented in Table 4, erlotinib and the novel compounds 14b, 17 and 18 shared the drawback of unwanted hepatotoxic actions. Erlotinib, 17 and 18 demonstrated high maximum tolerated dose. In contrast, 14b demonstrated lower maximum tolerated doses which involve the advantage of the broad therapeutic index of erlotinib, 17 and 18. Lastly, the oral acute toxic doses of the novel compounds 14b, 17 and 18 are higher than that of erlotinib. Moreover, the oral chronic toxic doses of the novel compounds 14b and 17 are nearly equal to that of erlotinib but higher than that of 18.
3. Conclusion
Fifteen new iodoquinazoline derivatives bearing different side chain moieties were designed and synthesized as dual EGFRWT/EGFRT790M inhibitors. The cytotoxic activities of the new compounds were screened in vitro against four different cancer cell lines (HepG2, MCF-7, HCT116 and A549) using an MTT assay. Their ability to bind with EGFRWT and EGFRT790M receptors was examined by molecular modeling. Compounds 18, 17 and 14b showed the highest anticancer effects with IC50 = 5.25, 6.46, 5.68 and 5.24 μM, 5.55, 6.85, 5.40 and 5.11 μM and 5.86, 7.03, 6.15 and 5.77 μM against the HepG2, MCF-7, HCT116 and A549 cell lines, respectively. Compounds 10, 13, 14a, 15 and 16 exhibited very good anticancer effects against the tested cancer cell lines. The eight highly effective derivatives 10, 13, 14a, 14b, 15, 16, 17 and 18 were examined against VERO normal cell line. These compounds possessed low toxicity against VERO normal cells with IC50 ranging from 43.44 to 52.11 μM. Additionally, all derivatives were evaluated for their ability as dual inhibitors of EGFRWT and EGFRT790M. The evaluated candidates showed excellent-to-low inhibitory activities.Compound 17 exhibited the same inhibitory activity as erlotinib. Compounds 10, 13, 14b, 16 and 18 remarkably inhibited EGFRWT activity with IC50 ranging from 0.17 to 0.50 μM. Candidates 5b, 6b, 8, 9, 14a and 15 significantly inhibited EGFRWT at an IC50 range of 0.60–1.00 μM. Moreover, compounds 18, 17, 14b and 16 excellently inhibited EGFRT790M activity with IC50 = 0.25, 0.30, 0.36 and 0.40 μM respectively. Candidates 8, 9, 10, 13, 14a and 15 significantly inhibited EGFRT790M at an IC50 range of 0.54–0.96 μM. Further, an ADMET study was conducted in silico for three best-performing compounds 14b, 17 and 18 in comparison with erlotinib. Our derivatives may be promising as prototypes for design, optimization, adaptation and investigation to construct further powerful selective dual EGFRWT/EGFRT790M inhibitors with higher antitumor activities.
4. Experimental
4.1. Chemistry
1H-NMR spectra were recorded with a Bruker 400 MHz NMR spectrophotometer at Microanalytical Center, Ain Shams University and/or a Mercury 300 MHz NMR spectrophotometer at Faculty of Science, Cairo University. 13C-NMR spectra were recorded with a Bruker 100 MHz NMR spectrophotometer at Microanalytical Unit, Faculty of Pharmacy, Cairo University.Alkyl 4-(2-chloroacetamido)benzoate (IIa–c), 4-(2-chloroacetamido)-N-alkylbenzamide (IIIa,b), 5-iodoanthranilic acid (2), 5-iodo-N-allylanthranilic acid (3), 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4), 5-iodo-N-benzylanthranilic acid (11), and 6-iodo-1-benzylquinazoline-2,4(1H,3H)-dione (12) were synthesized according to reported procedures.38–40
4.1.1.1. General method. A mixture of equimolar quantities of 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4) (3.28 g, 0.01 mol) and appropriate 4-(2-chloroacetamido)-N-alkylbenzamide (IIIa,b) (0.01 mol) in dry acetone (50 mL) was heated under reflux for 12 h in the presence of K2CO3 (1.38 g, 0.01 mol). The reaction mixture was filtered off while hot. The precipitated solids were filtered, dried and crystallized from ethanol to give corresponding target compounds 5a,b.
4.1.1.2. 4-{2-[1-Allyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}-N-methylbenzamide (5a). Yield, 75%; m.p. 290–2 °C; IRνmax (cm−1): 3280, 3250 (2NH), 1694, and 1655 (4C
O); 1H-NMR (400 MHz, DMSO-d6): 3.82 (s, 3H, NHCH3), 4.78–4.82 (m, 2CH2, CH2CO & CH2CH), 5.13–5.21 (m, 2H, CH2CH), 5.83–5.95 (m, 1H, CHCH2), 7.31–7.91 (m, 7H, 7Ar–H), 9.46 (s, 1H, NHCH3 (D2O exchangeable)), and 10.63 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 14.4, 50.8, 54.1, 94.4, 105.9, 115.0 (2), 120.9, 125.6 (3), 129.1, 131.4, 133.3, 134.5, 146.6, 151.2, 154.7, 165.2, 167.1, and 168.9; anal. calcd for C21H19IN4O4 (m.w. 518.31): C, 48.66; H, 3.70; and N, 10.81. Found: C, 48.74; H, 3.75; and N, 10.75.
4.1.1.3. 4-{2-[1-Allyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}-N-ethylbenzamide (5b). Yield, 76%; m.p. 292–4 °C; IRνmax (cm−1): 3308, 3180 (2NH), 1658, and 1632 (4C
O); 1H-NMR (400 MHz, DMSO-d6): 1.21–125 (t, 3H, CH2CH3), 3.82–3.88 (q, 2H, CH2CH3), 4.89 (s, 2H, CH2CO), 5.10–5.18 (m, 2CH2, CH2CH & CH2CH), 5.88–5.92 (m, 1H, CHCH2), 7.23–7.98 (m, 7H, 7Ar–H), 8.95 (s, 1H, NHCH2 (D2O exchangeable)), and 11.57 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 16.6, 29.7, 54.6, 58.3, 93.3, 115.0, 121.4 (2), 125.5, 127.0, 129.7 (3), 130.7, 134.2, 138.9, 143.6, 154.6, 156.3, 166.2, and 173.4; anal. calcd for C21H19IN4O4 (m.w. 518.31): C, 48.66; H, 3.70; and N, 10.81. Found: C, 48.55; H, 3.75; and N, 10.70.
4.1.2.1. General method. A mixture of equimolar quantities of 6-iodo-1-allylquinazoline-2,4(1H,3H)-dione (4) (3.28 g, 0.01 mol) and appropriate alkyl 4-(2-chloroacetamido)benzoate IIb,c (0.01 mol) in dry acetone (50 mL) was heated under reflux for 12 h in the presence of K2CO3 (1.38 g, 0.01 mol). The reaction mixture was filtered off while hot. The precipitated solids were filtered, dried and crystallized from ethanol to give corresponding target compounds 6a,b.
4.1.2.2. Methyl 4-{2-[1-allyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (6a). Yield, 72%; m.p. 263–5 °C; IRνmax (cm−1): 3481 (NH), 1725, 1672, and 1653 (4C
O); 1H-NMR (400 MHz, DMSO-d6): 3.78 (s, 3H, COOCH3), 4.75–4.78 (m, 2CH2, CH2CO & CH2CH), 5.12–5.14 (m, 2H, CH2CH), 5.87–5.90 (m, 1H, CHCH2), 7.38–7.90 (m, 7H, 7Ar–H), and 10.65 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 35.1, 53.4, 57.6, 95.6, 125.0 (2), 126.5, 128.3, 129.9 (3), 136.6, 137.9, 139.4, 149.0, 152.1, 155.8, 158.8, 162.9, 168.3, and 172.3; anal. calcd for C21H18IN3O5 (m.w. 519.30): C, 48.57; H, 3.49; and N, 8.09. Found: C, 48.55; H, 3.58; and N, 7.96.
4.1.2.3. Ethyl 4-{2-[1-allyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (6b). Yield, 75%; m.p. 256–8 °C; IRνmax (cm−1): 3290 (NH), 1718, 1701, 1685, and 1675 (4C
O); 1H-NMR (400 MHz, DMSO-d6): 1.35–1.38 (t, 3H, CH2CH3), 4.32–4.35 (q, 2H, CH2CH3), 4.80 (s, 2H, CH2CO), 4.96 (d, 2H, CH2CH), 5.22–5.29 (m, 2H, CH2CH), 5.90–5.98 (m, 1H, CHCH2), 7.19–8.25 (m, 7H, 7Ar–H), and 10.64 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 19.2, 33.3, 48.3, 51.6, 95.7, 121.4, 122.5 (2), 124.0, 126.8, 128.4, 129.6 (3), 131.9, 136.6, 137.8, 144.3, 148.9, 152.8, 158.7, and 167.8; anal. calcd for C22H20IN3O5 (m.w. 533.32): C, 49.55; H, 3.78; and N, 7.88. Found: C, 49.47; H, 3.86; and N, 7.79.
Yield, 80%; m.p. 297–9 °C; IRνmax (cm−1): 3250, 3200, 3117 (2NH & NH2), 1710, and 1685 (4CO); 13C-NMR (100 MHz, DMSO-d6): 52.5, 53.8, 95.7, 104.4, 122.6 (2), 124.1, 127.6, 128.1, 129.8 (3), 136.7, 138.6, 139.4, 149.0, 152.6, 158.6, 160.3, and 160.7; anal. calcd for C20H18IN5O4 (m.w. 519.30): C, 46.26; H, 3.49; and N, 13.49. Found: C, 46.30; H, 3.55; and N, 13.54.
Yield, 76%; m.p. 288–9 °C; IRνmax (cm−1): 3234, 3179 (2NH), 1685, 1670 (3CO), and 1255 (CS); 1H-NMR (400 MHz, DMSO-d6): 4.69 (s, 2H, CH2CO), 5.08–5.17 (m, 2CH2, CH2CH & CH2CH), 5.84–5.97 (m, 1H, CHCH2), 7.23–8.02 (m, 7H, 7Ar–H), 11.57 (s, 1H, NH, D2O exchangeable), and 13.79 (s, 1H, SH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 51.5, 53.2, 94.4, 112.4, 116.6, 120.0 (2), 125.6, 130.7 (3), 131.4, 133.2, 134.6, 142.9, 148.7, 154.9, 158.9, 165.4, 167.3, and 167.7; anal. calcd for C21H16IN5O4S (561.35): C, 44.93; H, 2.87; and N, 12.48. Found: C, 45.05; H, 2.80; and N, 12.54.
Yield, 78%; m.p. 295–7 °C; IRνmax (cm−1): 3315 (NH), 1703, and 1659 (3CO); 1H-NMR (400 MHz, DMSO-d6): 2.72 (s, 3H, CH3), 4.75–4.77 (s, 2CH2, CH2CO & CH2CH), 5.13–5.14 (m, 2H, CH2 = CH), 5.87–5.90 (m, 1H, CHCH2), 7.58–8.04 (m, 7H, 7Ar–H), and 10.50 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 22.7, 53.5, 55.2, 95.3, 110.3, 114.2, 117.9, 124.1 (2), 125.3, 130.5 (3), 130.9, 132.2, 133.5, 142.5, 147.5, 153.8, 159.8, 167.8, and 168.4; anal. calcd for C22H18IN5O4 (543.32): C, 48.63; H, 3.34; and N, 12.89. Found: C, 48.58; H, 3.30; and N, 12.96.
Yield, 72%; m.p. 297–9 °C; IRνmax (cm−1): 3316 (NH), 1734, 1701, and 1660 (3CO); 1H-NMR (400 MHz, DMSO-d6): 1.99 (s, 3H, CH3 at position-5 of pyrazole), 2.19 (s, 3H, CH3 at position-3 of pyrazole), 4.79–4.83 (s, 2CH2, CH2CO & CH2CH), 5.12–5.21 (m, 2H, CH2CH), 5.84–5.98 (m, 1H, CHCH2), 6.22 (s, 1H, CH pyrazole), 7.31–8.10 (m, 7H, 7Ar–H), and 10.67 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 12.7, 15.8, 50.8, 54.7, 97.3, 111.9, 118.3, 118.8, 120.3 (2), 126.9, 127.5, 129.6 (3), 134.1, 139.0, 142.6, 152.6, 157.9, 162.3, 156.3, 168.0, and 171.9; anal. calcd for C24H22IN5O3 (555.38): C, 51.90; H, 3.99; and N, 12.61. Found: C, 52.02; H, 4.04; and N, 12.58.
4.1.7.1. General method. A mixture of equimolar quantities of 1-benzyl-6-iodoquinazoline-2,4(1H,3H)-dione (12) (3.78 g, 0.01 mol) and 4-(2-chloroacetamido)-N-methyllbenzamide (IIIa) (2.26 g, 0.01 mol) in dry acetone (50 mL) was heated under reflux for 12 h in the presence of K2CO3 (1.38 g, 0.01 mol). The reaction mixture was filtered off while hot. The precipitated solids were filtered, dried and crystallized from ethanol to give the corresponding target compound 13.
Yield, 75%; m.p. 292–4 °C; IRνmax (cm−1): 3283, 3230 (2NH), 1692, 1658, and 1653 (4CO); 1H-NMR (400 MHz, DMSO-d6): 3.06 (s, 3H, NHCH3), 4.88 (s, 2H, CH2CO), 5.43 (s, 2H, NCH2), 7.03–8.12 (m, 12H, 12Ar–H), 9.75 (s, 1H, NHCH3 (D2O exchangeable)), and 10.61 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 24.7, 52.1, 55.9, 118.9, 120.0 (2), 121.4, 124.9 (3), 129.5 (2), 129.9, 130.5, 130.7, 131.3 (2), 133.3, 134.2 (2), 142.0, 144.2, 164.8, 165.8, and 167.6; anal. calcd for C25H21IN4O4 (m.w. 568.37): C, 52.83; H, 3.72; and N, 9.86. Found: C, 52.77; H, 3.68; and N, 9.90.
4.1.8.1. General method. A mixture of equimolar quantities of 1-benzyl-6-iodoquinazoline-2,4(1H,3H)-dione (12) (3.78 g, 0.01 mol) and appropriate alkyl 4-(2-chloroacetamido)benzoate IIb,c (0.01 mol) in dry acetone (50 mL) was heated under reflux for 12 h in the presence of K2CO3 (1.38 g, 0.01 mol). The reaction mixture was filtered off while hot. The precipitated solids were filtered, dried and crystallized from ethanol to give corresponding target compounds 14a,b.
4.1.8.2. Methyl 4-{2-[1-benzyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (14a). Yield, 70%; m.p. 272–4 °C; IRνmax (cm−1): 3385 (NH), 1751, 1734, 1697, and 1685 (4C
O); 1H-NMR (400 MHz, DMSO-d6): 3.79 (s, 3H, COOCH3), 4.84 (s, 2H, CH2CO), 5.39 (s, 2H, NCH2), 7.29–7.89 (m, 12H, 12Ar–H), and 10.68 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 33.0, 54.7, 57.1, 96.3, 109.4, 112.7, 117.1, 122.6 (2), 124.1 (3), 126.3, 129.7 (2), 132.3 (2), 137.1 (2), 141.2, 149.6, 153.1, 158.7, 166.1, and 170.7; anal. calcd for C25H20IN3O5 (m.w. 569.36): C, 52.74; H, 3.54; and N, 7.38. Found: C, 52.70; H, 3.51; and N, 7.44.
4.1.8.3. Ethyl 4-{2-[1-benzyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (14b). Yield, 75%; m.p. 277–9 °C; IRνmax (cm−1): 3262 (NH), 1722, 1697, 1666, and 1652 (4C
O); 1H-NMR (400 MHz, DMSO-d6): 1.26–1.29 (t, 3H, CH2CH3), 4.24–4.27 (q, 2H, CH2CH3), 4.84 (s, 2H, CH2CO), 5.38 (s, 2H, NCH2), 7.23–7.91 (m, 12H, 12Ar–H), and 10.67 (s, 1H, NH, D2O exchangeable); anal. calcd for C26H22IN3O5 (m.w. 583.38): C, 53.53; H, 3.80; and N, 7.20. Found: C, 53.50; H, 3.70; and N, 7.28.
Yield, 84%; m.p. 300–2 °C; IRνmax (cm−1): 3290, 3262, 3184 (2NH & NH2), 1733, and 1665 (4CO); 1H-NMR (400 MHz, DMSO-d6): 4.89 (s, 2H, CH2CO), 5.43 (s, 2H, NCH2), 6.65 (s, 2H, NH2), 7.26–8.12 (m, 12H, 12Ar–H), 10.63 (s, 1H, NHNH2, D2O exchangeable), and 12.03 (s, 1H, CONH, D2O exchangeable); anal. calcd for C24H20IN5O4 (m.w. 569.36): C, 50.63; H, 3.54; and N, 12.30. Found: C, 50.55; H, 3.60; and N, 12.42.
Yield, 78%; m.p. 297–9 °C; IRνmax (cm−1): 3313, 3255 (2NH), 1700, 1675, 1636 (3CO), and 1260 (CS); 1H-NMR (400 MHz, DMSO-d6): 4.89 (s, 2H, CH2CO), 5.42 (s, 2H, NCH2), 7.26–8.09 (m, 12H, 12Ar–H), 10.70 (s, 1H, NH, D2O exchangeable), and 14.60 (s, 1H, SH, D2O exchangeable); anal. calcd for C25H18IN5O4S (611.41): C, 49.11; H, 2.97; and N, 11.45. Found: C, 49.00; H, 3.04; and N, 11.38.
Yield, 74%; m.p. 305–7 °C; IRνmax (cm−1): 3318 (NH), 1733, 1686, and 1671 (3CO); 1H-NMR (400 MHz, DMSO-d6): 3.30 (s, 3H, CH3), 4.85 (s, 2H, CH2CO), 5.39 (s, 2H, NCH2), 7.33–8.11 (m, 12H, 12Ar–H), and 10.63 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 20.1, 52.4, 54.7, 93.4, 112.0, 116.8 (2), 121.4 (3), 124.3 (2), 126.9, 129.7 (2), 134.5 (2), 139.0, 144.1, 148.2, 152.0, 154.7, 158.4, 162.9, 167.2, and 171.8; anal. calcd for C26H20IN5O4 (593.38): C, 52.63; H, 3.40; and N, 11.80. Found: C, 52.58; H, 3.36; and N, 11.74.
Yield, 70%; m.p. 310–2 °C; IRνmax (cm−1): 3310 (NH), 1722, 1697, and 1666 (3CO); 1H-NMR (400 MHz, DMSO-d6): 2.17 (s, 3H, CH3 at position-5 of pyrazole), 2.53 (s, 3H, CH3 at position-3 of pyrazole), 4.89 (s, 2H, CH2CO), 5.42 (s, 2H, NCH2), 6.26 (s, 1H, CH pyrazole), 7.29–7.94 (m, 12H, 12Ar–H), and 10.69 (s, 1H, NH, D2O exchangeable); 13C-NMR (100 MHz, DMSO-d6): 14.4, 18.3, 51.5, 55.8, 95.8, 108.8, 112.3 (2), 115.8 (3), 119.7 (2), 122.7 (2), 126.7, 128.1, 129.8 (2), 130.5, 139.5, 141.9, 146.7, 149.1, 153.0, 162.1, 166.3, and 172.5; anal. calcd for C28H24IN5O3 (605.44): C, 55.55; H, 4.00; and N, 11.57. Found: C, 55.50; H, 3.94; and N, 11.49.
4.2. Docking studies
EGFRWT (PDB: 4HJO)41 and EGFRT790M (PDB: 3W2O)38 were used with the Molsoft program to carry out docking studies.4.3. Biological testing
Author contributions
K. El-Adl, Hanan Osman, Basmah Almohaywi, Samy Mohamady, Ahmed Aljohani, Ahmed El-morsy, and Marwa Alsulaimany were responsible for the conception and rational design of the work. K. El-Adl, Samy Mohamady, Ahmed Aljohani, Omar Alatawi and Marwa Alsulaimany were responsible for the data collection and synthesis of the new compounds. K. El-Adl and Majed Aljohani performed the molecular docking study. K. El-Adl, Hussam Alharbi, Basmah Almohaywi, Ahmed El-morsy, Ahmed Aljohani and Marwa Alsulaimany were responsible for spectral data analysis. Felemban Athary Abdulhaleem, Sara Almadani, Basmah Almohaywi, Hanan Osman and K. El-Adl conducted the cytotoxicity assay. Sara Almadani, Felemban Athary Abdulhaleem, Hanan Osman and K. El-Adl conducted the in silico pharmacokinetic study. All authors discussed the results and contributed to the writing and revision of the original manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors express their gratitude and admiration to Dr Mohamed Morsy, Faculty of Science, Al-Azhar University for supporting the pharmacological part.References
- M. T. Ibrahim, A. Uzairu and S. Uba, et al., Design of more potent quinazoline derivatives as EGFRWT inhibitors for the treatment of NSCLC: a computational approach, Future J. Pharm. Sci., 2021, 7, 140, DOI:10.1186/s43094-021-00279-3.
- S. Turker, H. Sahinli, P. Perkin, D. Yazilitas, N. O. Koklu, G. I. Imamoglu and C. Karacin, Altinbas M “Squamos cell lung cancer” case applying with dyspepsia complaints, J. Oncol. Sci., 2018, 4(3), 147–148 CrossRef.
- A. Arora and E. M. Scholar, Role of tyrosine kinase inhibitors in cancer therapy, J. Pharmacol. Exp. Ther., 2005, 315, 971–979 CrossRef CAS PubMed.
- S. S. Zahran, F. A. Ragab, M. G. El-Gazzar, A. M. Soliman, W. R. Mahmoud and M. M. Ghorab, Antiproliferative, antiangiogenic and apoptotic effect of new hybrids of quinazoline-4 (3H)-ones and sulfachloropyridazine, Eur. J. Med. Chem., 2023, 245, 114912 CrossRef CAS PubMed.
- B. Zhao, Z. Xiao, J. Qi, R. Luo, Z. Lan, Y. Zhang, X. Hu, Q. Tang, P. Zheng and S. Xu, Design, synthesis and biological evaluation of AZD9291 derivatives as selective and potent EGFRL858R/T790M inhibitors, Eur. J. Med. Chem., 2019, 163, 367–380, DOI:10.1016/j.ejmech.2018.11.069.
- B. A. Chan, Hughes BG Targeted therapy for non-small cell lung cancer: current standards and the promise of the future, Transl. Lung Cancer Res., 2015, 4(1), 36 CAS.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Jänne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos, EGFR mutation and resistance of non–small-cell lung cancer to gefitinib, N. Engl. J. Med., 2005, 352(8), 786–792, DOI:10.1056/NEJMoa044238.
- W. Zhou, D. Ercan, L. Chen, C.-H. Yun, D. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob and R. Padera, Novel mutant-selective EGFR kinase inhibitors against EGFR T790M, Nature, 2009, 462(7276), 1070–1074, DOI:10.1038/nature08622.
- S. A. Elmetwally, K. F. Saied, I. H. Eissa and E. B. Elkaeed, Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102944, DOI:10.1016/j.bioorg.2019.102944.
- A. A. Nasser, I. H. Eissa, M. R. Oun, M. A. El-Zahabi, M. S. Taghour, A. Belal, A. M. Saleh, A. B. M. Mehany, H. Luesch, A. E. Mostafa, W. M. Afifi, J. R. Rocca and H. A. Mahdy, Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFRWT and EGFRT790M, Org. Biomol. Chem., 2020, 18(38), 7608–7634, 10.1039/D0OB01557A.
- Y. Harada, A. Sato and H. Nakamura, et al., Anti-cancer effect of afatinib, dual inhibitor of HER2 and EGFR, on novel mutation HER2 E401G in models of patient-derived cancer, BMC Cancer, 2023, 3, 77, DOI:10.1186/s12885-022-10428-3.
- D. Lavacchi, F. Mazzoni and G. Giaccone, Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): current perspectives, Drug Des., Dev. Ther., 2019, 13, 3187–3198, DOI:10.2147/DDDT.S194231.
- D. A. Cross, S. E. Ashton, S. Ghiorghiu, C. Eberlein, C. A. Nebhan, P. J. Spitzler, J. P. Orme, M. R. Finlay, R. A. Ward, M. J. Mellor, G. Hughes, A. Rahi, V. N. Jacobs, M. Red Brewer, E. Ichihara, J. Sun, H. Jin, P. Ballard, K. Al-Kadhimi, R. Rowlinson, T. Klinowska, G. H. Richmond, M. Cantarini, D. W. Kim, M. R. Ranson and W. Pao, AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer, Cancer Discovery, 2014, 4(9), 1046–1061, DOI:10.1158/2159-8290.CD-14-0337.
- R. Pawara, I. Ahmad, D. Nayak, S. Belamkar, S. Surana, C. N. Kundu, C. Patil and H. Patel, Design and synthesis of the novel, selective WZ4002 analogue as EGFR-L858R/T790M tyrosine kinase inhibitors for targeted drug therapy in non-small-cell lung cancer (NSCLC), J. Mol. Struct., 2022, 1254, 132313, DOI:10.1016/j.molstruc.2021.132313.
- A. G. A. El-Helby, H. Sakr, I. H. Eissa, H. Abulkhair, A. A. Al-Karmalawy and K. El-Adl, Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2019, 352, 1900113 CrossRef CAS PubMed.
- M. A. Abdelgawad, K. El-Adl, S. S. El-Hddad, M. M. Elhady, N. M. Saleh, M. M. Khalifa, F. Khedr, M. Alswah, A. A. Nayl and M. M. Ghoneim, Design, molecular docking, synthesis, anticancer and anti-hyperglycemic assessments of thiazolidine-2, 4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists, Pharmaceuticals, 2022, 15, 226 CrossRef CAS PubMed.
- F. Khedr, M.-K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Phthalazine-based VEGFR-2 inhibitors: Rationale, design, synthesis, in silico, ADMET profile, docking, and anticancer evaluations, Arch. Pharm., 2021, e2100201, DOI:10.1002/ardp.202100201.
- A. G. A. El-Helby, R. R. Ayyad, H. Sakr, K. El-Adl, M. M. Ali and F. Khedr, design, synthesis, molecular docking, and anticancer activity of phthalazine derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2017, 350, 1700240 CrossRef PubMed.
- M. M. Ghorab, A. M. Soliman, K. El-Adl and N. S. Hanafy, New quinazoline sulfonamide derivatives as potential anticancer agents: Identifying a promising hit with dual EGFR/VEGFR-2 inhibitory and radiosensitizing activity, Bioorg. Chem., 2023, 140, 106791, DOI:10.1016/j.bioorg.2023.106791.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents, Bioorg. Chem., 2020, 104, 104218, DOI:10.1016/j.bioorg.2020.104218.
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors, Bioorg. Chem., 2020, 104, 104350, DOI:10.1016/j.bioorg.2020.104350.
- A. E. Abdallah, I. H. Eissa, A. B. M. Mehany, H. Sakr, A. Atwa, K. El-Adl and M. A. El-Zahabi, Immunomodulatory quinazoline-based thalidomide analogs: Design, synthesis, apoptosis and anticancer evaluations, J. Mol. Struct., 2023, 1281, 135164, DOI:10.1016/j.molstruc.2023.135164.
- N. S. Hanafy, N. A. A. M. Aziz, S. S. A. El-Hddad, M. A. Abdelgawad, M. M. Ghoneim, A. F. Dawood, S. Mohamady, K. El-Adl and S. Ahmed, Design, synthesis, and docking of novel thiazolidine-2,4-dione multitarget scaffold as new approach for cancer treatment, Arch. Pharm., 2023, e2300137, DOI:10.1002/ardp.202300137.
- D. Adel, K. El-Adl, T. Nasr, T. M. Sakr and W. Zaghary, Pyrazolo[3,4-d]pyrimidine derivatives as EGFRT790M and VEGFR-2 dual TK inhibitors: Design, synthesis, molecular docking, ADMET profile and anticancer evaluations, J. Mol. Struct., 2023, 1291, 136047, DOI:10.1016/j.molstruc.2023.136047.
- H. Elkady, K. El-Adl, H. Sakr, A. S. Abdelraheem, S. I. Eissa and M. A. El-Zahabi, Novel promising benzoxazole/benzothiazole-derived immunomodulatory agents: Design, synthesis, anticancer evaluation, and in silico ADMET analysis, Arch. Pharm., 2023, e2300097, DOI:10.1002/ardp.202300097.
- M. A. El-Zahabi, H. Elkady, H. Sakr, A. S. Abdelraheem, S. I. Eissa and K. El-Adl, Design, synthesis, anticancer evaluation, in silico docking and ADMET analysis of novel indole-based thalidomide analogs as promising immunomodulatory agents, J. Biomol. Struct. Dyn., 2023, 1–18, DOI:10.1080/07391102.2023.2187217.
- M. Alsulaimany, K. El-Adl, A. K. B. Aljohani, H. Y. Alharbi, O. M. Alatawi, M. S. Aljohani, A. El-Morsy, S. A. Almadani, A. A. Alsimaree, S. A. Salama, D. E. Keshek and A. A. Mohamed, Design, synthesis, docking, ADMET and anticancer evaluations of N-alkyl substituted iodoquinazoline derivatives as dual VEGFR-2 and EGFR inhibitors, RSC Adv., 2023, 13(51), 36301–36321, 10.1039/d3ra07700d.
- K. E. Anwer, S. S. A. El-Hddad, N. E. A. Abd El-Sattar, A. M. El-Morsy, F. Khedr, S. Mohamady, D. E. G. Keshek, S. A. Salama, K. El-Adl and N. SHanafy, Five and six membered heterocyclic rings endowed with azobenzene as dual EGFRT790M and VEGFR-2 inhibitors: design, synthesis, in silico ADMET profile, molecular docking, dynamic simulation and anticancer evaluations, RSC Adv., 2023, 13, 35321–35338 RSC , https://api.semanticscholar.org/CorpusID:265657522.
- M. S. A. El-Gaby, M. A. M. Abdel Reheim, Z. S. M. Akrim, B. H. Naguib, N. M. Saleh, A. A. A. M. El-Adasy, K. El-Adl and S. Mohamady, 2-Thioxo-3,4-dihydropyrimidine and Thiourea Endowed with Sulfonamide Moieties as Dual EGFRT790M and VEGFR-2 Inhibitors: Design, Synthesis, Docking and Anticancer Evaluations, Drug Dev. Res., 2024, 85, e22143, DOI:10.1002/ddr.22143.
- A. A. Mohamed, S. S. A. El-Hddad, A. K. B. Aljohani, F. Khedr, O. M. Alatawi, D. E. Keshek, S. Ahmed, M. Alsulaimany, S. A. Almadani, K. El-Adl and N. S. Hanafy, Iodoquinazoline-derived VEGFR-2 and EGFRT790M dual inhibitors: Design, synthesis, molecular docking and anticancer evaluations, Bioorg. Chem., 2024, 143, 107062, DOI:10.1016/j.bioorg.2023.107062.
- Y. T. Tseng, Y. H. Tsai, F. Fülöp, F. R. Chang and Y. C. Lo, 2-Iodo-4'-Methoxychalcone Attenuates Methylglyoxal-Induced Neurotoxicity by Activation of GLP-1 Receptor and Enhancement of Neurotrophic Signal, Antioxidant Defense and Glyoxalase Pathway, Molecules, 2019, 24(12), 2249, DOI:10.3390/molecules24122249.
- Y. Liu and N. S. Gray, Rational design of inhibitors that bind to inactive kinase conformations, Nat. Chem. Biol., 2006, 2, 358–364 CrossRef CAS PubMed.
- C.-W. Yu, P.-Y. Hung, H. Yang, Y.-H. Ho, H.-Y. Lai, Y.-S. Cheng and J.-W. Chern, Quinazolin-2,4-dione-Based Hydroxamic Acids as Selective Histone Deacetylase-6 Inhibitors for Treatment of Non-Small Cell Lung Cancer, J. Med. Chem., 2019, 62(2), 857–874, DOI:10.1021/acs.jmedchem.8b01590.
- M. El-Naggar, H. R. M. Rashdan and A. H. Abdelmonsef, Cyclization of Chalcone Derivatives: Design, Synthesis, In Silico Docking Study, and Biological Evaluation of New Quinazolin-2,4-diones Incorporating Five-, Six-, and Seven-Membered Ring Moieties as Potent Antibacterial Inhibitors, ACS Omega, 2023, 8(30), 27216–27230, DOI:10.1021/acsomega.3c02478.
- H. Chen, P. Li, R. Qin, H. Yan, G. Li and H. Huang, DMAP-Catalyzed One-Pot Synthesis of Quinazoline-2,4-diones from 2-Aminobenzamides and Di-tert-butyl Dicarbonate, ACS Omega, 2020, 5(16), 9614–9623, DOI:10.1021/acsomega.0c01104.
- T. Yamashiro, K. Tokushige and T. Abe, One-Pot Synthesis of Core Structure of Shewanelline C Using an Azidoindoline, J. Org. Chem., 2023, 88(6), 3992–3997, DOI:10.1021/acs.joc.3c00013.
- L. Zhang, Q. Chen, L. Li, N. Ma, J. Tian, H. Sun, Q. Xu, Y. Yang and C. Li, Synthesis of N-Unsubstituted and N3-Substituted Quinazoline-2,4(1H,3H)-diones from o-Aminobenzamides and CO2 at Atmospheric Pressure and Room Temperature, Org. Lett., 2023, 25(14), 2471–2475, DOI:10.1021/acs.orglett.3c00614.
- N. A. A. M. Aziz, R. F. George, K. El-Adl and W. R. Mahmoud, Design, synthesis, in silico docking, ADMET and anticancer evaluations of thiazolidine-2,4-diones bearing heterocyclic rings as dual VEGFR-2/EGFRT790M tyrosine kinase inhibitors, RSC Adv., 2022, 12(20), 12913–12931, 10.1039/D2RA01119K.
- N. A. A. M. Aziz, R. F. George, K. El-Adl and W. R. Mahmoud, Exploration of thiazolidine-2,4-diones as tyrosine kinase inhibitors: Design, synthesis, ADMET, docking, and antiproliferative evaluations, Arch. Pharm., 2023, 356(3), e2200465, DOI:10.1002/ardp.202200465.
- K. El-Adl, A.-G. A. El-Helby, H. Sakr and S. S. A. El-Hddad, Design, synthesis, molecular docking, and anticancer evaluations of 1-benzylquinazoline-2,4(1H,3H)-dione bearing different moieties as VEGFR-2 inhibitors, Arch. Pharm., 2020, 353(8), e2000068, DOI:10.1002/ardp.202000068.
- J. H. Park, Y. Liu, M. A. Lemmon and R. Radhakrishnan, Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain, Biochem. J., 2012, 448(3), 417–423 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- N. E. A. Abd El-Sattar, K. El-Adl, M. A. El-Hashash, S. A. Salama and M. M. Elhady, Design, synthesis, molecular docking and in silico ADMET profile of pyrano[2,3-d]pyrimidine derivatives as antimicrobial and anticancer agents, Bioorg. Chem., 2021, 115, 105186, DOI:10.1016/j.bioorg.2021.105186.
- Y. Jia, C. M. Quinn, A. I. Gagnon and R. Talanian, Homogeneous time-resolved fluorescence and its applications for kinase assays in drug discovery, Anal. Biochem., 2006, 356(2), 273–281 CrossRef CAS PubMed.
- S. Sogabe, Y. Kawakita, S. Igaki, H. Iwata, H. Miki, D. R. Cary, T. Takagi, S. Takagi, Y. Ohta and T. Ishikawa, Structure-based approach for the discovery of pyrrolo[3, 2-d]pyrimidine-based EGFR T790M/L858R mutant inhibitors, ACS Med. Chem. Lett., 2013, 4, 201–205 CrossRef CAS PubMed.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures, J. Med. Chem., 2015, 58, 4066, DOI:10.1021/acs.jmedchem.5b00104.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- A. Beig, R. Agbaria and A. Dahan, Oral delivery of lipophilic drugs: the tradeoff between solubility increase and permeability decrease when using cyclodextrin-based formulations, PLoS One, 2013, 8, e68237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00502c |
| This journal is © The Royal Society of Chemistry 2024 |