
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the influence of crosslinkers on the properties, response, and degradation of enzymatic hydrogels for electrochemical glucose biosensing through fluorescence analysis†
Jancarlo Diaz-Gonzalez ,
L. G. Arriaga and
Jannu R. Casanova-Moreno*
,
L. G. Arriaga and
Jannu R. Casanova-Moreno*
Centro de Investigación y Desarrollo Tecnológico en Electroquímica, Pedro Escobedo, Querétaro, 76703, Mexico. E-mail: jcasanova@cideteq.mx
First published on 20th March 2024
Abstract
Drop-cast crosslinked hydrogels are a common platform for enzymatic electrochemical biosensors. Despite the widespread use of these complex systems, there are still several questions about how their physicochemical properties affect their performance, stability, and reproducibility. In this work, first-generation faradaic biosensors composed of glucose oxidase and branched polyethyleneimine (BPEI) are prepared using either glutaraldehyde (GA) or ethylene glycol diglycidyl ether (EGDGE) as crosslinkers. While EGDGE gels present an increasing electrochemical response with increasing crosslinker concentration, the current of GA gels decreases at high crosslinker concentration probably due to the hampered diffusion on tightly networked gels. We compared different strategies to use fluorescence microscopy to gain insight into the gel structure either by labeling the gel components with fluorophores or taking advantage of the intrinsic fluorescence of the imines formed upon crosslinking with GA. By monitoring the fluorescence of the crosslinking bonds and the electrochemical response, we demonstrate that hydrolysis, a common hydrogel degradation mechanism, is not responsible for the loss of electrical current over time in gels prepared with glutaraldehyde. Most hydrogel-based electrochemical biosensor studies do not perform specific experiments to determine the cause of the degradation and instead just infer it from the dependence of the current on the preparation conditions (most commonly concentrations). We show that, by taking advantage of several analytical techniques, it is possible to gain more knowledge about the degradation mechanisms and design better enzymatic biosensors.
Introduction
Hydrogels are 3D polymeric networks with the remarkable ability to absorb and retain substantial amounts of water within their porous structure. These polymeric networks are typically insoluble and owe their water-absorbing characteristics to the presence of hydrophilic functional groups such as –OH, –COOH, and –NH2 along their polymer chains. This property enables them to swell significantly, increasing to more than 10 times their original weight, without dissolving in the process. Hydrogels exhibit diverse characteristics, leading to various classification schemes based on their physicochemical attributes. Among these, an important classification depends on the method of 3D structure formation, distinguishing between physical and chemical processes.1 When the crosslinking reaction involves the formation of covalent bonds, the resulting hydrogel is termed a chemical hydrogel. Conversely, if the crosslinking relies on weaker interactions (e.g. electrostatic or hydrogen bonds), the hydrogel is classified as a physical hydrogel.Incorporating biological molecules, such as enzymes, into these polymeric matrices confers hydrogels interesting properties that find utility in applications such as biosensors, drug delivery, energy conversion and even chemical synthesis. The combination of hydrogels and enzymes becomes especially relevant when used in biosensing in complex environments, such as body fluids, typically for glucose quantification, or in miniaturized cell culture platforms, where complex media are used to evaluate the consumption of this analyte for extended periods. It has been reported that the resulting three-dimensional polymeric matrices offer properties such as support and significant increase in surface area, reusability, biocompatibility, protection from the external environment and enzyme leaching and the generation of favorable environments for the optimal functioning of enzymes, among other aspects.2–4 On the other hand, the use of hydrogels in biosensing systems presents significant challenges, such as their low mechanical strength, long-term stability, and reproducibility. These aspects make their study more difficult due to the inherent complexity of the systems. In most cases, problems are multifactorial, which makes it challenging to isolate and resolve them.3,5
Polysaccharide-based polymers like chitosan and the polyethyleneimine (PEI) family have been used to prepare enzymatic hydrogels that display good stability, biocompatibility, surface area, and sensitivity.6–8 In the PEI group, the use of branched polyethyleneimines (BPEIs) holds special significance owing to the abundant presence of amino groups in their chemical structure, encompassing 25% primary, 50% secondary, and 25% tertiary amines.9 Together with the –NH2 groups present in the tertiary structure of the enzyme, BPEIs offer multiple bonding sites significantly enhancing network stability.
Dialdehydes and diepoxides have been commonly employed to carry out chemical crosslinking reactions with the amino groups present in the structures of polymers and enzymes to form enzymatic hydrogels. Historically, glutaraldehyde (GA) has been widely used. However, its use has decreased recently due to its toxicity and the complexity arising from the multiple forms it can generate in aqueous solutions.10,11 Today, ethylene glycol diglycidyl ether (EGDGE) and its longer derivatives (polyEGDGE) are common crosslinking agents due to their low cost and innocuity compared to GA. Additionally, the amide bond formed by the reaction between EGDGE and primary amines under neutral conditions (pH ≈ 7) has greater stability compared to the imine bond (Schiff base) formed between GA and primary amines, which can be hydrolyzed under certain pH conditions.11,12 In general, it has been reported that the characteristics of the support, as well as the immobilization method and conditions can have an important effect on enzymatic electrochemical biosensors, effectively tuning their selectivity, sensitivity and stability.13,14 In the case of the enzymatic hydrogels, the choice of crosslinker will affect the structure and properties of the electrochemical biosensors so prepared. Frequently, these first- or second-generation enzymatic biosensors are prepared by depositing the gels on the electrode surfaces by drop-casting. Although this electrode modification technique is not one of the most reproducible ones, its simplicity makes it very common.15 In these systems, the effect of the crosslinker is usually evaluated only in terms of the electrical current produced. Typically, studies vary the crosslinker concentration and find an optimal value that they employ in the rest of the experiments.16
Fluorescence microscopy is a highly effective imaging technique that has transformed the field of materials science. Researchers widely use it as a powerful tool for visualizing and analyzing the spatial distribution of specific molecules or structures within hydrogels in a non-invasive and highly precise manner.17 This technique relies on the fluorescence properties of certain molecules, known as fluorophores, which may be inherently present in the sample or deliberately introduced. A diverse selection of fluorescent dyes is available on the market, offering a broad range of photochemical properties that cover the UV-visible to near-infrared (NIR) spectrum. These dyes are ideal for labeling gels, or the molecules being studied within them. The most common reaction is toward primary amines (–NH2) with reactive groups such as isothiocyanates, active esters and carboxylates. Some of the more relevant fluorophores include carbocyanines, benzopyrylium dyes, push–pull dyes, xanthene dyes, difluoroboron (BODIPY) complexes, AIEgens, and NIR-II dyes. Xanthene dyes are the most popular and widely used due to their high chemical stability and brightness. Rhodamine and fluorescein, which emit in the red and green regions respectively, stand out within this group.18,19 Some types of gels exhibit intrinsic fluorescence without the need to incorporate an additional fluorescent dye. These gels are sometimes referred to as “fluorescent gels”. Intrinsic fluorescence often arises due to π–π conjugation of polymer chains or functional groups that are formed during polymerization. For example, Deng et al. reported the presence of strong intrinsic fluorescence of hydrogels based on acrylamide (AAm)/poly(itaconic acid) (PITAc) which responds to the presence of metal cations.20 Another common cause of intrinsic fluorescence occurs through the formation of Schiff bases in amine-rich gels in the presence of aldehydes. In this context, Christadore's group synthesized and analyzed hydrogels polymerized from lysine and aldehyde-terminated polyethylene glycol macromonomers for their application in cell detection. Using steady-state spectrofluorimetry and confocal fluorescence microscopy, they observed a unique fluorescent structure (Schiff base) within the hydrogel network. This Schiff base exhibited a relatively significant Stokes shift (65 nm) and a wide emission bandwidth with maximum excitation at 380 nm and maximum emission at 445 nm.21 Another example of interest is that of chitosan hydrogels crosslinked with genipin. In this case, genipin can fluoresce after the crosslinking process, although the mechanism behind this fluorescence is still limited in understanding.22,23 These fluorescent components within the hydrogel create a remarkable contrast, resulting in images of exceptional quality. Thus, fluorescence microscopy allows precise visualization and analysis of critical parameters such as porosity, degradation, and diffusion of molecules in these materials.17,24,25 However, the use of fluorescence microscopy in hydrogels has been mainly focused on the investigation of cell/tissue behavior26–29 and drug release,30–32 which has contributed significantly to scientific advances in these areas.
In this context, fluorescence microscopy can provide valuable insights in the study of hydrogel-based electrochemical biosensors. In this study, a comparison of the generated morphological structures and electrochemical responses in BPEI/GOx-based first-generation biosensors, using GA and EGDGE as crosslinking agents, is presented. Despite their limitations due to oxygen dependence, we chose first-generation biosensors because they are nevertheless commonly utilized today, and their simpler composition allowed us to test our proposed methodology. The effects of the crosslinker identity and concentration on the hydrogel-based biosensor performance and stability were evaluated. Two strategies were used for the fluorescence analysis either by labeling the gel components or by measuring the gel intrinsic fluorescence. We took advantage of the fluorescent nature of the crosslinking bonds when using glutaraldehyde to assess the state of the gel. The photopolymerization and photodegradation processes triggered by exposure to light of certain wavelengths were characterized. As well, by combining fluorescence microscopy and electrochemical measurements, we evaluated whether the signal loss of these first-generation enzymatic electrochemical biosensors was due to hydrolysis of the crosslinking bonds. Generally, studies on hydrogel-based biosensors focus on improving stability without analyzing the failure or degradation mechanisms associated with these devices. A better understanding of these mechanisms, however, will offer a basis for future improvements.
Materials and methods
Reagents and solutions
Branched poly(ethyleneimine) (BPEI, Mw ∼25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), fluorescein-5-isothiocyanate (FITC), tetramethylrhodamine-5-isothiocyanate, G-isomer (TRITC, G-isomer), glucose oxidase (GOx, Aspergillus niger type X-S; 100000–250000 U g−1), glutaraldehyde (GA, 50 wt% in water), and glucose (β-D-glucose ≥99.5%) were acquired from Sigma Aldrich (Saint Louis, USA). Ethylene glycol diglycidyl ether (EGDGE, 100 wt%) was purchased from Polysciences, Inc (Niles, USA). Ammonia 28–30% and hydrogen peroxide 30% were purchased from J. T. Baker (Phillipsburg, USA). A stock solution of phosphate buffer (PB) 0.1 M pH 7.4, was prepared from KH2PO4 (1.14 g/500 mL) and K2HPO4·12H2O (10.87 g/500 mL) salts, to achieve a total concentration of 0.1 M. The solutions were prepared in ultrapure water with a resistivity of 18.2 MΩ cm from a Merck Millipore Simplicity UV purification system (Billerica, U.S.A) with a 0.22 μm filter and stored at room temperature.
000), fluorescein-5-isothiocyanate (FITC), tetramethylrhodamine-5-isothiocyanate, G-isomer (TRITC, G-isomer), glucose oxidase (GOx, Aspergillus niger type X-S; 100000–250000 U g−1), glutaraldehyde (GA, 50 wt% in water), and glucose (β-D-glucose ≥99.5%) were acquired from Sigma Aldrich (Saint Louis, USA). Ethylene glycol diglycidyl ether (EGDGE, 100 wt%) was purchased from Polysciences, Inc (Niles, USA). Ammonia 28–30% and hydrogen peroxide 30% were purchased from J. T. Baker (Phillipsburg, USA). A stock solution of phosphate buffer (PB) 0.1 M pH 7.4, was prepared from KH2PO4 (1.14 g/500 mL) and K2HPO4·12H2O (10.87 g/500 mL) salts, to achieve a total concentration of 0.1 M. The solutions were prepared in ultrapure water with a resistivity of 18.2 MΩ cm from a Merck Millipore Simplicity UV purification system (Billerica, U.S.A) with a 0.22 μm filter and stored at room temperature.
Fluorescence labeling of glucose oxidase and branched poly(ethyleneimine)
FITC and TRITC solutions were prepared by dissolving 1 mg of the fluorophore in 2 mL of 0.1 M disodium hydrogen phosphate for 10 minutes in a water bath at 25 °C. After that, 2 mL of enzyme (5 mg mL−1) or polymer (10 mg mL−1) solution was mixed with 1 mL of the fluorophore of interest and allowed to react for 30 minutes avoiding light. The solution was incorporated into a GE Healthcare PD-10 Sephadex™ G-25 M (Uppsala, Sweden) chromatographic column following the supplier's protocol for purification.Optical and electrochemical cell design
For optical evaluations, glass slides (Henso Labware Manufacturing Co., Hangzhou, China) and gold-coated glass slides (deposited as described below) were used. The electrochemical cell consists of a drop-based sensing device reported previously.33 Briefly, the chip device contains two polycrystalline gold electrodes deposited on a glass substrate by e-beam evaporation and the lift-off technique. Electrodes were composed of two metal layers: 20 nm of titanium to improve the adhesion to the substrate and 200 nm of gold that will form the electroactive area. Additionally, an Ag|AgCl wire was added as a pseudo-reference electrode (RE) to obtain a three-electrode cell configuration.Gel preparation
A cleaning process with RCA-1 solution (ultrapure water, ammonia and H2O2 in a 5:1:1 v/v ratio, respectively) was carried out for each substrate before modification.34 Crosslinking with EGDGE or GA was used to create enzymatic hydrogels on the supports. We have previously compared the use of these two crosslinkers in second-generation biosensors.33,35 In this case, however, a first-generation system was preferred to avoid fluorescence quenching by the transition metals in the mediators. Solutions of 10 mg mL−1 BPEI in ultrapure water and 5 mg mL−1 GOx in 0.1 M pH 7.4 phosphate buffer (PB) were prepared and mixed in a 3.3:1 v/v ratio, respectively. Fresh solutions of EGDGE and GA crosslinkers were added to the BPEI/GOx mixture, to obtain a final concentration range from 3.33 to 33.3 mM. Immediately, a 0.15 μL aliquot was taken with a 0.1–2.5 μL micropipette and deposited by drop-casting on each surface to be evaluated. The solution was allowed to dry for 20 min at 30 °C to get the desired hydrogels and was subsequently, if applicable, rinsed with ultrapure water to remove the excess.
Additionally, a procedure was designed to obtain free-standing gel samples for evaluations of the swelling process without a support and infra-red (IR) spectroscopy. To this end, 2 and 5 μL aliquots of these hydrogels were deposited on a fluorinated ethylene propylene (FEP) film and detached after drying.
Optical evaluations
For optical experiments, a Nikon Eclipse Ti-U inverted microscope (Melville, USA), coupled with a QImaging OptiMOS camera (Surrey, Canada) controlled by the Ocular software, was employed to conduct the assessment of crosslinked hydrogels. The illumination source utilized was a Lumen Dynamics (Ottawa, Canada) 200 W mercury arc lamp with a spectrum between 340–800 nm. All acquired images were subjected to analysis using the ImageJ software (version 1.52a) with the Fiji processing package.36 Fluorescence and bright-field micrographs were captured to evaluate the physical changes before and after the swelling process in each system. To evaluate the expansion in the XY plane upon gel swelling, the outlines of the regions covered by the gels were defined and the area within such regions calculated. The ratio of the area after swelling to the one before swelling was used to calculate the expansion percentage. A white light (brightfield) set and three fluorescence filter sets (Chroma, Bellows Falls, USA), described in Table 1, were used. To enhance clarity, these were referred to as UV, blue, and green light, corresponding to their respective excitation wavelengths. Images were acquired using a 4× objective. Exposure times of 10 ms were employed for bright-field images, while fluorescence images were obtained with exposure times of 50 and 500 ms. BPEI/GOx hydrogels were crosslinked with different concentrations of EGDGE or GA on glass or gold surfaces. Additionally, EGDGE-based hydrogels were fluorescently labeled with the procedure mentioned above to evaluate their morphology.| Light | Set number | Excitation filter | Emission filter |
|---|---|---|---|
| UV | 39000 | 361–389 nm | 435–485 nm |
| Blue | 49002 | 450–490 nm | 500–550 nm |
| Green | 49005 | 530–560 nm | 590–650 nm |
Electronic absorption spectra (UV-vis) were obtained using a Hach (Loveland, USA) DR 6000 spectrophotometer. Fluorescence spectra were performed using an Agilent Technologies (Santa Clara, USA) Cary Eclipse fluorescence spectrophotometer, using a quartz cell with a path length of 1 cm. For both analyses, a 1:1 dilution of the BPEI/GOx/GA reaction mixture (using 33.3 mM GA) and 0.1 M PB pH 7.4 was prepared. Before analysis, this mixture was allowed to react for 2 days to allow the higher molecular weight aggregates formed by crosslinking to precipitate.
Electrochemical evaluations
For electrochemical evaluations, BPEI/GOx/GA and BPEI/GOx/EGDGE hydrogels were deposited on gold electrodes as described in the “Gel Preparation” section above. Glucose solutions with 0–10 mM concentration in 0.1 M PB pH 7.4 were prepared. Solutions were stored at 2–8 °C for 24 h so that the anomeric carbohydrate mixture reached equilibrium.37 First, cyclic voltammetry (CV) was carried out in 0.1 M PB pH 7.4 containing 10 mM H2O2, applying potentials from −0.1 to +0.55 V vs. pseudo-Ag|AgCl, to detect H2O2 oxidation on gold surfaces. Next, the working electrode (WE) was modified with 0.15 μL BPEI/GOx hydrogels crosslinked with different degrees of EGDGE or GA. A 35 μL drop of the solution to be evaluated was placed so that the three electrodes were in contact. Electrochemical evaluations were made by chronoamperometry following the H2O2 oxidation produced by the enzymatic oxidation of glucose. In this case, a potential step was applied from the open circuit potential to the anodic peak potential determined in the previous CVs. The electrochemical responses were obtained by plotting the current at two minutes vs. the glucose concentration.Profilometry
Contact profilometry measurements were performed on the EGDGE and GA crosslinked gels before the swelling process, to assess surface morphology. A Veeco Dektak® 6M profilometer (Plainview, USA) was used, applying a measurement force of 1 mg.IR analysis
The IR spectra of GOx, BPEI, and crosslinked gels with a concentration of 33.3 mM GA were analyzed before and after exposure to green light for 15 minutes. Additionally, a hydrogel exposed to 10 mM NaBH4 for 2 h was also analyzed, to study the reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group resulting from the crosslinking reaction. All analyses were carried out using a Shimadzu (Kyoto, Japan) IRAffinity-1S spectrophotometer equipped with the ATR accessory. Scans were performed in a spectral range of 4000–650 cm−1 with a wavenumber resolution of 2 cm−1 and an average of 45 scans per measurement.
N group resulting from the crosslinking reaction. All analyses were carried out using a Shimadzu (Kyoto, Japan) IRAffinity-1S spectrophotometer equipped with the ATR accessory. Scans were performed in a spectral range of 4000–650 cm−1 with a wavenumber resolution of 2 cm−1 and an average of 45 scans per measurement.
Results and discussion
Morphological characterization of hydrogels
In this section, we present bright-field and fluorescence evaluations of hydrogels crosslinked with EGDGE or GA on glass surfaces, which were selected to avoid the fluorescence quenching issues that gold surfaces can generate. The results assess the effect of different crosslinker concentrations on the morphologies/structures and component distribution in the hydrogels, utilizing two different fluorescence strategies: (I) using fluorophores for hydrogels crosslinked with EGDGE, or (II) exploiting intrinsic fluorescence due to imine formation when GA is used.Fig. 1 shows brightfield and fluorescence images of hydrogels, with diameter between 1–2 mm. Notably, brightfield images reveal a discernible peripheral ring surrounding the hydrogel droplets. Profilometry analysis confirms that this ring exhibits greater thickness compared to the central region of the droplet (Fig. S1, ESI†). This phenomenon, known as the “coffee-ring effect”, is a well-documented characteristic of the drop-casting technique attributed to drying kinetics.38,39 For EGDGE gels, variations in crosslinker concentration have limited impact on the height of the rings. However, the central region experiences a significant increase in thickness at the highest concentration. Therefore, at the highest concentration, both the ring and the central region exhibit comparable heights, rendering the ring nearly imperceptible in the profilometry (Fig. S1c, ESI†). At the low and middle concentrations (Fig. S1a and b, ESI†), some spikes are seen in the central region, with heights almost an order of magnitude greater than their surroundings.
 | ||
| Fig. 1 Brightfield (a–c, g–i) and fluorescence (d–f, j–l) images of hydrogels prepared with EGDGE (a–f) and GA at concentrations of 3.33 mM (a, d, g and j), 16.65 mM (b, e, h and k) and 33.3 mM (c, f, i and l) and deposited on a glass slide. Fluorescence images for EGDGE gels corresponds to green-labelled GOx and red-labelled BPEI, whereas for GA gels it corresponds to the intrinsic fluorescence of the crosslinking bonds. The lookup table of each image has been adjusted to show proper contrast and therefore intensities cannot be compared among them. Dashed lines refer to the location of the profiles shown in Fig. 2. | ||
Hydrogels prepared using EGDGE as a crosslinker were labeled with fluorescent dyes. Green-emitting FITC and red-emitting TRITC were employed to modify the GOx and BPEI respectively. Fig. 2 shows the profilometry and fluorescence of both channels across the same line (dashed line in Fig. 1a and d) of a gel prepared with 3.33 mM EGDGE. The general shape of the fluorescence signals clearly resembles the profilometry, being more intense in the ring than in the central region. This can easily be understood, since a thicker gel would have more fluorescent molecules and therefore higher intensity. The corresponding fluorescence image (Fig. 1d), shows a slight segregation between the enzyme, with a higher contribution in the ring, and the polymer with a larger relative contribution observed in the center of the drop. As well, some regions of localized high intensity (usually termed “hot spots”) are observed. Some of these hot spots appear yellow in the image indicating the presence of both enzyme and polymer and are localized preferentially closer to the ring. These are probably aggregates that form during the gelation of the reaction mixture. There is another type of hot spots that fluoresce preferentially in the red channel. These are aggregates containing mainly polymer and are probably formed in the polymer solution before the reaction mixture preparation. As the EGDGE concentration increases (Fig. 1e), the segregation and the number of these aggregates increase. At the highest EGDGE concentration (Fig. 1f), high segregation is observed, not only between the ring and the central regions but also in the different parts of the central region. Some parts are clearly dominated by the enzyme fluorescence while others present almost exclusively the signal corresponding to the labeled polymer. This might be created because, at high crosslinker concentrations, the reaction takes place more quickly not allowing for a homogeneous distribution of the components in the reaction mixture.
 | ||
| Fig. 2 Profilometries (a and c) and fluorescence intensities (b and d) of hydrogels prepared with 3.33 mM EGDGE (a and b) and GA (c and d) deposited on a glass slide, recorded across the dashed lines in Fig. 1. | ||
Furthermore, bright-field images reveal blister-like features in the central region of the low-concentration EGDGE gels. Profilometry (Fig. 2a) confirms that these features are convex and significantly higher than their surroundings, even exceeding the height of the ring. However, there is no corresponding increase in fluorescence, indicating that they are not simply thicker gel regions. Additionally, applying sufficient force during profilometry punctures these structures and releases fluid. Therefore, we conclude that these are thin hydrogel membranes that detach from the glass, forming what we call “reservoirs” and their frequency decreases as the crosslinker concentration increases. As we will discuss below, the presence of these reservoirs might be related to the deposit stability.
In contrast, analysis of the brightfield and fluorescence images in GA-based gels (Fig. 1g–l) revealed intrinsic fluorescence without the need for dyes. This fluorescence is associated with imine bond (Schiff base) formation during the crosslinking process between amino and aldehyde groups,40–42 that will be discussed further in subsequent sections. Due to this intrinsic fluorescence interfering with FITC and TRITC fluorescence, a similar strategy as in EGDGE gels could not be employed to evaluate segregation. Instead, fluorescence images in this section depict the spatial distribution of crosslinking bonds.
Compared with EGDGE-based gels, brightfield images of gels prepared with GA (Fig. 1g–i) present a more homogeneous structure. In Fig. 2c and d it can be observed that the intrinsic fluorescence more closely resembles the profilometry across a given line. This suggests that the fluorescing crosslinking bonds are homogeneously distributed across the hydrogel volume. Profilometry on the gels prepared with the three different tested GA concentrations shows an increasing heterogeneity in the central region as the concentration increases (Fig. S1d–f†). Similar to what was observed with EGDGE, the highest concentration yields gels with thicker central regions. This is clearly evidenced in the reduced contrast between the ring and the central regions in the fluorescence images of the highest concentration compared with the lowest one (Fig. 1l and j respectively). However, the increase in fluorescence is not simply proportional to the thickness of the hydrogel. For example, using both the fluorescence images and the profilometry, a ratio of 0.67 fluorescence a.u. nm−1 of thickness could be calculated on average for a 3.33 mM gel. In contrast, a similar calculation yields a ratio of 1.23 fluorescence a.u. nm−1 of thickness for a 33.3 mM gel. This shows that, besides being thicker, hydrogels prepared with higher GA concentrations present more crosslinking bonds per unit volume of gel. As well, a change in morphology can be clearly observed in the brightfield images. The 3.33 mM GA concentration yields a relatively homogeneous structure with a few blister-like “reservoir” structures, but significantly smaller than in EGDGE gels. The 16.6 mM concentration, on the other hand, presents a striated structure. Finally, the highest concentration consistently produces gels of smaller diameter regardless of the use of the same deposition volume. These gels block the light transmission in the brightfield images significantly more than the two other concentrations, therefore appearing darker. Also, in GA-based gels, the fluorescent hot spots are significantly higher than in the EGDGE case (Fig. 2c and d). This might be caused by the faster kinetics of the crosslinking reaction when using GA compared to EGDGE. Therefore, larger aggregates can form in the reaction mixture.
Swelling process characterization
Swelling is one of the main processes of hydrogels. When immobilized, however, the large increase in volume can, produce strains that affect the gel integrity and stability. Therefore, in this section we evaluate how the swelling process affects our systems. Initially, hydrogels based on EGDGE and GA were deposited separately onto glass and gold surfaces to assess the influence of each surface on swelling behavior. Water absorption induced morphological changes in hydrogels placed on glass surfaces, causing deformations in both gel types. Notably, EGDGE-based gels exhibited more significant deformations, particularly in the ring region, yielding folding and creasing structures (Fig. 3a–d). These structural deformations are typical failure modes in soft materials when exposed to fluids. Their magnitude depends on the tension–compression effects generated during swelling and the polymer chains' resilience to revert to their original state.43,44 Substantial swelling, particularly when the gel has adhered to a rigid surface, leads to non-uniform deformations, favoring distortions in areas with weaker surface attachment. Real-time assessments of a GA-based hydrogel swelling (Video S1, ESI†) provided evidence of this phenomenon. The higher swelling observed for EGDGE gels agrees with literature. For example, the use of EGDGE as a crosslinker for α,β-polyaspartahydrazide has been reported to present a higher swelling compared to similar gels prepared with GA.45 This difference has been rationalized in terms of the more hydrophilic nature of the EGDGE. As well, EGDGE is about twice as long as GA. Longer crosslinkers have been shown to contribute to a larger mesh size, lower crosslinking density and increased swelling.46,47 The observed failure mechanisms of the hydrogels deposited on glass might be related to the poor bonding between the gel and the glass surface. In contrast, when gels were deposited on gold surfaces, minimal changes were observed in their structures upon exposure to buffer solution (Fig. 3e–h). This could be attributed to enhanced interactions resulting from electrostatic and coordination bonds facilitated by the chelating properties of BPEI.48–50 | ||
| Fig. 3 Stability analysis of hydrogels upon swelling with buffer solution. Bright-field micrographs of BPEI/GOx hydrogels crosslinked with 33.3 mM EGDGE (a, b, e and f) and GA (c, d, g and h), before (a, c, e and g) and after (b, f, d and h) swelling on glass (a–d) and gold (e–h) surfaces. | ||
The adhesion of both gel types to various surfaces directly affected their stability. Stability tests were conducted on glass and gold surfaces, with gels exposed to 0.1 M PB at pH 7.4 for 15 and 30 days, as summarized in Table S1.† Results demonstrated more detachment in EGDGE-based gels compared to GA-based gels on both surfaces. This shows that, despite ether bonds being in general more stable than imine bonds, other factors (e.g. the observed “reservoirs” where gel is not adhered) reduce the stability of gels crosslinked with EGDGE. As well, hydrogels deposited on gold surfaces presented less adhesion failures compared to ones deposited on glass.
Subsequently, a more relevant system for electrochemical assessments was employed. Gels were deposited onto working electrodes (WE), consisting of gold structures on glass substrates, to account for the combined surface effects. Under these conditions, changes in morphology due to swelling were again observed, as evidenced in the previous evaluation for each type of surface. There was an expansion in the XY plane of 13.49 ± 7.83% and 0.30 ± 0.12% for representative gels (n = 3) based on 33.3 mM EGDGE and GA, respectively. Fig. 4a–f shows that with increasing EGDGE concentration, swelling led to a higher number of folds, particularly in regions in contact with glass. The reason behind this trend could be attributed to the epoxy crosslinking reaction products and their ability to absorb water.51,52 However, this effect could compromise gel mechanical properties, as observed at medium and high concentrations, resulting in significant structural damage after swelling (Fig. S2, ESI†). For GA-based gels, the degree of crosslinking has been inversely linked to swelling.53 Higher GA concentrations lead to reduced water retention within the gel. Fig. 4g–l illustrates how our gels behave accordingly, where medium and high concentrations showed no significant structural changes, despite the medium concentration gel partially peeling off the glass surface. In contrast, gels with the lowest concentration exhibited partial dissolution, as evidenced by the presence of material on the counter electrode (left) after the swelling. Overall, GA-gels exhibited greater stability compared to EGDGE-gels. These findings agree with the observations of Collins and Birkinshaw54 when exploring various crosslinkers for hyaluronic acid/polyethyleneimine-based gel formation. Among the five evaluated crosslinkers, GA and polyEGDGE stood out, with GA yielding more stable structures than polyEGDGE.
 | ||
| Fig. 4 Influence of EGDGE and GA concentration on the morphology and water absorption of gels formed from BPEI/GOx and deposited on Au/glass surfaces. Gels crosslinked with EGDGE (a–c) and GA (g–i) in their dehydrated state, and after hydration with ultrapure water (d–f and j–l). | ||
Similarly, both gels were evaluated for their swelling process when unsupported by immersing free-standing gels in the buffer solution. Images of the gels were taken after they were hydrated for an hour using 0.1 M PB pH 7.4. The physical changes resulting from the swelling process are shown in Fig. S3 (ESI).† The EGDGE-based gels showed remarkable degradation due to the presence of polar functional groups (amines, alcohols, ethers). Besides a few pieces on the surface, the solution appeared clear, suggesting complete dissolution or very small aggregate size. On the other hand, the GA-based gel with the lowest concentration exhibited partial dissolution when the concentration of the crosslinker is reduced significantly. Compared to the EGDGE gels, however, more aggregates were visible and the solution was turbid, indicating a larger aggregate size. The gels prepared with medium and high concentrations of GA showed considerable swelling compared to their dry state. These GA-based gels were recovered from the tubes and weighed to compare the hydrated mass with the dry mass. Upon swelling, the mass increase was 15.1, 14.4 and 2.6 times for 33.3, 16.65 and 3.33 mM GA. Despite increasing the hydrophobicity, crosslinking with glutaraldehyde allows to maintain the gel physical integrity resulting in a larger hydrated mass. In general, these results show lower stability compared with adhered hydrogels, demonstrating that stability is greatly influenced by attachment, particularly on gold surfaces. Bonds formed between the gel and surface restricted gel swelling in the XY plane, functioning as anchor points that limited the gel dissolution in most cases.
Overall, the performed hydrogel characterization showed that crosslinker identity and concentration influences the morphology, stability, and attachment of the gel to solid surfaces. Since in electrochemical biosensors these gels need to be immobilized on the electrode surface, it is expected that the observed differences will affect the performance of the biosensors. More crosslinking, for example, is expected to result in more immobilized enzyme. As well, gel disintegration or detachment will translate into a loss of signal. Therefore, the next section presents the analytical performance of gels prepared with the same compositions tested above.
Influence of crosslinker concentration and nature on the electrochemical response
The effect of the concentration and type of crosslinker on the electrochemical response of biosensors was investigated. The current vs. concentration calibration curves are discussed first, followed by the enzyme kinetic parameters extracted from them. Finally, analytical figures of merit are compared between the different compositions to decide on the most suitable for the following steps.Chronoamperometric measurements were carried out at 0.4 V vs. Ag|AgCl, previously selected for H2O2 oxidation in cyclic voltammetry. The amperometric response was measured as a function of the glucose concentration between 0 and 10 mM. The current densities recorded at 2 min are shown in the calibration curves in Fig. 5. The electrochemical response of the EGDGE-based hydrogels (Fig. 5a) shows that higher EGDGE concentrations produce higher current densities. This implies that the amount of immobilized enzyme in the gels depends on the EGDGE concentration and is the predominant factor in the electrochemical response, as reported by other authors.55,56 In simpler terms, an increase in crosslinker concentration leads to greater retention of GOx in the gel, resulting in higher production of H2O2 and a consequent increase in current.
 | ||
| Fig. 5 Glucose calibration curves (n = 3) of crosslinked gels with different concentrations of EGDGE (a) and GA (b) in 0.1 M PB pH 7.4. The dashed lines in the graph represent the non-linear fit for the apparent Michaelis–Menten constant (Kappm) and the maximum current density (Jmax) calculation. | ||
In comparison, GA-based gels (Fig. 5b) did not follow the trend observed with EGDGE. In this case, the middle concentration produced the highest current density. As discussed above in the fluorescence/profilometry characterization, a higher concentration of GA results in a higher number of crosslinking bonds per unit volume. This can create highly compact three-dimensional networks at high concentrations, which can affect the electrochemical response. At lower and medium concentrations, mass transport through the gels is not impeded significantly. Hence, the amount of immobilized enzyme in the gel (proportional to the GA concentration) could be the determining factor, as in the case of EGDGE-based gels, yielding the lowest current for the lowest concentration and a higher current for the medium concentration. However, this trend breaks when evaluating the gel with the highest concentration of GA. For this concentration, we hypothesize that a tenfold increase in GA concentration reduces the mesh size enough to affect the mass transport of glucose through the gel. This limitation leads to a decrease in the current. Additionally, another factor that might contribute to the reduction in current is the potential alteration of the enzyme structure due to the high GA concentration,57,58 compromising its catalytic activity and thereby affecting the biosensor's response. Nevertheless, given the high complexity of the systems evaluated in this study, it would not be appropriate to attribute these effects to a single cause, as multiple phenomena could be concurrently influencing the measured signals.
The catalytic activity of immobilized GOx within the gels was assessed using the Michaelis–Menten equation.59 In enzymatic electrochemical biosensors, it is possible to approach enzymatic kinetics by correlating the current density (J) with the substrate concentration, as the number of electrons transferred to the electrode is proportional to the number of molecules produced by the enzymatic reaction. This leads to the determination of an apparent Michaelis–Menten constant (Kappm) and a maximum current density (Jmax). These parameters were estimated by a non-linear fit using the data from the calibration curves after subtracting the current of the blank (without glucose). The Jmax and Kappm values for EGDGE and GA-based gels are presented in Fig. 6a and b, respectively and in Table S2 (ESI).† Jmax values show, in general, the trend discussed above for the currents. Kappm values show a clear inverse dependence with the EGDGE concentration, starting at ∼13.5 mM for the lowest crosslinker content and decreasing to ∼1.5 mM for the gel with the most concentrated EGDGE. In all cases, Kappm values are lower than the Km values reported for free GOx (26–30 mM). It has been reported that, upon immobilization, GOx presents a lower Kappm.59 This difference is because Kappm does not only reflect enzyme kinetics but is also influenced by other factors, such as substrate partition and diffusion through the polymeric network, oxygen concentration, or even conformational changes. The reduction in Kappm values with increasing EGDGE concentration is likely caused by substrate preconcentration within the gel resulting from partition equilibria35 as the chemical environment changes with the degree of crosslinking. In contrast, gels crosslinked with GA present a low value of Kappm even at the lowest concentration. This reflects the higher crosslinking present using this molecule. In fact, a slight increase in Kappm can be observed at the highest GA concentration, as expected for a system where diffusion of the substrate is noticeably impeded.
 | ||
| Fig. 6 (a) Maximum current densities (Jmax) and (b) apparent Michaelis–Menten constants (Kappm) determined for crosslinked gels with different concentrations of EGDGE and GA. | ||
Finally, Table 2 compares the analytical figures of merit for the different concentrations of EGDGE and GA-based gels. The highest concentration of both gels achieved the best stability losing only ∼23% of its initial Jmax. The observed similarity between the two crosslinkers contrasts with the poorer structural stability for EGDGE gels discussed in the previous section. This discrepancy can be attributed to the duration of the experiments, during which the hydrogels were immersed in the solution for only 2 h (equivalent to 3 consecutive calibration curves), in contrast to the measurement period of 15 and 30 days for the structural stability evaluation on glass and gold surfaces independently. The performance of 33.3 mM EGDGE gels surpassed the other two concentrations of EGDGE-based gels, exhibiting the best LOD (0.11 mM) and LOQ (0.39 mM) values, along with a sensitivity of 10.76 μA cm2 mM−1. For GA-crosslinked gels, the highest concentration demonstrated the greatest stability. However, both LOD and LOQ were impacted, presenting higher values compared to the lower and medium concentrations. On the other hand, the gel with the medium GA concentration displayed the best analytical performance, with values of 0.03 mM and 0.12 mM for LOD and LOQ, respectively. Despite this, its stability and linear range were compromised, losing 57.88% of its initial Jmax and limiting its linear range between 0–1 mM. The linear range of most of the prepared hydrogels is not sufficiently large for common glucose sensing applications (e.g. blood). Nevertheless, this is a frequent consequence of GOx immobilization that has been solved industrially by incorporating diffusion-limiting membranes.60 The electrochemical behavior and stability of the highest concentrations of both crosslinkers (33.3 mM) suggests that they could be considered promising options to be evaluated and even integrated into glucose detection systems in the future. Overall, however, GA-based gels show more mechanical stability on the electrode surfaces. Therefore, in the following sections, we limit the study to these hydrogels. This also means that all the following fluorescence images correspond to intrinsic hydrogel fluorescence and not to labeled components.
| Gel | Linear range (mM) | R2 | LOD (mM) | LOQ (mM) | Stabilitya (%) | Sensitivity (μA cm2 mM−1) |
|---|---|---|---|---|---|---|
| a Stability is reported as the percentage loss from the initial Jmax after three calibration curves. | ||||||
| BPEI/GOx/GA 3.33 mM | 0–2.5 | 0.96 | 0.06 | 0.21 | 57.17 | 14.05 |
| BPEI/GOx/GA 16.65 mM | 0–1 | 0.96 | 0.03 | 0.12 | 57.88 | 28.96 |
| BPEI/GOx/GA 33.3 mM | 0–2.5 | 0.96 | 0.39 | 1.32 | 23.27 | 14.50 |
| BPEI/GOx/EGDGE 3.33 mM | 0–10 | 0.98 | 1.51 | 5.04 | 49.13 | 1.12 |
| BPEI/GOx/EDGGE 16.65 mM | 0–2.5 | 0.98 | 0.60 | 2.03 | 39.09 | 3.96 |
| BPEI/GOx/EGDGE 33.3 mM | 0–2.5 | 0.95 | 0.11 | 0.39 | 23.39 | 10.76 |
Characterization of the intrinsic fluorescence of GA-based gels
To characterize the intrinsic fluorescence of GA-based hydrogels, we initially conducted electronic absorption (UV-vis) spectroscopy on the reaction mixture to identify potential excitation wavelengths for the fluorophore. Fig. S4a† shows that the absorption spectrum presents maxima at 325, 423, 536, and 644 nm. Subsequently, fluorescence excitation and emission spectra were acquired in each of these regions, except for the shortest wavelength, where no significant emission was detected. Fig. S4b–d† show the normalized excitation and emission spectra, alongside the fluorescence filter set employed, if applicable. Excitation maxima were observed at 466, 540 and 647 nm, corresponding to emission maxima at 500, 559 and 663 nm, respectively. The relative intensity of these three fluorescence signals was determined to be 1:13.2:4.2, with the most intense signal closely matching the fluorescence observed by Ling et al. in BPEI-GA nanoparticles.41 Therefore, we used the fluorescence signal characterized by green excitation and red emission signal (λexc = 540 nm, λem = 559 nm) for all subsequent gel crosslinking characterizations with GA.
After characterizing the fluorescence signals, we investigated the possible effect on crosslinking bonds by illuminating them with light of different wavelengths, using UV, blue or green excitation filters. The reader needs to take into account and distinguish that each captured image presents two stages of illumination: (I) excitation with green light (λexc = 540 nm, λem = 559 nm), which is essential for capturing images before and after exposure to the different wavelengths, and (II) the additional illumination, whose effect we want to explore and which can be UV, blue or green.
Fig. 7a shows the base fluorescence intensity (blank) of a fresh hydrogel crosslinked with 33.3 mM GA, while Fig. 7b presents a similar image after exposing small regions (approximately 130 μm in diameter) with green (left), blue (center) and UV (right) lights for 15 minutes. This image shows that the area exposed to blue light does not present changes in fluorescence intensity compared to the surrounding (unexposed) areas. In contrast, the areas exposed to green and UV light presented a decrease and an increase in fluorescence intensity, respectively. The change in fluorescence intensity can best be observed by dividing the intensity after exposure by the intensity before exposure (Fig. 7c and d).
 | ||
| Fig. 7 Effect of exposure to light of different wavelength on the red fluorescence of hydrogels crosslinked with 33.3 mM GA. (a and b) Fluorescence micrographs of a hydrogel (a) before and (b) after exposure of selected regions to green (left), blue (center) and UV light (right). (c) Normalized fluorescence image obtained by dividing (b) by (a). (d) Intensity profile at the dashed line in subfigure (c). (e) Red fluorescence intensity variation when exposed to green (circles) or UV light (triangles and squares). Solid lines in circles correspond to nonlinear fitting to mono- and bi-exponential decays, respectively. | ||
To better understand these processes, two freshly prepared hydrogels were irradiated with UV or green light, recording images every minute. Fig. 7e (filled circles) shows that, when irradiated with green light, the fluorescence intensity decreased. This change can be appropriately described by a monoexponential decay (R2 = 0.9680), with a decay time constant τ = 1.97 min associated with a photobleaching process and possibly with a degradation of the bonds formed during crosslinking. In contrast, the hydrogel irradiated with UV light (Fig. 7e, filled triangles) started at a comparable intensity but showed a 5.4-fold increase in fluorescence after being exposed for 20 min. After reaching this maximum, further exposure to UV light caused a decrease in the fluorescence intensity so that after a total exposure time of 60 min, the intensity decreased by 41% with respect to the maximum. We hypothesized, that the decrease in fluorescence could be caused by the inevitable exposure to green light necessary to perform the measurements (5 s per measurement). To confirm this hypothesis, the experiment was repeated in a fresh hydrogel reducing the measurement frequency to once every two minutes (Fig. 7e, squares). Indeed, a similar increase was initially observed but the decay after the maximum was proportionally slower, losing only 22% of the maximum intensity at 60 min of total exposure.
Old hydrogels (several months since their preparation) were also exposed to green or UV light under the same conditions as the freshly prepared hydrogels (Fig. 7e, open symbols). We observed an initial intensity 5.8 times higher compared with the initial fluorescence of the fresh gels, and comparable to the maximum fluorescence achieved on the fresh gels. Upon illumination with green light, we observed a decrease in intensity that was not appropriately described by a monoexponential decay (Fig. 7e, open circles). Instead, a biexponential decay was required (R2 = 0.9997) with τ1 = 2.83 min and τ2 = 15.74 min. This behavior is frequently found in photobleaching curves and is generally explained in terms of the presence of two populations of fluorophores with two different photodegradation rates.61 These results suggest that the environment gets more complex as the crosslinking reaction proceeds. When exposing the old hydrogel to UV light, no corresponding increase in fluorescence intensity is observed (Fig. 7e, open triangles). Instead, a slow decrease is observed, which can be attributed to the exposure to green light during measurements as discussed above for the fresh gels.
Considering all the evidence, then, we suggest that the exposure to UV light increases the energy of the reaction mixture promoting the formation of the fluorescent crosslinking bonds. In fresh hydrogels, even after the initial drying time, the crosslinking reaction has not reached completeness. Therefore, the effect of UV light is easily observed. In old hydrogels, complete crosslinking has taken place. This is evidenced by both the observed high fluorescence intensity and the absence of an increase in fluorescence when exposed to UV light. These results show the usefulness of monitoring the Schiff base fluorescence to assess the progress of the crosslinking reaction between amines and GA. Furthermore, these results suggest that, in the absence of UV light, the same levels of fluorescence (and therefore degree of crosslinking) can be attained given that enough time is given for the crosslinking reaction to reach completion. As well, results show that UV light can be employed to reduce the drying/reaction time for electrode modification. This can be of relevance in the industrial manufacturing of this type of electrodes.
On the other hand, when hydrogels are irradiated with green light, the excitation of the fluorophore moieties results in their degradation through photobleaching. Interestingly, it has been reported that Schiff bases can degrade through hydrolysis when exposed to UV light.62 We think that this discrepancy can be explained in terms of the absorption properties of the different Schiff bases. While most of the reported ones absorb in the UV or violet range,41,63,64 the main excitation of the fluorophores in our gels takes place in the green region of the spectrum. Similar behavior has been observed by Ling et al. in nanoparticles formed by PEI and GA.41 Therefore, the main photodegradation of the fluorophore takes place when exposed to green light instead of UV. It is this characteristic of the gels that allows UV light to enhance crosslinking without damaging them.
As a result of these experiments, we assumed the degradation upon exposure to green light to be caused by the rupture of the imine bond (Schiff base). To test this hypothesis, ATR-IR spectra were acquired. Peaks observed in the ATR-IR spectrum of GOx and BPEI (Fig. 8, curves a and b, respectively) at 3361 and 3280 cm−1 are attributed to the N–H stretching of primary and secondary amines.42,65 When the hydrogel is formed (Fig. 8, curve c), these peaks change from a sharp form to a wider peak, due to the OH groups produced in the crosslinking reaction and the presence of absorbed water. The peak present at 1525 cm−1 in the GOx and BPEI spectra, was attributed to the N–H bending vibrations of primary and secondary amines. When the GA-based hydrogel is formed, this peak disappears and a new peak is observed at 1566 cm−1. This peak was associated with CN stretching of the imine formed by the reaction between GA and primary amines. This assignment was confirmed by the disappearance of the peak at 1566 cm−1, when the GA-based hydrogel was immersed in a 10 mM NaBH4 solution for 2 h, to reduce the imine groups (Fig. 8, curve e).66,67 However, when analyzing the ATR-IR spectrum of the hydrogel degraded with green light (Fig. 8, curve d), there is no decrease or absence of the peak at 1566 cm−1, indicating that there was no breaking or degradation of the CN bond. These results suggest that the fluorophore in these gels is more complex than often described in the literature. All the authors who crosslink glutaraldehyde with amine-containing molecules (such as GOx and/or BPEI) directly attribute the fluorescence to the Schiff base.42,65,68,69 However, the Schiff base could be a part of a larger fluorophore and not the fluorophore by itself. It is known that GA can form at least ten different forms in solution, some of them cyclic or polymeric.11 It is possible that some of these forms, together with the Schiff base, form a larger fluorophore. This could also explain why the absorbance and emission wavelengths are significantly red-shifted compared to similar systems prepared with simpler aldehydes.41
 | ||
| Fig. 8 FT-IR spectra of GOx (a), BPEI (b), hydrogels crosslinked with 33.3 mM GA before (c) and after (d) exposure to green light for 15 minutes and of a hydrogel exposed to 10 mM of NaBH4 for 2 h (e). | ||
Fluorescence monitoring of gel degradation during electrochemical tests
Finally, 33.3 mM GA-based gels were evaluated both in terms of their electrochemistry (via calibration curves as described above) and fluorescence signals. As stated above, hydrolysis is a known degradation mechanism for hydrogels crosslinked with imime-based chemistry.70–72 By comparing the fluorescence of the crosslinking bonds and the electric current from peroxide oxidation, it is possible to assess whether the loss of electrochemical signal is due to this hydrolysis. Gels were prepared under different conditions: a gel was kept in the dark while two others were exposed to UV or green light for 15 minutes after the drying step and before the initial rinse. Fig. 9a shows the results of the electrochemical response, where the gel kept in the dark initially recorded the highest current density, followed by the gels exposed to UV light and green light, with values of Jmax = 48.79, 31.67 and 15.79 μA cm−2, respectively. In the second electrochemical evaluation (EC 2), it was observed that all the gels displayed a significant decrease in current, losing more than 50% of their initial Jmax. In the subsequent third evaluation (EC 3), an even more substantial signal decrease was observed, exceeding 90% of the initial current density. In the fluorescence evaluations (Fig. 9b), the 15 minutes exposures produced changes resembling what was described in the previous section. After the hydration/swelling process that takes place during the rinsing step, all the gels showed an increase in fluorescence intensity. This change could derive from the change in the refractive index of the medium (0.1 M PB pH 7.4 vs. air). As well, any possible self-quenching due to the proximity of fluorophores can decrease due to the longer separation between fluorophore molecules in the hydrated state compared to the dehydrated state. The swelling process then magnifies the differences in intensity between the three gels. The initial degradation of the gel exposed to green light resulted in a much lower fluorescence intensity than the other two gels during all the remaining evaluations. After the swelling process, there is a noticeable difference between the UV-exposed and unexposed gels, which becomes smaller as the crosslinking reaction takes place in the gel kept in the dark. In contrast to the current density, there was an increase in fluorescence intensity in both gels between the first and the second electrochemical evaluations. Even after the third electrochemical evaluation, when most of the current has been lost, the fluorescence is still comparable to the one obtained during the first electrochemical evaluation. This rules out the hypothesis that the observed degradation is due to the loss or cleavage of the imine crosslinking bonds, suggesting that the degradation mechanism of the gels is directly associated with the enzyme. Future work will include the monitoring of the enzyme amount and activity to further clarify which is the predominant mechanism that limits the stability of these and other enzymatic hydrogels when used as faradaic biosensors.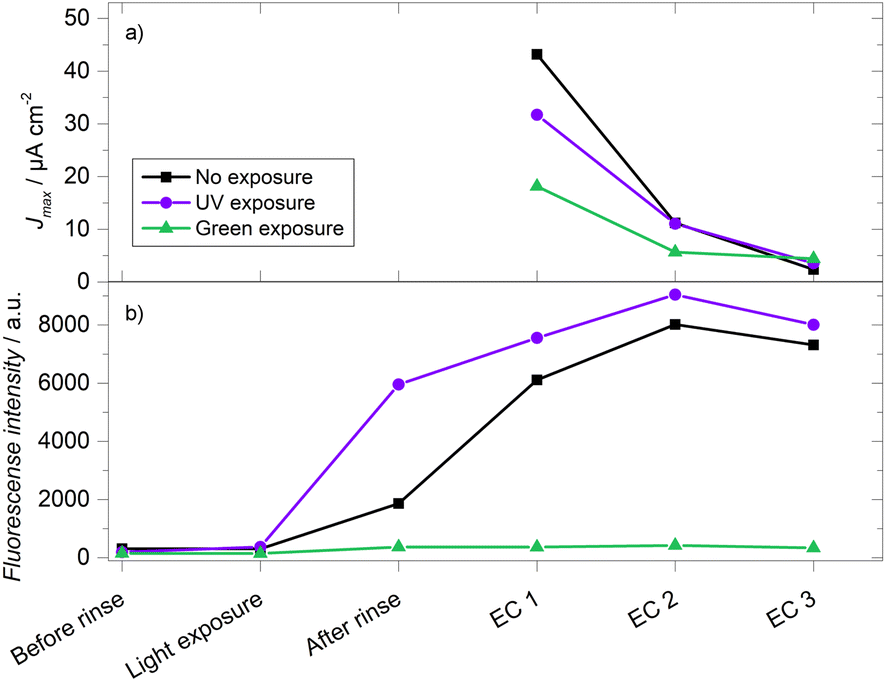 | ||
| Fig. 9 Simultaneous evaluation of the electrochemical response (a) and fluorescence (b) of crosslinked gels with 33.3 mM GA exposed to light of different wavelength. All evaluations were performed in 0.1 M PB pH 7.4. Each electrochemical (EC) evaluation represents a calibration curve with a glucose concentration range from 0 to 10 mM. | ||
Conclusions
This work compared the morphology, stability, and electrochemical response of enzymatic hydrogels, focusing on the application of fluorescence microscopy as a complement to electrochemical evaluation. By varying the crosslinker identity and concentration their influence could be analyzed with a variety of techniques. In general, hydrogels prepared with GA presented better long-term mechanical stability compared with gels prepared with EGDGE. Fluorescence microscopy employing labeled enzyme and polymer showed greater segregation of the components with increasing concentrations of EGDGE. On the other hand, the intrinsic fluorescence of the crosslinking bonds when using GA reflected a homogeneous distribution of these bonds across the hydrogel volume.The analytical performance of the different compositions revealed that in EGDGE-based gels, the highest crosslinker concentration tested resulted in higher currents, and therefore better sensitivity and LOD. This is indicative of the amount of immobilized enzyme limiting the overall response. On GA-based gels, on the other hand, the middle crosslinker concentration performed better, as the highest concentration likely produced a mesh size too small for proper diffusion of glucose, as well as possible effects on the enzyme conformation.
The intrinsic fluorescence of the GA-based gels was characterized. It was observed that the photoactivity of the reaction mixture and the formed hydrogel can be used to characterize the polymerization and degradation processes. IR spectroscopy showed that the fluorophore responsible for the red emission in GA/BPEI gels might be more complex than the usually reported Schiff base. By monitoring fluorescence intensity and electrochemical response, it was established that the decrease in the electrochemical response of GA-based gels upon repeated use is not caused by the hydrolysis of the crosslinking bonds.
The approach described in this work allowed us to obtain a better understanding of the degradation/failure mechanisms in BPEI/GOx hydrogel-based biosensors crosslinked with EGDGE or GA. When comparing to other studies characterizing enzymatic hydrogels for glucose biosensing (Table 3), one can observe that fluorescence is underutilized. Furthermore, most of the reported works focus only on the electrocatalytic evaluation and stability of the devices, which suggests a lack of depth in the understanding of the possible degradation mechanisms that affect their performance and in the identification of areas for improvement. Further exploration of these evaluations of the presence and function of the enzymatic hydrogel components is expected to translate into a better understanding of the stability and degradation as fundamental aspects in the future application of these materials in biosensor platforms.
| Hydrogel material | Biorecognition element | Immobilization method | Characterization techniques | Measured parameters | Describes degradation mechanism or associates the electrochemical response with the techniques used? | Ref. |
|---|---|---|---|---|---|---|
| a The following abbreviations are used in this table: lactate oxidase (LOx), horseradish peroxidase (HRP), transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDX), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), atomic force microscopy (AFM), Brunauer–Emmett–Teller analysis (BET). | ||||||
| Poly(ethylene glycol) diacrylate (PEG-DA) | GOx, LOx | Crosslinking/entrapment | Electrochemistry | Electrocatalytic activity, stability | No | 73 |
| Chitosan | GOx | Entrapment | Electrochemistry | Electrocatalytic activity, stability | No | 74 |
| Chitosan | GOx | Electrostatic/entrapment | FT-IR ATR, electrochemistry | Chemical structure, electrocatalytic activity, stability | No | 75 |
| Poly(ethylene glycol) diacrylate (PEG-DA) | GOx | Electrostatic/entrapment | Electrochemistry | Electrocatalytic activity | No | 76 |
| DNA hydrogel | HRP, GOx | Encapsulation | TEM, SEM, EDX, XPS, XRD, Raman, fluorescence microscopy, AFM, photoelectrochemistry | Chemical structure, morphology, composition, photoelectrocatalytic activity, stability | No | 77 |
| Polyacrylic acid (PAA) | GOx | Crosslinking | SEM, BET, UV-vis, FT-IR, electrochemistry | Chemical structure, morphology, electrocatalytic activity, stability | Yes | 78 |
| Polyaniline (PANI) | GOx | Crosslinking | SEM, TEM, XPS, electrochemistry | Chemical structure, morphology, electrocatalytic activity | No | 79 |
| BPEI | GOx | Crosslinking | Fluorescence and bright field microscopy, profilometry, FT-IR ATR, electrochemistry | Chemical structure, morphology, electrocatalytic activity | Yes | This work |
Author contributions
Conceptualization, J. D.-G. and J. R. C.-M.; methodology, J. D.-G. and J. R. C.-M.; validation, J. D.-G.; formal analysis, J. D.-G. and J. R. C.-M; investigation, J. D.-G.; resources, J. R. C.-M. and L. G. A.; writing—original draft preparation, J. D.-G., and J. R. C.-M.; writing—review and editing, L. G. A.; visualization, J. D.-G., and J. R. C.-M.; supervision, L. G. A. and J. R. C.-M.; project administration, L. G. A. and J. R. C.-M.; funding acquisition, L. G. A. and J. R. C.-M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the support of the Mexican National Science and Technology Council (CONACYT) through the reaccreditation of the National Micro and Nanofluidics Laboratory. We also thank Yussemi Mixtega-Cruz for the acquisition of some of the microscopy images, Montserrat Rueda-Becerril for useful discussions on IR spectroscopy, as well as Luis Antonio Ortiz Frade, Janet Ocampo Hernandez and Yunny Meas Vong for access to the IR, UV-vis and fluorescence spectroscopy equipment.References
- Md. S. Rahman, Md. M. Islam, Md. S. Islam, A. Zaman, T. Ahmed, S. Biswas, S. Sharmeen, T. U. Rashid and M. M. Rahman, in Cellulose-Based Superabsorbent Hydrogels, ed. Md. I. H. Mondal, Springer International Publishing, Cham, 2019, pp. 819–863 Search PubMed.
- Y. Li, H. Zhang, Y. Qi and C. You, Foods, 2023, 12, 4405 CrossRef CAS PubMed.
- A. Herrmann, R. Haag and U. Schedler, Adv. Healthcare Mater., 2021, 10, 2100062 CrossRef CAS PubMed.
- J. Meyer, L. Meyer and S. Kara, Eng. Life Sci., 2022, 22, 165–177 CrossRef CAS PubMed.
- A. Barhoum, O. Sadak, I. A. Ramirez and N. Iverson, Adv. Colloid Interface Sci., 2023, 317, 102920 CrossRef CAS PubMed.
- K. Pal, A. K. Banthia and D. K. Majumdar, Des. Monomers Polym., 2009, 12, 197–220 CrossRef CAS.
- Dhanjai, A. Sinha, P. K. Kalambate, S. M. Mugo, P. Kamau, J. Chen and R. Jain, TrAC, Trends Anal. Chem., 2019, 118, 488–501 CrossRef CAS.
- K. Völlmecke, R. Afroz, S. Bierbach, L. J. Brenker, S. Frücht, A. Glass, R. Giebelhaus, A. Hoppe, K. Kanemaru, M. Lazarek, L. Rabbe, L. Song, A. Velasco Suarez, S. Wu, M. Serpe and D. Kuckling, Gels, 2022, 8, 768 CrossRef PubMed.
- C. Lungu, M. Diudea, M. Putz and I. Grudziński, Int. J. Mater. Sci., 2016, 17, 555 Search PubMed.
- X. Lu, Y. Xu, C. Zheng, G. Zhang and Z. Su, J. Chem. Technol. Biotechnol., 2006, 81, 767–775 CrossRef CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790–802 CrossRef CAS PubMed.
- J. Leach, J. Wolinsky, P. Stone and J. Wong, Acta Biomater., 2005, 1, 155–164 CrossRef PubMed.
- S. C. Perry, S. M. Gateman, J. Sifakis, L. Pollegioni and J. Mauzeroll, J. Electrochem. Soc., 2018, 165, G3074–G3079 CrossRef CAS.
- D. Semenova, T. Pinto, M. Koch, K. V. Gernaey and H. Junicke, Biosens. Bioelectron., 2020, 170, 112702 CrossRef CAS PubMed.
- A. Kaliyaraj Selva Kumar, Y. Zhang, D. Li and R. G. Compton, Electrochem. Commun., 2020, 121, 106867 CrossRef CAS.
- J. Radomski, L. Vieira and V. Sieber, Bioelectrochemistry, 2023, 151, 108398 CrossRef CAS PubMed.
- J. Zhong, T. Zhao and M. Liu, NPG Asia Mater., 2022, 14, 38 CrossRef CAS.
- S. P. Zustiak, H. Boukari and J. B. Leach, Soft Matter, 2010, 6, 3609 RSC.
- S. Bou, A. S. Klymchenko and M. Collot, Adv. Mater., 2021, 2, 3213–3233 RSC.
- J. Deng, H. Wu, W. Xie, H. Jia, Z. Xia and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 39967–39975 CrossRef CAS PubMed.
- L. Christadore, M. Grinstaff and S. Schaus, Molecules, 2018, 23, 936 CrossRef PubMed.
- S. Matcham and K. Novakovic, Polymers, 2016, 8, 385 CrossRef PubMed.
- M. F. Butler, Y.-F. Ng and P. D. A. Pudney, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3941–3953 CrossRef CAS.
- N. Russ, B. I. Zielbauer, K. Koynov and T. A. Vilgis, Biomacromolecules, 2013, 14, 4116–4124 CrossRef CAS PubMed.
- F. Fergg, F. J. Keil and H. Quader, Colloid Polym. Sci., 2001, 279, 61–67 CrossRef CAS.
- W. Su, R. Wang, C. Qian, X. Li, Q. Tong and T. Jiao, J. Chem., 2020, 2020, 1–17 CrossRef.
- X. Li, S. Chen, J. Li, X. Wang, J. Zhang, N. Kawazoe and G. Chen, Polymers, 2016, 8, 269 CrossRef PubMed.
- L. Zhu, C. Shao, H. Chen, Z. Chen and Y. Zhao, Research, 2021, 2021, 2021–9845679 CrossRef PubMed.
- W. Wang, Y. H. Chan, S. Kwon, J. Tandukar and R. Gao, Nano Convergence, 2022, 9, 30 CrossRef CAS PubMed.
- J. Cardellini, A. Balestri, C. Montis and D. Berti, Pharmaceutics, 2021, 13, 861 CrossRef CAS PubMed.
- M. Vigata, C. Meinert, D. W. Hutmacher and N. Bock, Pharmaceutics, 2020, 12, 1188 CrossRef CAS PubMed.
- C. Wei, X. Dong, Y. Zhang, J. Liang, A. Yang, D. Zhu, T. Liu, D. Kong and F. Lv, Sens. Actuators, B, 2018, 273, 264–275 CrossRef CAS.
- J. M. Díaz-González, R. A. Escalona-Villalpando, L. G. Arriaga, S. D. Minteer and J. R. Casanova-Moreno, Electrochim. Acta, 2020, 337, 135782 CrossRef.
- J. Casanova-Moreno, J. To, C. W. T. Yang, R. F. B. Turner, D. Bizzotto and K. C. Cheung, Sens. Actuators, B, 2017, 246, 904–909 CrossRef CAS.
- L. Navarro-Nateras, J. Diaz-Gonzalez, D. Aguas-Chantes, L. L. Coria-Oriundo, F. Battaglini, J. L. Ventura-Gallegos, A. Zentella-Dehesa, G. Oza, L. G. Arriaga and J. R. Casanova-Moreno, Biosensors, 2023, 13, 582 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- H. D. Belitz, W. Grosch and P. Schieberle, in Food Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2009, pp. 248–339 Search PubMed.
- D. Mampallil and H. B. Eral, Adv. Colloid Interface Sci., 2018, 252, 38–54 CrossRef CAS PubMed.
- M. Rey, J. Walter, J. Harrer, C. M. Perez, S. Chiera, S. Nair, M. Ickler, A. Fuchs, M. Michaud, M. J. Uttinger, A. B. Schofield, J. H. J. Thijssen, M. Distaso, W. Peukert and N. Vogel, Nat. Commun., 2022, 13, 2840 CrossRef CAS PubMed.
- S. A. Poursamar, A. N. Lehner, M. Azami, S. Ebrahimi-Barough, A. Samadikuchaksaraei and A. P. M. Antunes, Mater. Sci. Eng., C, 2016, 63, 1–9 CrossRef CAS PubMed.
- Y. Ling, F. Qu, Q. Zhou, T. Li, Z. F. Gao, J. L. Lei, N. B. Li and H. Q. Luo, Anal. Chem., 2015, 87, 8679–8686 CrossRef CAS PubMed.
- N. Nematidil, M. Sadeghi, S. Nezami and H. Sadeghi, Carbohydr. Polym., 2019, 222, 114971 CrossRef CAS PubMed.
- J. Yoon, J. Kim and R. C. Hayward, Soft Matter, 2010, 6, 5807–5816 RSC.
- S. S. Velankar, V. Lai and R. A. Vaia, ACS Appl. Mater. Interfaces, 2012, 4, 24–29 CrossRef CAS PubMed.
- F. Castelli, G. Pitarresi and G. Giammona, Biomaterials, 2000, 21(8), 821–833 CrossRef CAS PubMed.
- S. Bhattacharya and R. Shunmugam, ACS Omega, 2020, 5, 2800–2810 CrossRef CAS PubMed.
- H. Salimi-Kenari, F. Mollaie, E. Dashtimoghadam, M. Imani and B. Nyström, Carbohydr. Polym., 2018, 181, 141–149 CrossRef CAS PubMed.
- S. Kobayashi, K. Hiroishi, M. Tokunoh and T. Saegusa, Macromolecules, 1987, 20, 1496–1500 CrossRef CAS.
- Y. R. Dangi, J. K. Bediako, X. Lin, J.-W. Choi, C.-R. Lim, M.-H. Song, M. Han and Y.-S. Yun, Sci. Rep., 2021, 11, 17836 CrossRef CAS PubMed.
- Y. R. Dangi, X. Lin, J.-W. Choi, C.-R. Lim, M.-H. Song, M. Han, J. K. Bediako, C.-W. Cho and Y.-S. Yun, Environ. Technol. Innovation, 2022, 28, 102605 CrossRef CAS.
- J. Zhou and J. P. Lucas, Polymer, 1999, 40, 5505–5512 CrossRef CAS.
- L. Li, Y. Yu, Q. Wu, G. Zhan and S. Li, Corros. Sci., 2009, 51, 3000–3006 CrossRef CAS.
- S. Khan and N. M. Ranjha, Polym. Bull., 2014, 71, 2133–2158 CrossRef CAS.
- M. N. Collins and C. Birkinshaw, J. Appl. Polym. Sci., 2008, 109, 923–931 CrossRef CAS.
- D. MacAodha, M. L. Ferrer, P. Ó. Conghaile, P. Kavanagh and D. Leech, Phys. Chem. Chem. Phys., 2012, 14, 14667 RSC.
- N. Vasylieva, B. Barnych, A. Meiller, C. Maucler, L. Pollegioni, J.-S. Lin, D. Barbier and S. Marinesco, Biosens. Bioelectron., 2011, 26, 3993–4000 CrossRef CAS PubMed.
- H. Chen, Q. Zhang, Y. Dang and G. Shu, Adv. J. Food Sci. Technol., 2013, 5, 932–935 CrossRef CAS.
- Y. Witazora, Yandri, T. Suhartati, H. Satria and S. Hadi, J. Phys.: Conf. Ser., 2021, 1751, 012097 CrossRef CAS.
- N. Vasylieva and S. Marinesco, in Microelectrode Biosensors, ed. S. Marinesco and N. Dale, Humana Press, Totowa, NJ, 2013, vol. 80, pp. 95–114 Search PubMed.
- A. Maines, D. Ashworth and P. Vadgama, Anal. Chim. Acta, 1996, 333, 223–231 CrossRef CAS.
- D. L. Kolin, S. Costantino and P. W. Wiseman, Biophys. J., 2006, 90, 628–639 CrossRef CAS PubMed.
- Z. Huang, D. Wan and J. Huang, Chem. Lett., 2001, 30, 708–709 CrossRef.
- R. M. Issa, A. M. Khedr and H. F. Rizk, Spectrochim. Acta, Part A, 2005, 62, 621–629 CrossRef PubMed.
- R. M. Issa, A. M. Khedr and H. Rizk, J. Chin. Chem. Soc., 2008, 55, 875–884 CrossRef CAS.
- K.-S. Hwang, H.-Y. Park, J.-H. Kim and J.-Y. Lee, Korean J. Chem. Eng., 2018, 35, 798–804 CrossRef CAS.
- Sudirman, W. Z. Lubis, Mujamilah, G. Tj. Sulungbudi and N. Rahmayani, AIP Conf. Proc., 2021, 2349, 020005 CrossRef CAS.
- X. Wang, M. Ding, Z. Liu and D. Wang, RSC Adv., 2015, 5, 19541–19551 RSC.
- J. Vernengo, G. W. Fussell, N. G. Smith and A. M. Lowman, J. Biomed. Mater. Res., 2010, 93B, 309–317 CrossRef CAS PubMed.
- N. Islam, H. Wang, F. Maqbool and V. Ferro, Molecules, 2019, 24, 1271 CrossRef CAS PubMed.
- S. Wang, S. Tavakoli, R. P. Parvathaneni, G. N. Nawale, O. P. Oommen, J. Hilborn and O. P. Varghese, Biomater. Sci., 2022, 10, 6399–6412 RSC.
- R. Yu, E. Petit, M. Barboiu, S. Li, W. Sun and C. Chen, Mater. Sci. Eng., C, 2021, 127, 112210 CrossRef CAS PubMed.
- N. Boehnke, C. Cam, E. Bat, T. Segura and H. D. Maynard, Biomacromolecules, 2015, 16, 2101–2108 CrossRef CAS PubMed.
- J. Yan, V. A. Pedrosa, A. L. Simonian and A. Revzin, ACS Appl. Mater. Interfaces, 2010, 2, 748–755 CrossRef CAS PubMed.
- P. Murugan, J. Annamalai, R. Atchudan, M. Govindasamy, D. Nallaswamy, D. Ganapathy, A. Reshetilov and A. K. Sundramoorthy, Micromachines, 2022, 13, 304 CrossRef PubMed.
- W. Lipińska, K. Siuzdak, J. Karczewski, A. Dołęga and K. Grochowska, Sens. Actuators, B, 2021, 330, 129409 CrossRef.
- A. Mugweru, B. L. Clark and M. V. Pishko, J. Diabetes Sci. Technol., 2007, 1, 366–371 CrossRef PubMed.
- R. Zeng, Z. Huang, Y. Wang and D. Tang, ChemElectroChem, 2020, 7, 1537–1541 CrossRef CAS.
- H. Al-Sagur, S. Komathi, M. A. Khan, A. G. Gurek and A. Hassan, Biosens. Bioelectron., 2017, 92, 638–645 CrossRef CAS PubMed.
- D. Zhai, B. Liu, Y. Shi, L. Pan, Y. Wang, W. Li, R. Zhang and G. Yu, ACS Nano, 2013, 7, 3540–3546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental results of profilometry, absorption and fluorescence spectra, mechanical stability evaluation, and enzyme kinetic parameters. See DOI: https://doi.org/10.1039/d4ra00265b |
| This journal is © The Royal Society of Chemistry 2024 |