
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew pyrazolylindolin-2-one based coumarin derivatives as anti-melanoma agents: design, synthesis, dual BRAFV600E/VEGFR-2 inhibition, and computational studies†
Ahmed Sabt a,
Mohammed A. Khedrbc,
Wagdy M. Eldehnad,
Abdelsamed I. Elshamya,
Mohamed F. Abdelhameede,
Rasha M. Allame and
Rasha Z. Batran*a
a,
Mohammed A. Khedrbc,
Wagdy M. Eldehnad,
Abdelsamed I. Elshamya,
Mohamed F. Abdelhameede,
Rasha M. Allame and
Rasha Z. Batran*a
aChemistry of Natural Compounds Department, Pharmaceutical and Drug Industries Research Institute, National Research Centre, Dokki, Cairo, 12622, Egypt. E-mail: rasha_batran@yahoo.com
bDepartment of Pharmaceutical Chemistry, College of Pharmacy, Kuwait University, Safat 13110, Kuwait
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Helwan University, 11795, Egypt
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, 33516, Egypt
ePharmacology Department, Medical and Clinical Research Institute, National Research Centre, Dokki, Cairo 12622, Egypt
First published on 16th February 2024
Abstract
Malignant melanoma is the most invasive skin cancer with the highest risk of death. The inhibition of BRAFV600E appears relevant for overcoming secondary resistance developed during melanoma treatment. BRAFV600E triggers angiogenesis via modification of the expression of angiogenic inducers, which play a crucial role in the metastasis of melanoma. Accordingly, the dual inhibition of the BRAFV600E/VEGFR-2 signaling pathway is considered a rational approach in the design of anti-melanoma candidates. In this study, a new class of pyrazolylindolin-2-one linked coumarin derivatives as dual BRAFV600E/VEGFR-2 inhibitors targeting A375 melanoma cells was designed. Target compounds were tailored to occupy the pockets of BRAFV600E and VEGFR-2. Most of the synthesized compounds demonstrated potent mean growth inhibitory activity against A375 cells. Compound 4j was the most active cytotoxic derivative, displaying an IC50 value at a low micromolar concentration of 0.96 μM with a significant safety profile. Moreover, 4j showed dual potent inhibitory activity against BRAFV600E and VEGFR-2 (IC50 = 1.033 and 0.64 μM, respectively) and was more active than the reference drug sorafenib. Furthermore, derivative 4j caused significant G0/G1 cell cycle arrest, induced apoptosis, and inhibited the migration of melanoma cells. Molecular docking showed that compound 4j achieved the highest ΔG value of −9.5 kcal mol−1 against BRAFV600E and significant ΔG of −8.47 kcal mol−1 against VEGFR-2. Furthermore, the structure–activity relationship study revealed that TPSA directly contributed to the anticancer activity of the tested compounds.
1. Introduction
Melanoma is one of the most aggressive forms of skin malignancy and considered the leading cause of death among other skin cancers.1–3 Dysregulation of the RAS-RAF-MEK-ERK cascade, a mitogen-activated protein kinase (MAPK) pathway, is a critical step in melanoma pathogenesis.4RAF family proteins involve serine–threonine kinases composed of A-RAF, B-RAF, and C-RAF, which regulate intracellular signal transduction. Among them, the BRAF oncogene shows better biochemical potencies than A-RAF and C-RAF isoforms owing to its higher basal kinase activity and ease of activation by RAS, which explains the observed frequent mutational activation of the BRAF gene in human tumors.5–7 Approximately 90% of the detected BRAF mutations in human cancer are amino acid substitution at the nucleotide residue 600 (V600E) of valine by glutamic acid.8 The BRAFV600E mutation leads to constitutive kinase activation, which is 500–700-fold greater than wild-type BRAF, resulting in the amplification of the MAPK signaling pathway and stimulation of vascular endothelial growth factor (VEGF), thus inducing metastasis.9,10 This mutation is mostly associated with melanoma.11–13
The successful inhibition of BRAF was a turning point in the development of anti-melanoma agents. Targeted therapy with BRAF inhibitors represents a milestone in the treatment of metastatic melanoma that harbors BRAFV600E mutations. However, resistance to the treatment was identified in patients receiving these drugs within months, which makes the discovery of new targeted anti-melanoma therapeutics that can selectively block tumor signals in the RAS-RAF-MAPK complex network a substantial therapeutic challenge.14–16
Various drug development programs have been initiated, such as the dual inhibition of BRAFV600E and VEGFR-2, which has been considered a rational approach in the design of anti-melanoma candidates.17,18 This approach aims to seek alternative chemical backbones that avoid the resistance and side effects acquired by current anti-melanoma FDA approved therapies such as vemurafenib,19 dabrafenib,20 and encorafenib,21 which may be due to paradoxical MAPK activation in cells expressing wild-type BRAF.22–25 Dual inhibitors such as sorafenib and RAF265, which are type II inhibitors, were approved for or entered clinical trials (Fig. 1).26,27 Sorafenib, i.e., BAY 43e9006, is a multi-kinase inhibitor with dual inhibitory properties against VEGFR and RAF kinases. It suppresses MAPK signaling via RAF inhibition in various cancer cell lines including melanoma.28 Regorafenib, i.e., BAY 73-4506, effectively inhibits a wide range of angiogenic kinases such as VEGFR-1/2/3 and intracellular signaling kinases, including BRAF and CRAF.29 Furthermore, RAF265 is a strong dual RAF/VEGFR-2 inhibitor that efficiently suppresses the growth and survival of cancer cells by blocking tumor development and preventing blood supply to cancerous cells (Fig. 1).30
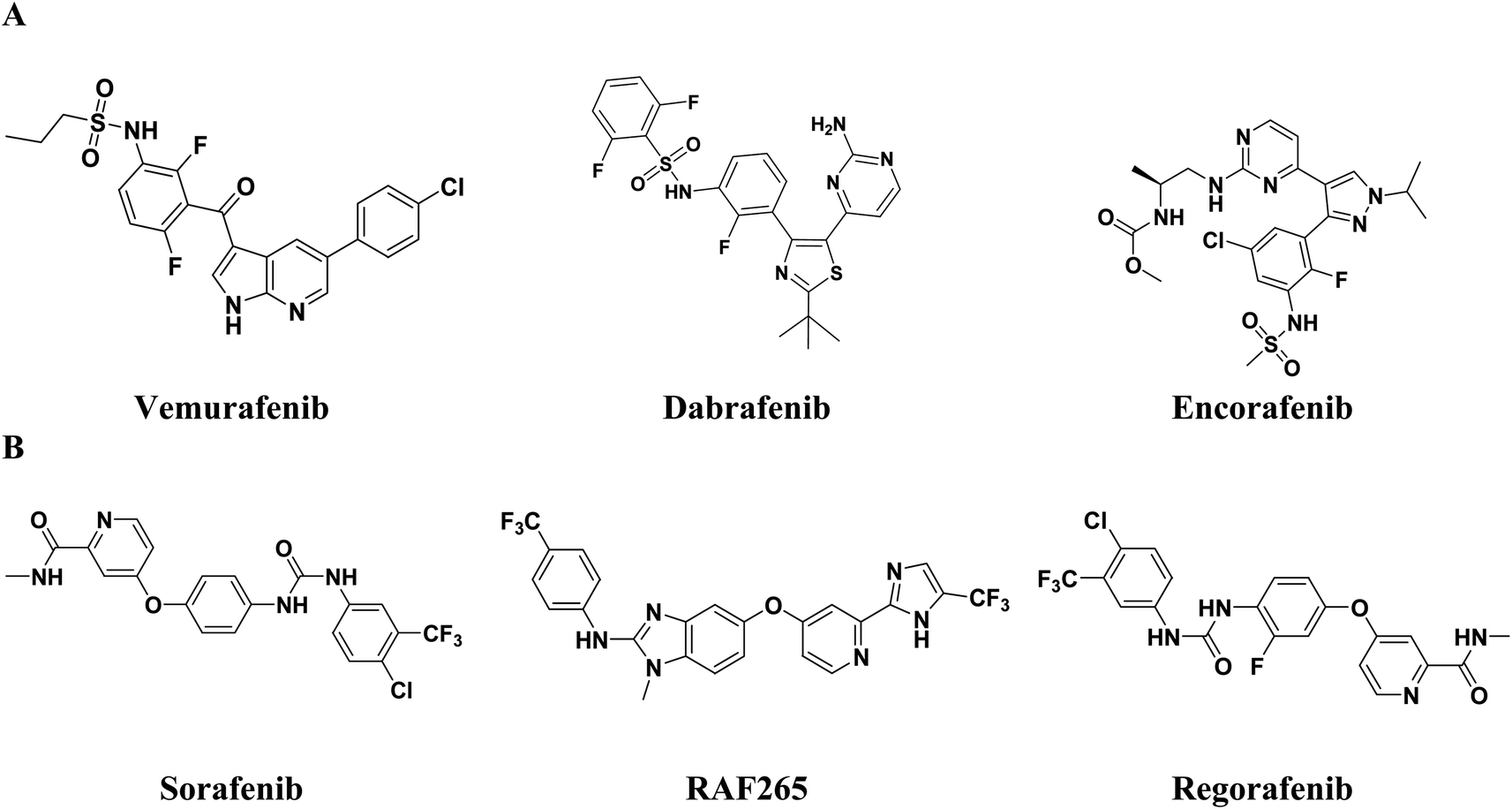 | ||
| Fig. 1 (A) Examples of FDA approved anti-melanoma drugs. (B) Dual inhibitors of BRAF/VEGFR-2. | ||
Coumarin derivatives are known for their potent anticancer activity, displaying remarkable activity against human malignant melanoma and preventing its recurrence.31–33 Moreover, coumarin analogues exhibit selective RAF inhibiting efficacies, making the coumarin nucleus a prominent bioactive structural motif in the search for new anti-melanoma cancer molecules.34,35 Furthermore, 4-hydoxycoumarin derivatives have been extensively used as a valuable core in the synthesis of VEGFR-2 inhibitors.36,37 Moreover, several FDA approved drugs and inhibitors in clinical trials containing pyrazole (e.g., encorafenib and GDC0879) and indole or indole isosteres (e.g., vemurafenib (azaindole), SB-590885 (indene) and XL281(indole)38) have been investigated for potent and selective anti-BRAF activity. Alternatively, pyrazole and indole derivatives have also been reported for their potent VEGFR-2 inhibitory activity (Fig. 1 and 2).30–43
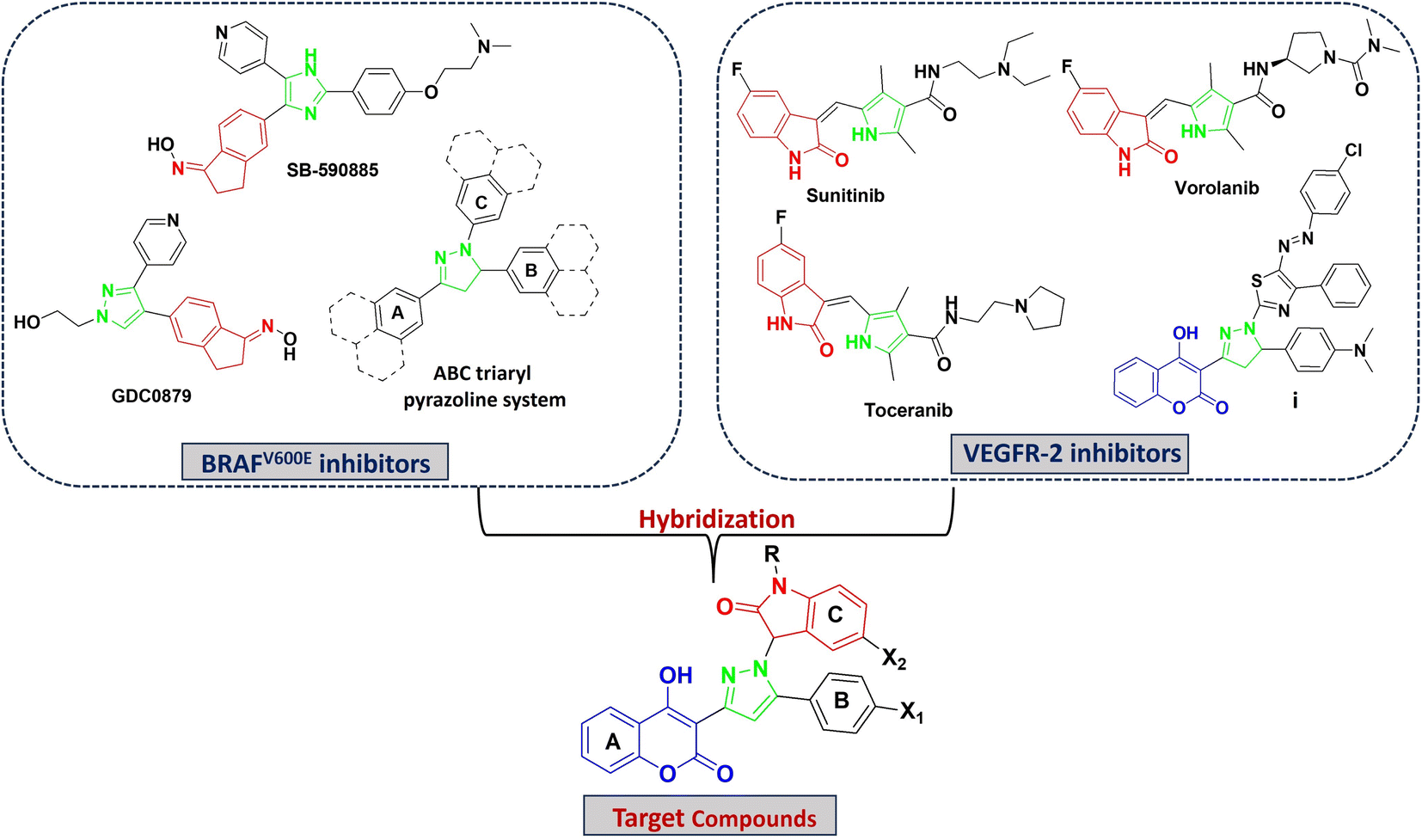 | ||
| Fig. 2 Rationale for the design of target compounds. | ||
In this work, we aim to discover dual BRAFV600E and VEGFR-2 inhibitors that were rationally designed based on the ABC triaryl pyrazole system,44–46 simultaneously displaying the pharmacophoric features of BRAFV600E and VEGFR-2 inhibitors (Fig. 2). Based on the above-mentioned studies, a new series of pyrazolyl indoline-based coumarin derivatives was designed, synthesized, and biologically assessed for their cytotoxic activity against melanoma cells. Furthermore, the promising derivatives were evaluated for dual inhibition towards BRAFV600E and VEGFR-2, cell migration, cell cycle analysis and apoptosis. The molecular modeling study was carried out to investigate the binding mode of the promising candidates in the binding pockets of BRAFV600E and VEGFR-2.
2. Results and discussion
2.1. Chemistry
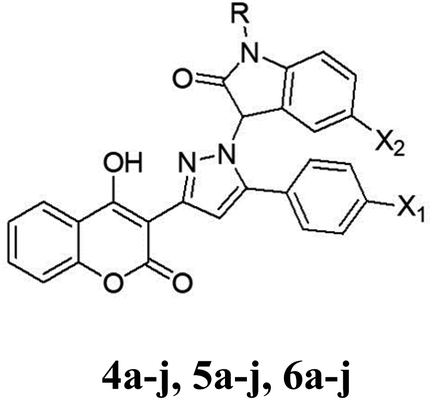
The synthetic procedures for the synthesis of the target pyrazolylindoline-2-one-coumarin hybrids 4a–j, 5a–j and 6a–j are demonstrated in Scheme 1. Intermediate chalcone derivatives 2a–c were prepared through the Claisen–Schmidt condensation reaction of 3-acetyl-4-hydroxycoumarin 1 and the appropriate aromatic aldehydes, namely, 4-fluorobenzaldehyde, 4-chlorobenzaldehyde and/or 4-bromobenzaldehyde according to the reported procedures.47,48 The cyclo–addition reaction of hydrazine hydrate with chalcone intermediates 2a–c furnished the corresponding pyrazolines 3a–c, respectively. The target pyrazolylindolin-2-one derivatives 4a–j, 5a–j and 6a–j were afforded by reacting pyrazoline compounds 3a–c with isatin derivatives via the elimination of a water molecule (Scheme 1). The structures of the newly synthesized compounds were assigned via elemental and spectral analyses including 1H NMR and 13C NMR.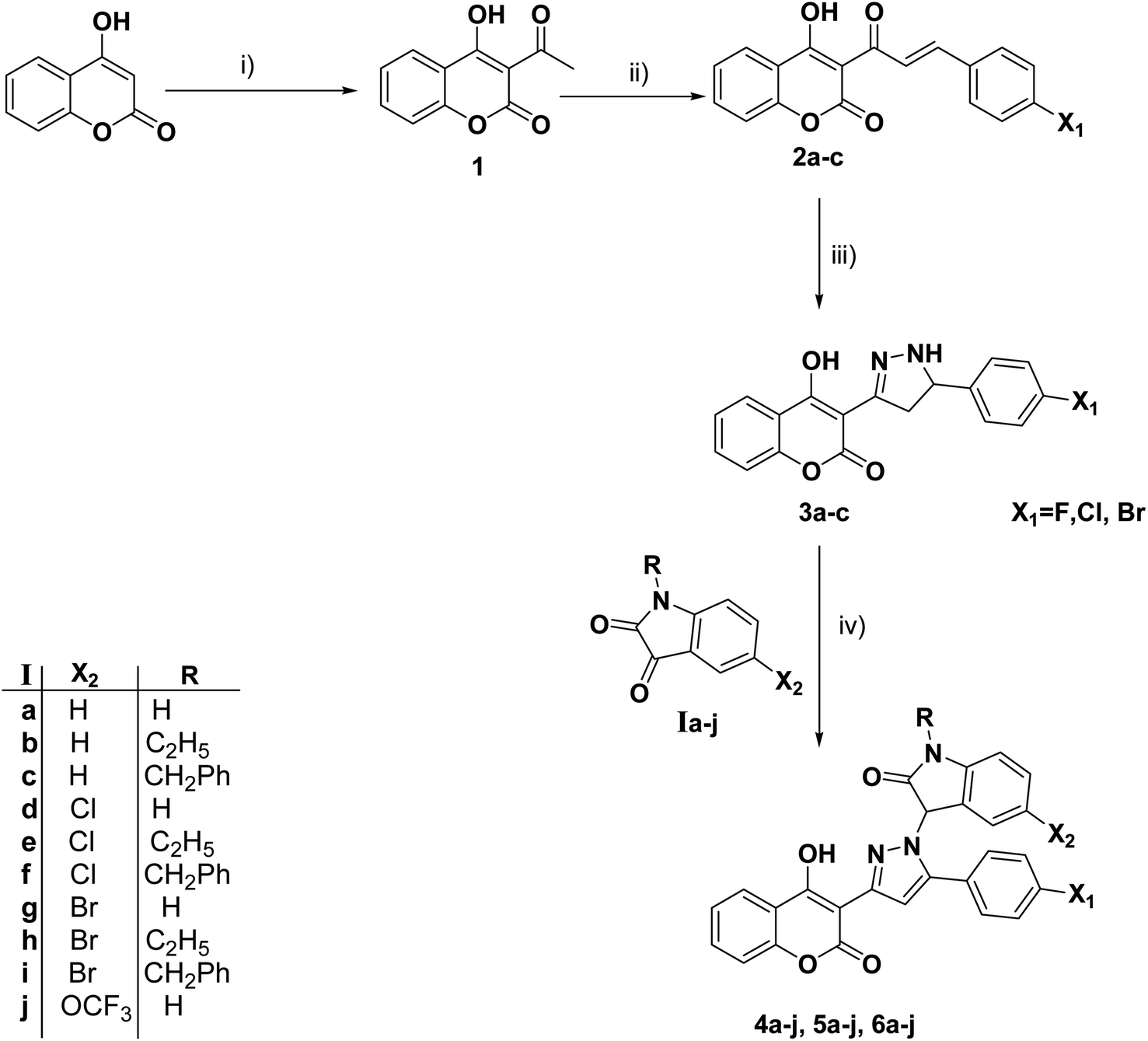 | ||
| Scheme 1 Reagents and conditions: (i) POCl3/AcOH/reflux. (ii) ArCHO/piperidine/EtOH/reflux. (iii) NH2NH2·H2O/EtOH/reflux. (vi) Isatin derivatives/EtOH/reflux. | ||
2.2. Biology
|
||||
|---|---|---|---|---|
| Compound | R | X1 | X2 | (IC50, μM) |
| a The cytotoxic activity was determined after treatment of A375 with serial dilution of the target compounds for 72 h, where data are expressed as mean ± SD (n = 3). | ||||
| 4a | H | F | H | 6.25 ± 0.75 |
| 4b | CH2CH3 | F | H | 79.47 ± 3.60 |
| 4c | CH2PH | F | H | 4.6 ± 0.32 |
| 4d | H | F | Cl | 4.3 ± 0.38 |
| 4e | CH2CH3 | F | Cl | 82.63 ± 2.65 |
| 4f | CH2Ph | F | Cl | 1.114 ± 0.02 |
| 4g | H | F | Br | 5.85 ± 0.95 |
| 4h | CH2CH3 | F | Br | 38.65 ± 2.60 |
| 4i | CH2Ph | F | Br | 1.28 ± 0.02 |
| 4j | H | F | OCF3 | 0.96 ± 0.03 |
| 5a | H | Cl | H | 7.13 ± 0.44 |
| 5b | CH2CH3 | Cl | H | 33.76 ± 1.80 |
| 5c | CH2Ph | Cl | H | 3.8 ± 0.22 |
| 5d | H | Cl | Cl | 5.43 ± 0.82 |
| 5e | CH2CH3 | Cl | Cl | 7.32 ± 0.69 |
| 5f | CH2Ph | Cl | Cl | 1.09 ± 0.09 |
| 5g | H | Cl | Br | 6.18 ± 0.73 |
| 5h | CH2CH3 | Cl | Br | 5.44 ± 0.41 |
| 5i | CH2Ph | Cl | Br | 1.02 ± 0.07 |
| 5j | H | Cl | OCF3 | 4.17 ± 0.36 |
| 6a | H | Br | H | 8.45 ± 0.60 |
| 6b | CH2CH3 | Br | H | 68.38 ± 3.50 |
| 6c | CH2Ph | Br | H | 4.84 ± 0.30 |
| 6d | H | Br | Cl | 6.25 ± 0.72 |
| 6e | CH2CH3 | Br | Cl | 8.99 ± 0.78 |
| 6f | CH2Ph | Br | Cl | 4.36 ± 0.22 |
| 6g | H | Br | Br | 7.32 ± 0.80 |
| 6h | CH2CH3 | Br | Br | 47.22 ± 1.90 |
| 6i | CH2Ph | Br | Br | 5.47 ± 0.20 |
| 6j | H | Br | OCF3 | 4.37 ± 0.24 |
| Sorafenib | — | — | — | 3.2 ± 0.67 |
| Compound | A375 IC50 (μM) | HSF IC50 (μM) | Selectivity index (SI) |
|---|---|---|---|
| 4f | 1.114 ± 0.02 | 4.18 ± 0.25 | 3.6 |
| 4i | 1.28 ± 0.021 | 3.26 ± 0.29 | 2.5 |
| 4j | 0.96 ± 0.03 | 9.58 ± 0.73 | 9.9 |
| 5f | 1.09 ± 0.09 | 3.59 ± 0.32 | 3.3 |
| 5i | 1.02 ± 0.07 | 3.63 ± 0.76 | 3.5 |
The cancerous specificity and the safety of the most active derivatives were evaluated on normal human skin fibroblast cells (HSF) and their selectivity index values were calculated and shown in Table 2. The five most potent derivatives (4j, 4f, 5i, 5f and 4i) showed relatively low toxicity with IC50 values on HSF cells of 9.58, 4.18, 3.63, 3.59 and 3.26 μM, respectively. Among them, compound 4j was the safest hybrid (SI = 9.9), displaying the highest selectivity and potency towards melanoma cells A375.
| Compound | BRAFV600E (IC50, μM) | VEGFR-2 (IC50, μM) |
|---|---|---|
| 4j | 1.03 ± 0.71 | 0.64 ± 0.08 |
| Sorafenib | 2.86 ± 0.45 | 1.94 ± 0.05 |
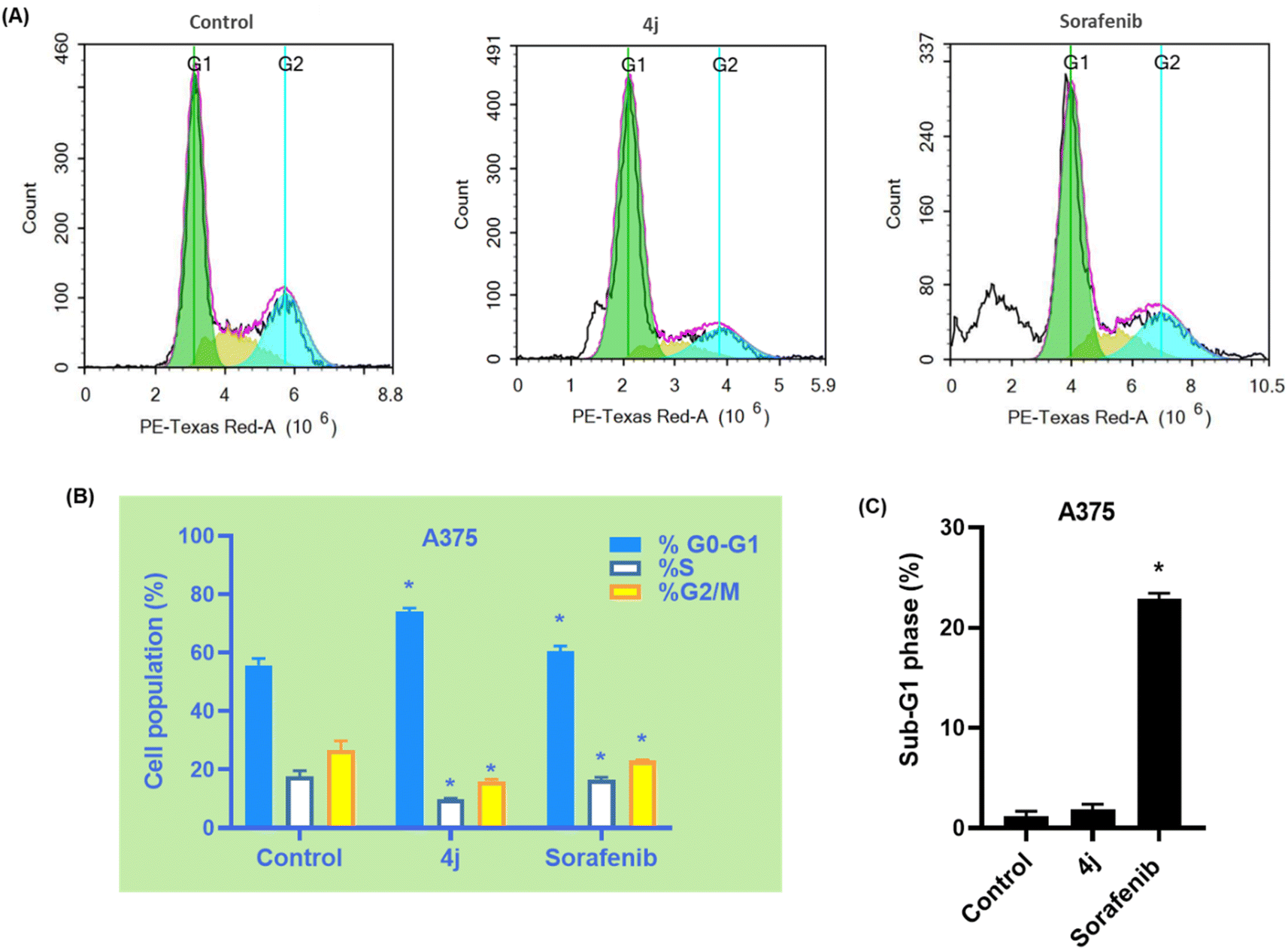 | ||
| Fig. 3 (A) Cell cycle distribution in A375 cells after treatment with 4j and sorafenib for 48 h determined using DNA cytometry analysis compared with a control (untreated cells). (B) Quantification of the percentage of cells in each phase of the cell cycle is depicted as a bar graph of mean ± SD; n = 3. (C) Sub-G1 phase was plotted alone as a proportion of total events. * Statistically significant difference from the control (P < 0.05). | ||
Treatment with sorafenib resulted in observable cell cycle arrest at the G0/G1 phase from 55.71% to 60.43%, with almost the same alterations in the S phase from 17.62% to 16.55% and G2/M phase from 26.67% to 23.02% compared to the control cells (Fig. 3A and B), respectively. However, treatment with sorafenib resulted in significant cell death (P < 0.05) manifested by a considerable increase in the cell population of the Pre-G phase from 1.21% to 22.88% compared with the control cells (Fig. 3C). These results are consistent with that reported for sorafenib, which significantly inhibits the growth of cancer cells through the accumulation of cells in the pre-G1 phase.51
These findings shed light on the mode of action of coumarin derivatives as antiproliferative agents (inducing G0/G1 arrest), which differs from the cytotoxic effect of sorafenib (inducing pre-G1 arrest) and highlight the potential use of coumarins as effective anticancer agents in melanoma treatment. This can be explained by the fact that the G1 phase is a phase that precedes DNA replication, in which the cellular conditions influence cell cycle progression, causing DNA to either rebuild in a cell or initiate apoptosis, and several studies address the role of G0/G1 phase arrest in intensifying cellular apoptosis.52
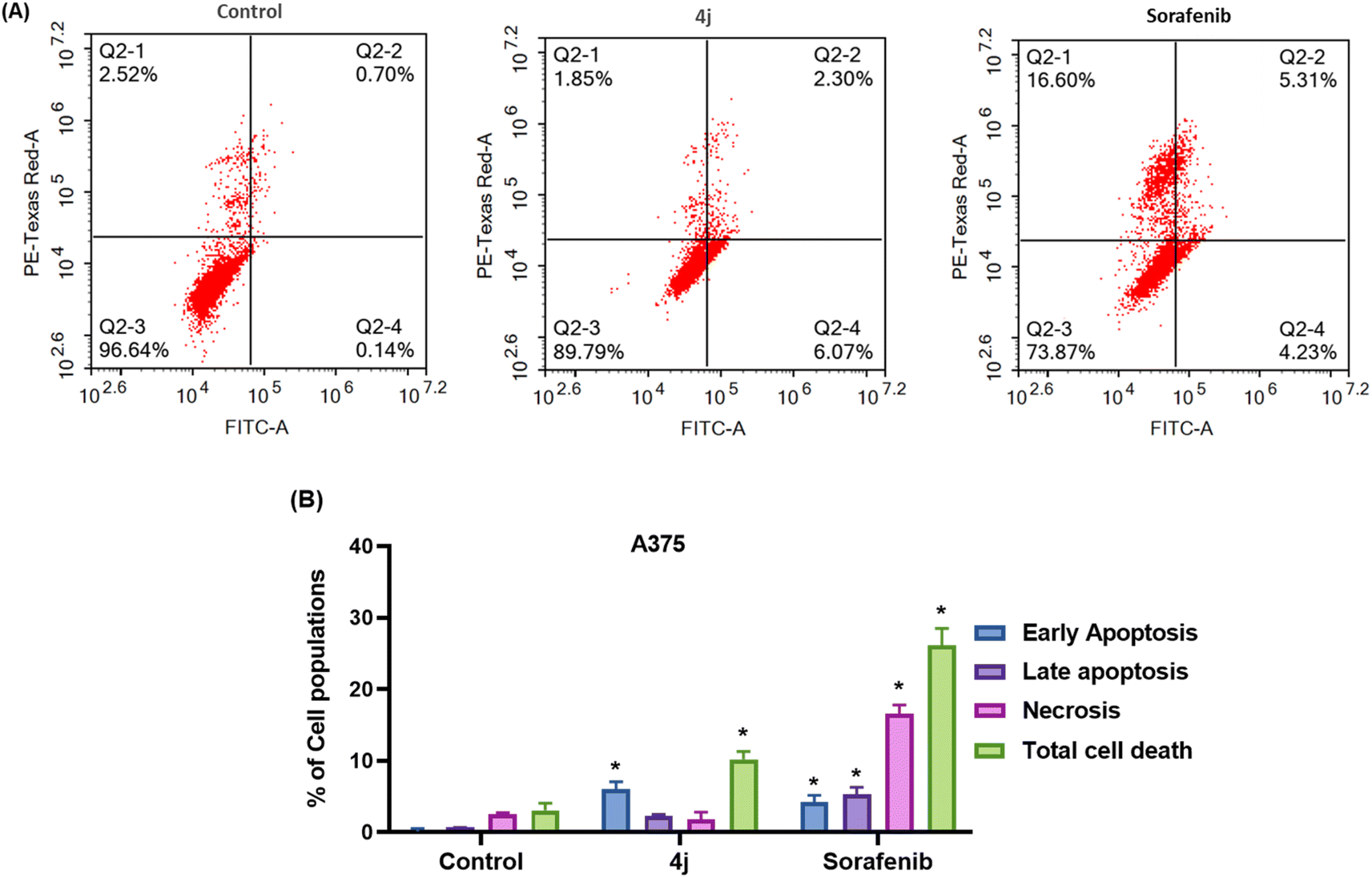 | ||
| Fig. 4 (A) Evaluation of cell death modality (apoptosis/necrosis) after treatment with 4j and sorafenib for 48 h in A375 cells, followed by double staining with Annexin-FITC/PI. (B) Different cell populations plotted as the percentage of total events. Data are presented as mean ± SD; n = 3. Total cell death calculated by combining apoptosis and necrosis. * Statistically significant difference from the control (P < 0.05). | ||
Compound 4j induced significant (p < 0.05) total cell death of A375 cells (nearly 10.22%) by mainly apoptotic death with early apoptosis of 6.07% and late apoptosis of 2.3% compared with the control untreated A375 cells with 0.14% early and 0.7% late apoptosis, respectively (Fig. 4A). These findings show that most of the cells underwent cell death by the apoptotic mode, suggesting that 4j exerted its anticancer activity through the apoptosis route. This is consistent with the studies that reported apoptosis as a mechanism of cell death in cancer treatment by coumarin compounds.48 However, treatment with 4j did not induce notable necrotic cell death compared to the control cells (2.52% and 1.85%, respectively). This indicates that 4j at its IC50 concentration can induce apoptosis, mainly early apoptosis, of A375 melanoma cells but not necrosis. Furthermore, it arrested cells in the G1 phase, which promoted cell apoptosis in melanoma cells. Meanwhile, treatment with sorafenib showed a dramatic decrease in cell viability. Sorafenib treatment produced greater cell death than 4j in A375, which was mainly necrotic (16.6%) (p < 0.05), and nearly the same percentage of apoptotic cell death (9.6%) compared with the 4j-treated cells. This can be related to the sorafenib dose, which only modestly boosted apoptosis relative to necrosis.53 It is also worth noting that the amount of necrosis did not correlate with apoptosis, indicating that distinct mechanisms are involved.54
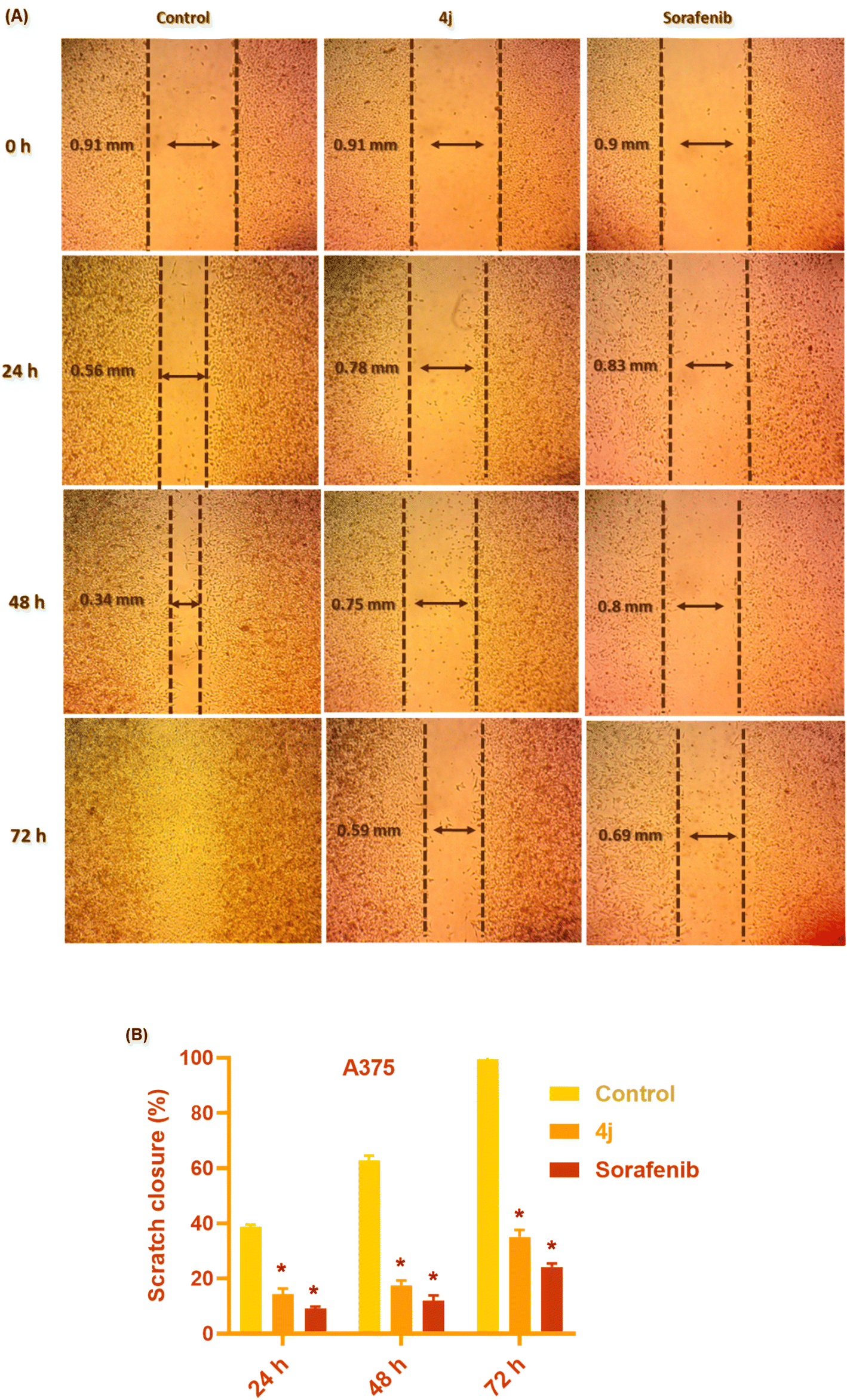 | ||
| Fig. 5 Effect of 4j and sorafenib on the migration of A375 cells. (A) Scratch widths were measured after 4j and sorafenib treatment at 24, 48, and 72 h. (B) Data are plotted as scratch closure % at each time interval and presented as triplicates. * Statistically significant difference from the control (P < 0.05). | ||
Compound 4j displayed a significant (P < 0.05) anti-metastasis effect by inhibiting A375 cell migration at all time intervals compared with the control untreated cells. Also, it showed a comparable effect to sorafenib anti-migratory activity. After 24 h, 4j treatment postponed the migration tendency of A375 cancer cells, as evidenced by the 14.28% ± 2.13% delay in cell-free zone closure compared to 38.68% ± 0.89% for the untreated cells. After 48 h of 4j treatment, a similar pattern was observed, with a delayed scratch closure of 17.57% ± 1.72% compared to 62.77% ± 1.78% in the control cells, demonstrating that 4j may exert an anti-migratory effect in melanoma cancer cells. The following day (72 h), complete scratch closure was observed in the control group (100%); however, the scratch remained open in the 4j-treated cells, with the scratch closure of 35.16% ± 2.45%, indicating that 4j could exert a potent anti-migratory effect at this concentration (0.5 μM, nearly 1/2 IC50 value). Similarly, compared to the untreated cells, the sorafenib-treated melanoma cells demonstrated delayed migration, which was slightly more potent than the 4j-treated cells, with scratch closure of 9.15% ± 0.64%, 12.07% ± 1.8%, and 24.17% ± 1.32% at 24, 48, and 72 h, respectively.
2.3. Molecular modeling study
| Compound | ΔG (kcal mol−1) | GBVI/WSA ΔG (kcal mol−1) | RMSD (Å) | TPSA (Å2) | Interacted residues |
|---|---|---|---|---|---|
| 4a | −8.16 | −38.3 | 1.6 | 93.4 | Ile463, Asp594, Glu501 |
| 4b | −6.45 | −30.75 | 2.75 | 84.6 | Trp531, Asp594 |
| 4c | −8.77 | −38.91 | 2.59 | 84.6 | Asp594, Ile463 |
| 4d | −8.57 | −42.5 | 1.2 | 93.4 | Cys532 |
| 4e | −6.37 | −30.89 | 2.7 | 84.6 | Trp531 |
| 4f | −9.04 | −41.35 | 1.44 | 84.6 | Phe593, Cys532 |
| 4g | −8.82 | −39.05 | 1.41 | 93.4 | Cys532, Ala481, Lys483 |
| 4h | −7.54 | −35.62 | 2.35 | 84.6 | Trp531, Cys532 |
| 4i | −9.1 | −41.39 | 1.37 | 84.6 | Trp531, Phe583 |
| 4j | −9.50 | −44.62 | 1.26 | 102.6 | Asp594, Thr529 |
| 5a | −8.17 | −37.08 | 1.26 | 93.4 | Ile463, Glu501 |
| 5b | −7.66 | −35.21 | 2.4 | 84.6 | Asn581, Asp594 |
| 5c | −9.09 | −41.45 | 1.79 | 84.6 | Ile463 |
| 5d | −8.52 | −38.46 | 1.04 | 93.4 | Cys532, Trp531, Asp594 |
| 5e | −8.07 | −37.5 | 2.5 | 84.6 | Ile563, Ala481 |
| 5f | −9.08 | −41.72 | 1.68 | 84.6 | Cys532, Trp531 |
| 5g | −8.26 | −38.02 | 1.86 | 93.4 | Cys532, Ala481, Lys483 |
| 5h | −8.83 | −38.55 | 1.41 | 84.6 | Trp531, Cys532 |
| 5i | −9.11 | −41.98 | 0.82 | 84.6 | Phe593, Cys532 |
| 5j | −8.92 | −41.36 | 2.72 | 102.6 | Cys532, Ile463, Lys481 |
| 6a | −8.09 | −34.05 | 1.6 | 93.4 | Ile463, Asp594 |
| 6b | −6.85 | −30.55 | 2.76 | 84.6 | Asn581 |
| 6c | −8.59 | −38.87 | 1.28 | 84.6 | Ile463, Asp594 |
| 6d | −8.20 | −38.42 | 1.52 | 93.4 | Cys532, Ala481, Lys483 |
| 6e | −8.03 | −34.35 | 1.44 | 84.6 | Trp531, Cys532 |
| 6f | −8.75 | −42.11 | 1.65 | 84.6 | Trp531, Phe583 |
| 6g | −8.04 | −37.66 | 1.27 | 93.4 | Cys532, Ala481, Lys483 |
| 6h | −7.46 | −34.79 | 2.5 | 84.6 | Trp531, Cys532 |
| 6i | −8.72 | −39.11 | 1.27 | 84.6 | Trp531, Phe583 |
| 6j | −8.65 | −38.87 | 1.22 | 102.6 | Ile463, Lys481 |
| SB-590885 | −8.66 | −43.54 | 0.95 | 86.6 | Cys532, Ile463, Glu501 |
| Sorafenib | −8.26 | −42.93 | 0.97 | 92.36 | Cys532, Asp594, Glu501 |
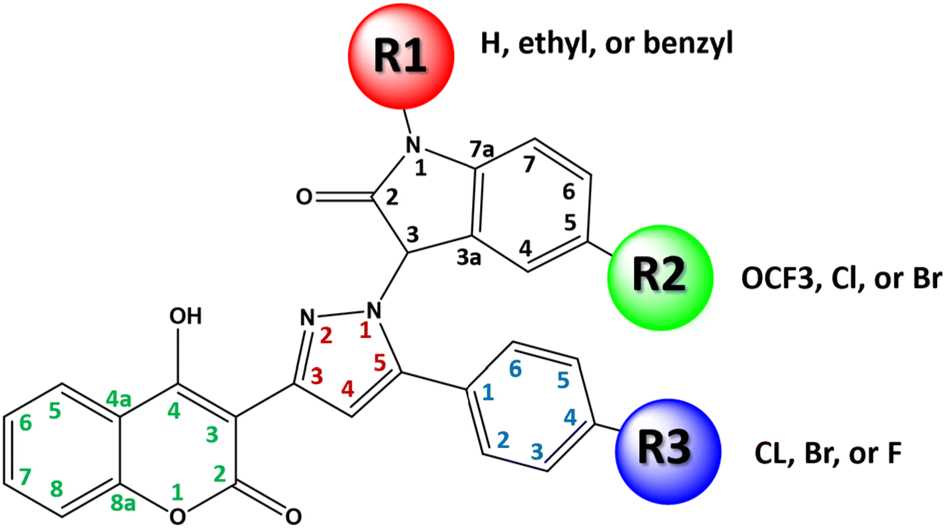 | ||
| Fig. 6 Three main positions R1, R2, and R3 directly correlated with the anti-melanoma activity. | ||
(i) Compounds with TPSA = 102.6 Å2, this group includes all compounds with trifluoromethoxy group at R2, halo atom (F, Cl, Br) at R3, and R1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, e.g., 4j, 5j and 6j.
H, e.g., 4j, 5j and 6j.
(ii) Compounds with TPSA = 93.4 Å2, this value is close to TPSA of sorafenib (92.36 Å2), e.g., compounds 4d, 5d, 4g, 5g, 4a, 6d, 5a, 6g and 6a. All these compounds showed potent anticancer activity.
(iii) Compounds with TPSA = 84.6 Å2, this value is close to that of the reference inhibitor SB-590885 (86.6 Å2), e.g., 5i, 5f, 4f, 4i, 5c, 6f, 4c, 6c, 5h, 6i, 5e and 6e. All these compounds also showed potent anticancer activity.
2.3.2.1 Class I: compounds with no substitution at N1-indolin (R1 = H) and halo substitution at R2 and/or R3. It was observed that the number of fluoro atom substitutions at R2 and R3 is very important for cytotoxic activity. The most active compound 4j (IC50 = 0.96 μM) with the highest number of F atoms (4 atoms) exhibited the highest TPSA value = 102.6 Å2 among the compounds and the reference drugs given that the fluoro atom increases the lipophilicity and TPSA value. The replacement of F in compound 6 with Br or Cl yielded compound 6j and 5j with a decrease in cytotoxic activity (IC50 = 4.37 and 4.17 μM, respectively). A further decrease in the number of F atoms resulted in a decrease in activity, for example, when the OCF3 in compound 4j was replaced with Br it yielded compound 4g (IC50 = 5.85 μM). This confirmed that the number of F atoms is directly proportional to the cytotoxic activity. The type of halo substitution at R2 and R3 can be ranked according to activity, following the order of (Cl&F) > (Cl&Cl) > (Br&Br) and this can be observed in compound 4d with Cl at R2 and F at R3 (IC50 = 4.3 μM), which is more active than compound 5d with Cl substitution at R2 and R3 (IC50 = 5.43 μM), while compound 6g with a Br atom at R2 and R3 was less active (IC50 = 7.32 μM). In the case of compounds 4a, 5a, and 6a having only one halo atom at R3 in the phenylpyrazolyl moiety, it was deduced that the presence of F substitution increased the activity, following the order of compound 4a (IC50 = 6.25 μM) > compound 5a (IC50 = 7.13 μM) > compound 6a (IC50 = 8.45 μM), which confirmed the importance of the presence of fluoro substitution, as discussed in the previous section. The compounds of this class (4a, 5a and 6a) exhibited the same TPSA value = 93.4 Å2, which is close to that of sorafenib (92.36 Å2).
2.3.2.2 Class II: compounds with benzyl group at N1-indolin (R1 = benzyl) and halo substitution at R2 and/or R3. The presence of a benzyl group in compounds 4c, 5c and 6c was very important due to the hydrophobic interactions provided by the phenyl ring, which were oriented between hydrophobic residues Trp531, Phe583, and Ile463. Also, the methylene –CH2 of the benzyl group was important for van der Waals formation to support the fitting of these compounds. The benzyl group increased the lipophilicity and decreased the TPSA value of these derivatives to reach 84.6 Å2, which is close to that of SB-590885 = 86.6 Å2 (Table 4) and may be correlated with their activity. The activity of 4c, 5c and 6c was improved compared to the compounds in the previous class with IC50 values of 3.8, 4.6, and 4.84 μM, respectively. Moreover, the presence of bromo substitution at R2 and R3 decreased the activity, as observed in compound 6i (IC50 = 5.47 μM), where the presence of fluoro substitution at the 4-position of the phenyl ring (R3) with either chloro or bromo substitution at the 5-position of the indolin-2-one scaffold (R2) improved the activity, as in compound 4f (IC50 = 1.1 μM) and compound 4i (IC50 = 1.28 μM). The substitution with two chloro atoms at both R2 and R3 retained the improved activity as in compound 5f (IC50 = 1.09 μM). The presence of chloro substitution at R2 and bromo substitution at R3 decreased the activity as in compound 6f (IC50 = 4.36 μM). The TPSA value of these compounds was 84.6 Å2, which is close to that of SB-590885 = 86.6 Å2 (Table 4).
2.3.2.3 Class III: compounds with ethyl at N1-indolin (R1 = ethyl). This class showed some deviation from the previous observations, where the best activity was achieved by compound 5h (R2
Br and R3Cl) with an IC50 value of 5.44 μM, which may need future investigation. In addition, dichloro substitution at R2 and R3 in compound 5e showed good activity (IC50 = 7.32 μM). However, the TPSA of these compounds was 84.6 Å2, which is close to that of SB-590885.
N of the pyridine ring, between the dimethyl amino group in the side chain and Ile463 and between the hydroxyl group of oxime and Glu501 (Fig. 7A). Sorafenib also interacted by three hydrogen bonds, where one was formed between –NH of the urea group and Cys532, the other bond between the acetamide side chain and Asp594 and the last one with Glu501 (Fig. 7B). The molecular docking of 4j with the highest ΔG value of −9.50 kcal mol−1 against BRAFV600E, revealed that the 4-hydroxy group formed a hydrogen bond with the –COOH group of Asp594 and the –NH of indoline formed a hydrogen bond with CO of Thr529. The 5-(trifluoromethoxy) indolin-2-one scaffold was oriented and surrounded by hydrophobic residues, i.e., Phe583, Ile463, and Trp531, with the possible formation of hydrophobic interactions (Fig. 7C and Table 4).
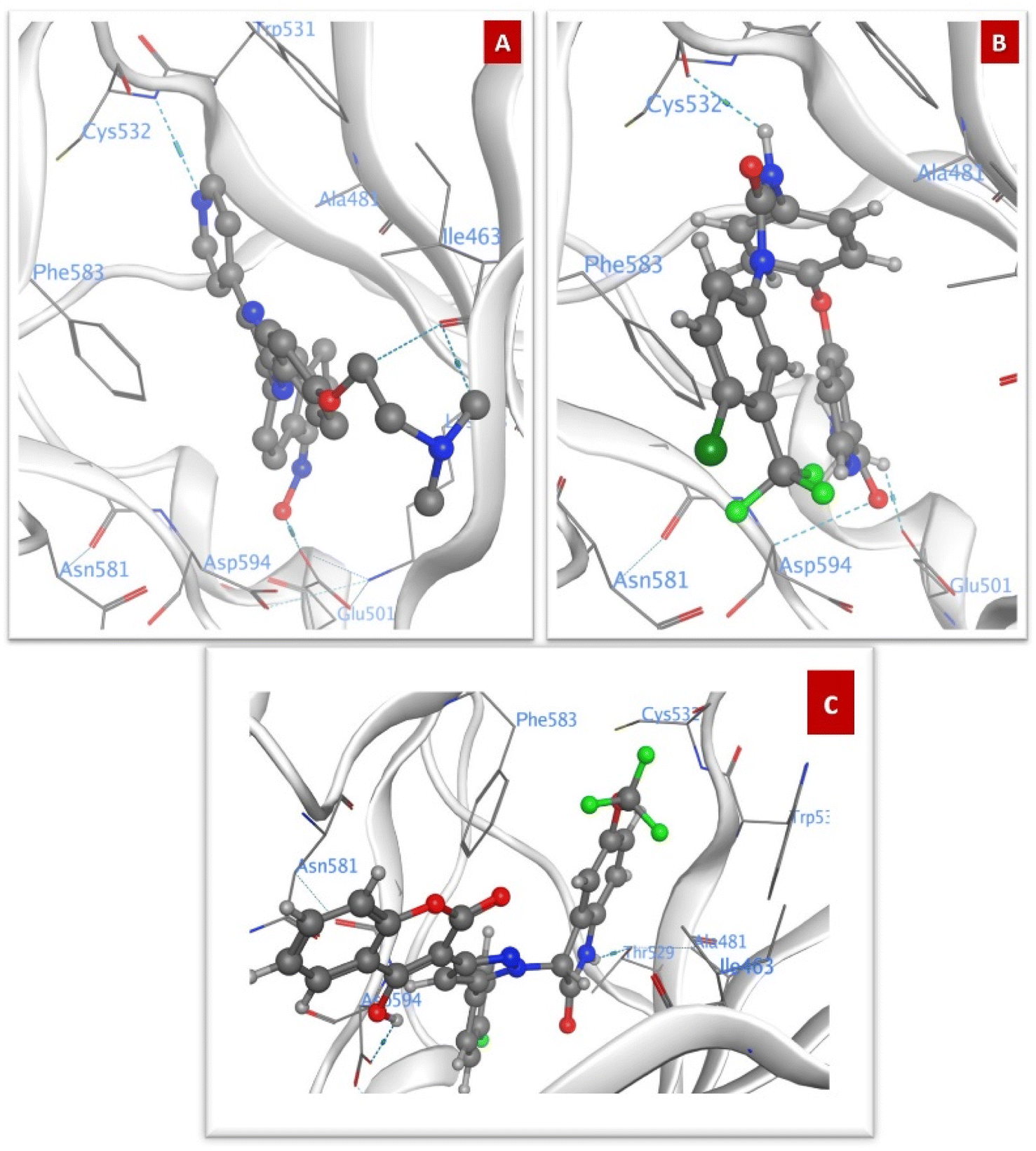 | ||
| Fig. 7 Docking poses against BRAFV600E (A) SB590885, (B) Sorafenib, and (C) 4j. | ||
Compound 5j showed the same orientation of the 5-(trifluoromethoxy) indolin-2-one scaffold surrounded by all the hydrophobic residues. It formed hydrogen bonds with Cys532, Ile463, and Lys481. Compound 6j also formed the same interactions as 5j with Ile463 and Lys481. Compound 4d formed one hydrogen bond with Cys532, while compound 5d formed three hydrogen bonds with Cys532, Trp531, and Asp594. Compound 6g showed three hydrogen bonds with Cys532, Ala481 and Lys483. The coumarin ring in compound 4a formed two hydrogen bonds with Asp594 and Glu501. The 4-bromophenyl ring in compound 5a showed hydrophobic interactions with Ile463. The –NH of the pyrazole in compounds 4a, 5a, and 6a interacted by a hydrogen bond with CO of Ile463 and the CO of coumarin in compound 6a showed a hydrogen bond with Asp594 (Fig. S1A–I†).
The –CH2 of benzyl moiety in compounds 4c and 6c formed van der Waals interactions with Ile463, while the same group in compound 4f formed hydrophobic interactions with Phe583. The phenyl ring of the benzyl moiety of compound 6i showed π–π interactions with Trp531 and Phe583. The –CH2 of the benzyl moiety in compound 5f formed hydrophobic interactions with Trp531 and Cys532 (Fig. S2A–F†). The docking of compounds with R1 = ethyl and R3 = halo substitution showed that compound 5b formed two hydrogen bonds with Asn581 and Asp594. Compound 6b showed a hydrogen bond with Asn581. The ethyl group in compound 4b contributed to van der Waals interactions with Trp531 and the CO of coumarin formed a hydrogen bond with Asp594. The pyrazole ring in compound 5e contributed to van der Waals interactions with Ile463 and the CO of coumarin formed a hydrogen bond with Ala481. The ethyl group in compound 6h formed two van der Waals interactions with Trp531 and Cys532. Compound 4e showed one van der Waals interaction between the ethyl group at R1 and Trp531 (Fig. S3A–F†).
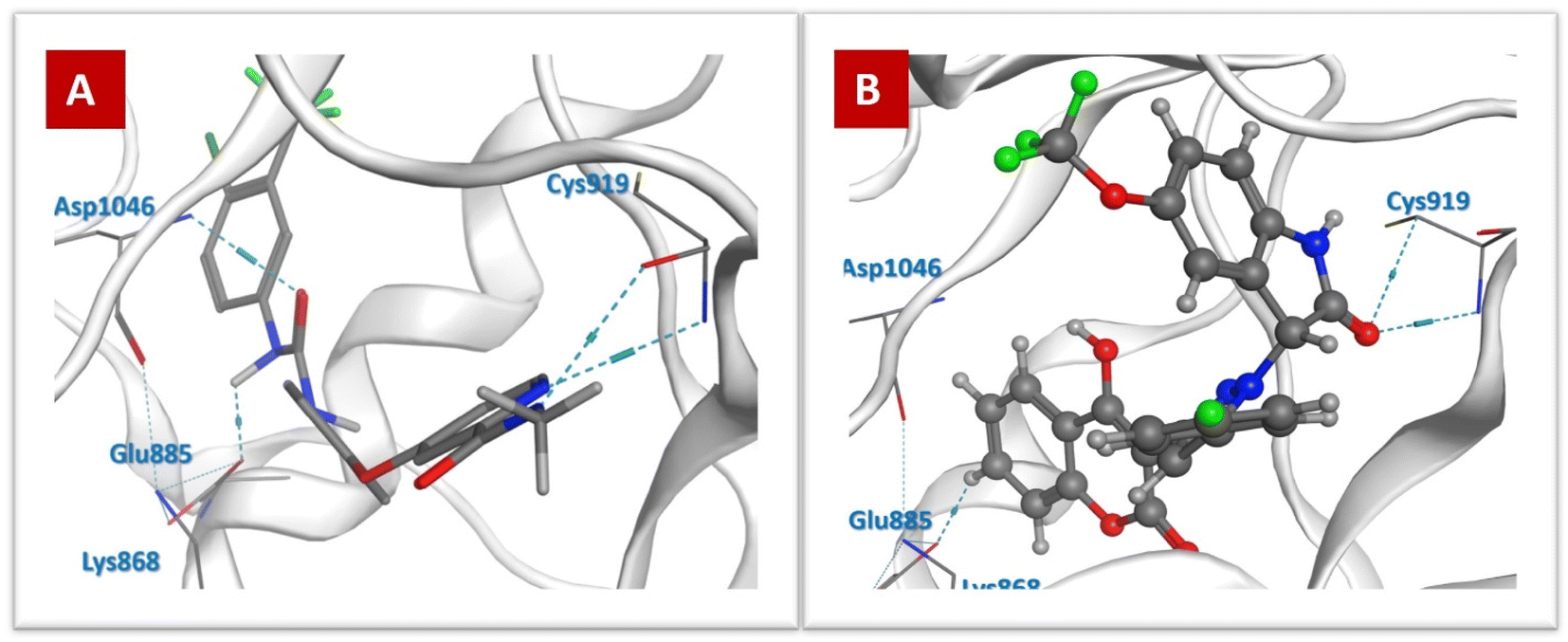 | ||
| Fig. 8 Docking poses against VEGFR-2: (A) Sorafenib and (B) 4j. | ||
| Compound | ΔG (kcal mol−1) | GBVI/WSA ΔG (kcal mol−1) | RMSD (Å) | Interacted residues |
|---|---|---|---|---|
| 4a | −7.65 | −36.99 | 1.35 | Glu885 |
| 4b | −7.04 | −36.61 | 2.05 | Lys868 |
| 4c | −8.23 | −36.6 | 1.83 | Cys919, Lys868 |
| 4d | −7.42 | −39.21 | 1.57 | Cys919 |
| 4e | −7.18 | −36.57 | 1.74 | Lys868 |
| 4f | −8.21 | −33.3 | 1.95 | Cys919 |
| 4g | −7.98 | −38.44 | 1.92 | Cys919 |
| 4h | −7.17 | −35.63 | 1.82 | Lys868, Glu885 |
| 4i | −7.86 | −40.31 | 1.81 | Cys919 |
| 4j | −8.47 | −37.79 | 1.61 | Cys919, Glu885 |
| 5a | −7.88 | −39.58 | 1.05 | Glu885 |
| 5b | −7.17 | −37.35 | 2.33 | Lys868 |
| 5c | −8.02 | −34.73 | 1.31 | Cys919, Lys868 |
| 5d | −7.53 | −35.11 | 1.39 | Cys919 |
| 5e | −7.71 | −34.67 | 2.12 | Lys868, Glu885 |
| 5f | −7.62 | −37.01 | 1.70 | Asp1046 |
| 5g | −7.27 | −36.17 | 1.42 | Cys919 |
| 5h | −7.44 | −36.83 | 1.76 | Lys868, Glu885 |
| 5i | −7.79 | −35.65 | 1.57 | Cys919, Lys868 |
| 5j | −7.75 | −36.29 | 2.32 | Cys919, Glu885 |
| 6a | −7.85 | −37.38 | 1.44 | Glu885 |
| 6b | −7.15 | −32.09 | 1.54 | Lys868, Glu885 |
| 6c | −7.10 | −35.68 | 1.11 | Cys919, Lys868 |
| 6d | −7.54 | −38.08 | 2.77 | Cys919 |
| 6e | −7.36 | −37.52 | 1.87 | Lys868, Glu885 |
| 6f | −7.18 | −37.34 | 2.1 | Cys919, Lys868 |
| 6g | −7.64 | −32.51 | 1.44 | Cys919 |
| 6h | −7.60 | −37.66 | 1.29 | Lys868, Glu885 |
| 6i | −7.95 | −35.55 | 1.72 | Cys919, Lys868 |
| 6j | −7.70 | −35.81 | 1.93 | Cys919, Glu885 |
| Sorafenib | −9.44 | −46.54 | 1.47 | Asp1046, Cys919, Glu885 |
The aforementioned information showed that the results of the in silico studies were consistent with that of the biological assays, where the most active compound 4j with the IC50 value of 0.96 μM against A375 cells and dual inhibition properties against BRAFV600E/VEGFR-2 (IC50 = 1.033 and 0.64 μM, respectively), revealed the highest ΔG value of −9.5 kcal mol−1 against BRAFV600E and showed a significant ΔG of −8.47 kcal mol−1 towards VEGFR-2. Moreover, compound 4j displayed different types of interactions with essential catalytic sites in the active region of BRAFV600E and VEGFR-2.
3. Conclusion
In this study, a series of new coumarin derivatives bearing a pyrazolylindolin-2-one core was synthesized and evaluated for their anti-proliferative effects against human melanoma A375 cells as dual BRAFV600E and VEGFR-2 inhibitors. Most of the compounds displayed potent antiproliferative activity against A375 cells with IC50 values in the range of 0.96–8.99 μM. Among them, compounds 4j, 4f, 4i, 5f and 5i demonstrated potent cytotoxic activity (IC50 = 0.96–1.28 μM) and good safety profile. Furthermore, compound 4j exerted greater potent dual inhibitory activity against BRAFV600E/VEGFR-2 than the reference drug sorafenib. The anticancer effect of 4j was correlated with the inhibition of cell proliferation, boosting cell cycle arrest at the G0/G1 phase, induction of apoptosis, and inhibition of migration. Docking simulation was carried out for the active compounds to predict their binding modes in the BRAFV600E and VEGFR-2 protein active sites, revealing various interactions with the binding residues in the active site. The SAR study showed the correlation between the number of fluoro atoms and both TPSA and cytotoxic activity.4. Experimental
4.1. Chemistry
Melting points were determined on an Electrothermal IA 9000 apparatus and were uncorrected. Elemental analyses were carried out at the Micro-analytical Central Services Laboratory, Faculty of Science, Cairo University, Egypt. 1H NMR and 13C NMR spectra were measured using a Bruker Avance II™ 400 MHz spectrometer (Bruker Biospin AG, Fällanden, Switzerland) in Prague, Czech Republic. 13C NMR spectra of compounds 4e, 4h and 5h could not be acquired due to their precipitation from the solvent used (DMSO). The reactions were monitored by TLC (silica gel, aluminium sheets 60 F254, Merck) using chloroform/methanol (9.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 v/v) as the eluent and sprayed with iodine–potassium iodide solution. The key intermediates 3a–c47,48 and isatin derivatives Ia–j59,60 and 4a47 were previously prepared.
:0.5 v/v) as the eluent and sprayed with iodine–potassium iodide solution. The key intermediates 3a–c47,48 and isatin derivatives Ia–j59,60 and 4a47 were previously prepared.
4.1.1.1 1-Ethyl-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 4b. Yellow crystals, m.p. 277–278 °C, yield (0.35 g, 73%). 1H NMR (400 MHz, DMSO-d6): δ = 1.15 (t, 3H, J = 7.2 Hz, CH3), 3.72 (q, 2H, J = 6.4 Hz, CH2), 6.32 (s, 1H, CH-indole), 7.06–7.09 (m, 1H, H–Ar), 7.18–7.28 (m, 4H, H–Ar), 7.34–7.44 (m, 5H, H–Ar), 7.64–7.70 (m, 2H, H–Ar), 7.84 (d, 1H, J = 8.4 Hz, H–Ar), 13.06 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 12.79, 34.95, 60.71, 95.10, 106.89, 109.85, 115.49, 116.44, 116.66, 116.78, 123.21, 124.04, 124.87, 125.06, 125.24, 130.59, 132.06, 132.15, 143.20, 145.46, 147.77, 152.43, 159.70, 162.10, 163.41, 164.56, 171.26. Anal. calcd for C28H20FN3O4 (481.47): C, 69.85; H, 4.19; N, 8.73. Found: C, 70.00; H, 4.37; N, 8.89.
4.1.1.2 1-Benzyl-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 4c. Yellow crystals, m.p. 220–221 °C, yield (0.4 g, 74%). 1H NMR (400 MHz, DMSO-d6): δ = 4.91 (d, 1H, J = 15.6 Hz, CHa-benzyl), 5.01 (d, 1H, J = 16.0 Hz, CHb-benzyl), 6.47 (s, 1H, CH-indole), 6.99 (d, 1H, J = 8.0 Hz, H–Ar), (t, 1H, J = 7.6 Hz, H–Ar), 7.26 (s, 1H, H–Ar). 7.30–7.46 (m, 11H, H–Ar). 7.67–7.71 (m, 1H, H–Ar), 7.79 (brs, 2H, H–Ar), 7.87 (d, 1H, J = 6.8 Hz, H–Ar), 13.02 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.42, 60.42, 95.36, 106.83, 110.44, 115.52, 116.57, 116.79, 116.83, 123.48, 124.01, 124.99, 125.09, 125.13, 127.49, 127.99, 129.14, 130.48, 132.10, 132.19, 133.70, 136.15, 143.50, 145.86, 148.13, 152.55, 160.10, 162.17, 163.62, 164.62, 171.99. Anal. calcd for C33H22FN3O4 (543.15): C, 72.92; H, 4.08; N, 7.73. Found: C, 73.09; H, 4.22; N, 7.91.
4.1.1.3 5-Chloro-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 4d. Pale yellow powder, m.p. 294–295 °C, yield (0.38 g, 78%). 1H NMR (400 MHz, DMSO-d6): δ = 6.24 (s, 1H, CH-indole), 6.96 (d, 1H, J = 8.0 Hz, H–Ar), 7.22 (s, 1H, H–Ar), 7.36–7.44 (m, 6H, H–Ar), 7.66–7.70 (m, 1H, H–Ar), 7.76–7.79 (m, 2H, H–Ar), 7.88 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 10.98 (s, 1H, NH), 13.13 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.70, 95.56, 106.73, 112.17, 115.50, 116.47, 116.69, 116.81, 124.11, 124.90, 125.11, 124.15, 125.55, 126.69, 127.72, 130.36, 132.16, 132.32, 133.69, 142.09, 145.84, 148.04, 152.53, 160.09, 163.61, 173.09 Anal. calcd for C26H15ClFN3O4 (487.86): C, 64.01; H, 3.10; N, 8.61. Found: C, 64.19; H, 3.24; N, 8.77.
4.1.1.4 5-Chloro-1-ethyl-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 4e. Yellow powder, m.p. 297–298 °C, yield (0.37 g, 72%). 1H NMR (400 MHz, DMSO-d6): δ = 1.13 (t, 3H, J = 7.2 Hz, CH3), 3.71 (q, 2H, J = 7.2 Hz, CH2), 6.32 (s, 1H, CH-indole), 7.22–7.26 (m, 2H, H–Ar), 7.36–7.45 (m, 4H, H–Ar), 7.48–7.51 (m, 2H, H–Ar), 7.67–7.75 (m, 3H, H–Ar), 7.88 (dd, 1H, J = 1.2 and 7.6 Hz, H–Ar), 13.03 (s, 1H, OH). Anal. calcd for C28H19ClFN3O4 (515.91): C, 65.18; H, 3.71; N, 8.14. Found: C, 65.36; H, 3.90; N, 8.32.
4.1.1.5 1-Benzyl-5-chloro-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 4f. Violet crystals, m.p. 223–225 °C, yield (0.45 g, 78%). 1H NMR (400 MHz, DMSO-d6): δ = 4.90 (d, 1H, J = 16.0 Hz, CH-benzyl), 5.02 (d, 1H, J = 16.0 Hz, CH-benzyl), 6.47 (s, 1H, CH-indole), 7.01 (d, 1H, J = 8.8 Hz, H–Ar), 7.26 (s, 1H, H–Ar). 7.29–7.45 (m, 10H, H–Ar). 7.53 (brs, IH, H–Ar) 7.66–7.70 (m, 1H, H–Ar), 7.80–7.83 (m, 2H, H–Ar), 7.87 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 12.97 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.49, 60.17, 95.36, 106.89, 111.87, 115.47, 116.53, 116.74, 116.81, 124.03, 124.98, 125.07, 125.10, 125.49, 127.06, 127.45, 127.65, 128.06, 129.17, 130.33, 132.22, 132.30, 133.71, 135.81, 142.49, 146.10, 148.35, 152.54, 160.09, 162.21, 163.64, 164.66, 171.75. Anal. calcd for C33H21ClFN3O4 (577.12): C, 68.57; H, 3.66; N, 7.27. Found: C, 68.75; H, 3.83; N, 7.44.
4.1.1.6 5-Bromo-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 4g. White powder, m.p. 295–296 °C, yield (0.36 g, 68%). 1H NMR (400 MHz, DMSO-d6): δ = 6.23 (s, 1H, CH-indole), 6.91 (d, 1H, J = 8.4 Hz, H–Ar), 7.21 (s, 1H, H–Ar), 7.35–7.43 (m, 4H, H–Ar), 7.52 (s, 2H, H–Ar), 7.65–7.69 (m, 1H, H–Ar), 7.77(brt, 2H, H–Ar), 7.87 (d, 1H, J = 7.6 Hz, H–Ar), 10.98 (s, 1H, NH), 13.14 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.61, 95.41, 106.72, 112.67, 114.30, 115.50, 116.46, 116.68, 116.81, 124.12, 124.90, 125.15, 128.09, 128.22, 132.16, 132.25, 133.21, 133.69, 142.51, 145.85, 148.05, 152.53, 162.16, 163.62, 172.97. Anal. calcd for C26H15BrFN3O4 (532.31): C, 58.66; H, 2.84; N, 7.89. Found: C, 58.85; H, 3.01; N, 8.09.
4.1.1.7 5-Bromo-1-ethyl-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 4h. Yellow crystals, m.p. 287–289 °C, yield (0.4 g, 71%). 1H NMR (400 MHz, DMSO-d6): δ = 1.03 (t, 3H, J = 6.0 Hz, CH3), 3.66 (q, 2H, J = 7.2 Hz, CH2), 6.29 (s, 1H, CH-indole), 7.09 (s, 1H, H–Ar), 7.16–7.25 (m, 4H, H–Ar), 7.39–7.46 (m, 2H, H–Ar), 7.69 (d, 1H, J = 2.0 Hz, CH2), 7.80–7.84 (m, 2H, H–Ar), 7.94–7.98 (m, 2H, H–Ar), 13.31 (s, 1H, OH). Anal. calcd for C28H19BrFN3O4 (559.05): C, 60.01; H, 3.42; N, 7.50. Found: C, 60.12; H, 3.51; N, 7.67.
4.1.1.8 1-Benzyl-5-bromo-3-(5-(4-fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 4i. Yellow powder, m.p. 247–249 °C, yield (0.41 g, 66%). 1H NMR (400 MHz, DMSO-d6): δ = 4.90 (d, 1H, J = 16.0 Hz, CHa-benzyl), 5.02 (d, 1H, J = 16.00 Hz, CHb-benzyl), 6.47 (s, 1H, CH-indole), 6.96 (d, 1H, J = 8.4 Hz, H–Ar), 7.26 (s, 1H, H–Ar), 7.29–7.45 (m, 9H, H–Ar), 7.54 (dd, 1H, J = 1.6 and 8.4 Hz, H–Ar), 7.63 (brs, 1H, H–Ar), 7.68–7.72 (m, 1H, H–Ar), 7.80–7.83 (m, 2H, H–Ar), 7.88 (dd, 1H, J = 1.8 and 8.0 Hz, H–Ar), 13.00 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.45, 60.09, 95.37, 106.87, 112.37, 115.31, 115.49, 116.53, 116.75, 116.84, 124.06, 125.01, 125.10, 127.39, 127.43, 128.07, 128.15, 129.18, 132.23, 132.32, 133.20, 133.75, 135.80, 142.92, 148.34, 152.56, 160.10, 162.21, 163.66, 164.66, 171.65. Anal. calcd for C33H21BrFN3O4 (621.06): C, 63.68; H, 3.40; N, 6.75. Found: C, 63.85; H, 3.58; N, 6.86.
4.1.1.9 3-(5-(4-Fluorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-5 (trifluoromethoxy) indolin-2-one 4j. Yellow crystals, m.p. 293–294 °C, yield (0.4 g, 74%). 1H NMR (400 MHz, DMSO-d6): δ = 6.32 (s, 1H, CH-indole), 7.02 (d, 1H, J = 8.4 Hz, H–Ar), 7.22 (s, 1H, H–Ar), 7.35–7.44 (m, 6H, H–Ar), 7.66–7.70 (m, 1H, H–Ar), 7.76 (brs, 2H, H–Ar), 7.87 (dd, 1H, J = 0.8 and 7.6 Hz, H–Ar), 11.02 (s, 1H, NH), 13.11 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.96, 95.40, 106.88, 111.62, 115.49, 116.39, 116.61, 116.82, 119.56, 121.85, 123.78, 124.07, 124.92, 125.17, 127.30, 132.13, 132.22, 133.70, 142.38, 143.85, 147.94, 152.53, 160.09, 162.14, 163.60, 173.50. Anal. calcd for C27H15F4N3O5 (537.41): C, 60.34; H, 2.81; N, 7.82. Found: C, 60.51; H, 2.87; N, 7.99.
4.1.1.10 3-(5-(4-Chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 5a. Rose crystals, m.p. 285–287 °C, yield (0.34 g, 72%). 1H NMR (400 MHz, DMSO-d6): δ = 6.21 (s, 1H, CH-indole), 6.92 (d, 1H, J = 7.6 Hz, H–Ar), 6.95(td, 1H, J = 0.8 and 6.8 Hz, H–Ar), 7.17 (d, 1H, J = 7.6 Hz, H–Ar), 7.20 (s, 1H, H–Ar), 7.28–7.39 (m, 3H, H–Ar), 7.58–7.65 (m, 3H, H–Ar), 7.70 (d, 2H, J = 8.0 Hz, H–Ar), 7.81 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 10.84 (s, 1H, NH), 13.11 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.76, 94.81, 106.75, 110.36, 115.02, 116.30, 122.56, 123.90, 124.73, 125.08, 125.56, 127.41, 129.47, 130.35, 131.33, 133.50, 134.94, 142.90, 145.28, 147.75, 152.27, 159.92, 163.41, 173.07. Anal. calcd for C26H16ClN3O4 (469.87): C, 66.46; H, 3.43; N, 8.94. Found: C, 66.62; H, 3.55; N, 9.08.
4.1.1.11 3-(5-(4-Chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-1-ethylindolin-2-one 5b. Off white powder, m.p. 245–246 °C, yield (0.37 g, 74%). 1H NMR (400 MHz, DMSO-d6): δ = 1.15 (t, 3H, J = 7.2 Hz, CH3), 3.72 (q, 2H, J = 8.0 Hz, CH2), 6.34 (s, 1H, CH-indole), 7.06–7.09 (m, 1H, H–Ar), 7.19–7.29 (m, 5H, H–Ar), 7.35–7.45 (m, 4H, H–Ar), 7.60–7.71 (m, 2H, H–Ar), 7.85 (d, 1H, J = 7.2 Hz, H–Ar), 13.08 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 12.61, 35.15, 60.68, 95.49, 107.06, 110.07, 115.31, 116.18, 123.12, 124.07, 124.90, 125.10, 125.21, 127.51, 129.52, 131.54, 133.68, 135.12, 137.08, 143.40, 147.94, 152.51, 160.08, 163.60, 171.23. Anal. calcd for C28H20ClN3O4 (497.92): C, 67.54; H, 4.05; N, 8.44. Found: C, 67.72; H, 4.19; N, 8.55.
4.1.1.12 1-Benzyl-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 5c. Yellow crystals, m.p. 235–237 °C, yield (0.41 g, 73%). 1H NMR (400 MHz, DMSO-d6): δ = 4.87 (d, 1H, J = 16.0 Hz, CHa-benzyl), 4.99 (d, 1H, J = 15.6 Hz, CHb-benzyl), 6.45 (s, 1H, CH-indole), 6.96 (d, 1H, J = 8.0 Hz, H–Ar), 7.00 (t, 1H, J = 7.6 Hz, H–Ar), 7.25 (s, 1H, H–Ar). 7.26–7.40 (m, 9H, H–Ar). 7.60–7.66 (m, 3H, H–Ar), 7.73–7.75 (m, 2H, H–Ar), 7.82 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 13.00 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.43, 60.45, 95.34, 106.89, 110.45, 115.48, 116.82, 123.50, 124.01, 124.99, 125.07, 125.12, 127.48, 127.52, 127.99, 129.14, 129.70, 130.50, 131.55, 133.71, 135.19, 136.12, 143.49, 145.72, 148.22, 152.54, 160.09, 163.61, 171.96. Anal. calcd for C33H22ClN3O4 (559.12): C, 70.78; H, 3.96; N, 7.50. Found: C, 70.91; H, 4.12; N, 7.66.
4.1.1.13 5-Chloro-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 5d. Pale yellow powder, m.p. > 300 °C, yield (0.34 g, 67%). 1H NMR (400 MHz, DMSO-d6): δ = 6.26 (s, 1H, CH-indole), 6.97 (d, 1H, J = 8.0 Hz, H–Ar), 7.24 (s, 1H, H–Ar), 7.36–7.46 (m, 4H, H–Ar), 7.63–7.71 (m, 3H, H–Ar), 7.74 (d, 2H, J = 8.4 Hz, H–Ar), 7.88–7.90 (m, 1H, H–Ar), 10.99 (s, 1H, NH), 13.11 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.75, 95.40, 106.79, 112.19, 115.49, 116.82, 124.13, 124.92, 125.59, 126.70, 127.53, 127.68, 129.60, 130.38, 131.62, 133.72, 135.14, 142.10, 145.69, 148.13, 152.54, 160.09, 163.63, 173.06. Anal. calcd for C26H15Cl2N3O4 (504.32): C, 61.92; H, 3.00; N, 8.33. Found: C, 62.09; H, 3.16; N, 8.45.
4.1.1.14 5-Chloro-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-1-ethylindolin-2-one 5e. Off white powder, m.p. 259–260 °C, yield (0.37 g, 69%). 1H NMR (400 MHz, DMSO-d6): δ = 1.13 (t, 3H, J = 6.8 Hz, CH3), 3.72 (q, 2H, J = 7.2 Hz, CH2), 6.34 (s, 1H, CH-indole), 7.25 (s, 1H, H–Ar). 7.36–7.40 (m, 1H, H–Ar), 7.42 (d, 1H, J = 8.0 Hz, H–Ar), 7.49–7.52 (m, 2H, H–Ar), 7.62–7.77 (m, 6H, H–Ar), 7.87 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 13.00 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 12.68, 35.20, 60.33, 95.39, 106.99, 111.36, 115.48, 116.82, 124.14, 124.91, 125.49, 127.15, 127.32, 127.48, 129.58, 130.41, 131.66, 133.73, 135.16, 142.45, 145.59, 148.17, 152.54, 160.10, 163.64, 171.00. Anal. calcd for C28H19Cl2N3O4 (531.07): C, 63.17; H, 3.60; N, 7.89. Found: C, 63.26; H, 3.77; N, 8.05.
4.1.1.15 1-Benzyl-5-chloro-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 5f. Violet crystals, m.p. 207–209 °C, yield (0.39 g, 66%). 1H NMR (400 MHz, DMSO-d6): δ = 4.91 (d, 1H, J = 15.6 Hz, CH-benzyl), 5.03 (d, 1H, J = 15.6 Hz, CH-benzyl), 6.49 (s, 1H, CH-indole), 7.02 (d, 1H, J = 8.8 Hz, H–Ar), 7.28 (s, 1H, H–Ar). 7.30–7.44 (m, 8H, H–Ar). 7.53(brs, 1H, H–Ar) 7.65–7.80 (m, 5H, H–Ar), 7.87 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 13.10 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.49, 60.21, 95.34, 106.95, 111.90, 115.49, 116.83, 124.06, 124.99, 125.54, 127.02, 127.38, 127.44, 127.65, 128.07, 129.13, 129.18, 129.66, 130.35, 131.68, 133.75, 135.22, 135.82, 142.50, 145.97, 148.44, 152.57, 160.09, 163.68, 171.72. Anal. calcd for C33H21Cl2N3O4 (593.09): C, 66.68; H, 3.56; N, 7.07. Found: C, 66.79; H, 3.69; N, 7.22.
4.1.1.16 5-Bromo-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 5g. Buff powder, m.p. 296–297 °C, yield (0.35 g, 64%). 1H NMR (400 MHz, DMSO-d6): δ = 6.26 (s, 1H, CH-indole), 6.92 (d, 1H, J = 8.8 Hz, H–Ar), 7.24 (s, 1H, H–Ar), 7.35 (t, 1H, J = 7.6 Hz, H–Ar), 7.41 (d, 1H, J = 8.4 Hz, H–Ar), 7.53 (s, 2H, H–Ar), 7.63–7.69 (m, 3H, H–Ar), 7.74 (d, 2H, J = 7.6 Hz, H–Ar), 7.87 (d, 1H, J = 7.6 Hz, H–Ar), 10.99 (s, 1H, NH), 13.09 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.65, 95.39, 106.79, 112.68, 114.32, 115.48, 116.80, 124.12, 124.89, 127.53, 128.04, 128.26, 129.04, 129.59, 131.62, 133.22, 133.70, 135.14, 142.51, 145.69, 148.13, 152.53, 160.07, 163.61, 172.93. Anal. calcd for C26H15BrClN3O4 (548.77): C, 56.90; H, 2.76; N, 7.66. Found: C, 57.10; H, 2.92; N, 7.77.
4.1.1.17 5-Bromo-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-1-ethylindolin-2-one 5h. Brown powder, m.p. > 300 °C, yield (0.45 g, 78%). 1H NMR (400 MHz, DMSO-d6): δ = 1.12 (t, 3H, J = 6.8 Hz, CH3), 3.68 (q, 2H, J = 6.8 Hz, CH2), 6.31 (s, 1H, CH-indole), 7.16–7.87 (m, 12H, H–Ar), 12.99 (s, 1H, OH). Anal. calcd for C28H19BrClN3O4 (575.02): C, 58.30; H, 3.32; N, 7.28. Found: C, 58.48; H, 3.51; N, 7.45.
4.1.1.18 1-Benzyl-5-bromo-3-(5-(4-chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 5i. Buff powder, m.p. 228–229 °C, yield (0.52 g, 82%). 1H NMR (400 MHz, DMSO-d6): δ = 4.90 (d, 1H, J = 15.6 Hz, CHa-benzyl), 5.02 (d, 1H, J = 15.6 Hz, CHb-benzyl), 6.49 (s, 1H, CH-indole), 6.97 (d, 1H, J = 8.8 Hz, H–Ar), 7.29 (s, 1H, H–Ar), 7.34–7.45 (m, 7H, H–Ar), 7.55 (d, 1H, J = 8.4 Hz, H–Ar), 7.64 (brs, 1H, H–Ar), 7.67–7.73 (m, 3H, H–Ar), 7.79–7.81 (m, 2H, H–Ar), 7.88 (d, 1H, J = 6.8 Hz, H–Ar), 12.93 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.46, 60.11, 95.35, 106.88, 112.39, 115.32, 115.47, 116.83, 123.97, 124.06, 125.00, 125.08, 127.34, 127.43, 127.85, 128.06, 128.20, 129.18, 131.77, 131.91, 132.58, 133.21, 133.76, 135.80, 142.93, 146.06, 148.46, 152.56, 160.08, 163.65, 171.61. Anal. calcd for C33H21BrClN3O4 (637.04): C, 62.04; H, 3.31; N, 6.58. Found: C, 62.19; H, 3.49; N, 6.77.
4.1.1.19 3-(5-(4-Chlorophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-5-(trifluoromethoxy)indolin-2-one 5j. Yellow powder, m.p. 299–300 °C, yield (0.34 g, 61%). 1H NMR (400 MHz, DMSO-d6): δ = 6.34 (s, 1H, CH-indole), 7.03 (d, 1H, J = 8.4 Hz, H–Ar), 7.24 (s, 1H, H–Ar), 7.36–7.44 (m, 4H, H–Ar), 7.61–7.70 (m, 3H, H–Ar), 7.74–7.83 (m, 2H, H–Ar), 7.86 (dd, 1H, J = 1.2 and 8.0 Hz, H–Ar), 11.04 (s, 1H, NH), 13.00 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.98, 95.38, 106.94, 111.65, 115.47, 116.82, 119.32, 119.60, 121.85, 123.80, 124.08, 124.92, 127.25, 127.56, 129.53, 131.59, 133.71, 135.11, 142.39, 143.86, 145.61, 148.02, 152.53, 160.09, 163.60, 173.46. Anal. calcd for C27H15ClF3N3O5(553.87): C, 58.55; H, 2.73; N, 7.59. Found: C, 58.68; H, 2.91; N, 7.78.
4.1.1.20 3-(5-(4-Bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 6a. Rose powder, m.p. 291–292 °C, yield (0.42 g, 81%). 1H NMR (400 MHz, DMSO-d6): δ = 6.25 (s, 1H, CH-indole), 6.96 (d, 1H, J = 8.0 Hz, H–Ar), 6.99 (td, 1H, J = 0.4 and 3.6 Hz, H–Ar), 7.21 (d, 1H, J = 7.6 Hz, H–Ar), 7.24 (s, 1H, H–Ar), 7.32–7.39 (m, 2H, H–Ar), 7.41 (d, 1H, J = 8.0 Hz, H–Ar), 7.65–7.69 (m, 3H, H–Ar), 7.76 (d, 2H, J = 8.4 Hz, H–Ar), 7.86 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 10.87 (s, 1H, NH), 13.11 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.92, 95.37, 106.69, 110.73, 115.49, 116.81, 122.72, 123.85, 124.07, 124.90, 125.21, 125.73, 127.93, 130.51, 131.71, 132.56, 133.67, 143.03, 145.52, 147.93, 152.52, 160.03, 163.57, 173.29. Anal. calcd for C26H16BrN3O4 (514.32): C, 60.72; H, 3.14; N, 8.17. Found: C, 60.92; H, 3.33; N, 8.32.
4.1.1.21 3-(5-(4-Bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-1-ethylindolin-2-one 6b. Yellow powder, m.p. 273–275 °C, yield (0.41 g, 76%). 1H NMR (400 MHz, DMSO-d6): δ = 1.11 (t, 3H, J = 6.8 Hz, CH3), 3.68 (q, 2H, J = 7.2 Hz, CH2), 6.30 (s, 1H, CH-indole), 7.02–7.06 (m, 1H, H–Ar), 7.15–7.24 (m, 3H, H–Ar), 7.30–7.41 (m, 3H, H–Ar), 7.58–7.64 (m, 3H, H–Ar), 7.69 (d, 2H, J = 8.0 Hz, H–Ar), 7.80 (dd, 1H, J = 1.2 and 8.0 Hz, H–Ar), 12.97 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 12.74, 35.00, 60.54, 95.33, 106.86, 109.83, 115.42, 116.75, 123.20, 123.82, 124.02, 124.85, 125.06, 125.17, 127.82, 129.61, 130.58, 131.70, 132.47, 133.63, 143.36, 145.34, 147.93, 152.46, 160.04, 163.51, 171.17. Anal. calcd for C28H20BrN3O4 (542.38): C, 62.00; H, 3.72; N, 7.75. Found: C, 62.12; H, 3.90; N, 7.89.
4.1.1.22 1-Benzyl-3-(5-(4-bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 6c. Off white powder, m.p. 239–241 °C, yield (0.47 g, 78%). 1H NMR (400 MHz, DMSO-d6): δ = 4.91 (d, 1H, J = 16.0 Hz, CHa-benzyl), 5.02 (d, 1H, J = 16.0 Hz, CHb-benzyl), 6.49 (s, 1H, CH-indole), 7.00 (d, 1H, J = 8.0 Hz, H–Ar), 7.04 (t, 1H, J = 6.8 Hz, H–Ar), 7.29 (s, 1H, H–Ar). 7.30–7.45 (m, 9H, H–Ar). 7.67–7.72 (m, 3H, H–Ar), 7.78–7.80 (m, 2H, H–Ar), 7.86 (dd, 1H, J = 1.2 and 8.0 Hz, H–Ar), 12.97 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.42, 60.44, 95.33, 106.84, 110.46, 115.51, 116.83, 121.65, 123.49, 123.92, 124.02, 124.99, 125.09, 125.12, 127.48, 127.89, 127.99, 128.86, 129.14, 130.49, 131.77, 132.63, 133.72, 136.13, 140.34, 143.50, 145.79, 148.25, 152.56, 160.10, 163.63, 171.95. Anal. calcd for C33H22BrN3O4 (603.07): C, 65.57; H, 3.67; N, 6.95. Found: C, 65.70; H, 3.84; N, 7.12.
4.1.1.23 3-(5-(4-Bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-5-chloroindolin-2-one 6d. White powder, m.p. > 300 °C, yield (0.42 g, 77%). 1H NMR (400 MHz, DMSO-d6): δ = 6.26 (s, 1H, CH-indole), 6.92 (d, 1H, J = 8.8 Hz, H–Ar), 7.24 (s, 1H, H–Ar), 7.36–7.44 (m, 3H, H–Ar), 7.52–7.55 (m, 2H, H–Ar), 7.66–7.69 (m, 2H, H–Ar), 7.77 (d, 2H, J = 8.4 Hz, H–Ar), 7.87–7.89 (m, 1H, H–Ar), 11.00 (s, 1H, NH), 13.09 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.69, 95.26, 106.46, 114.34, 115.38, 116.52, 123.88, 124.12, 124.90, 127.89, 128.03, 128.24, 131.83, 132.17, 132.52, 133.18, 133.71, 142.54, 148.15, 152.44, 160.01, 163.58, 173.00. Anal. calcd for C26H15BrClN3O4 (548.77): C, 56.90; H, 2.76; N, 7.66. Found: C, 57.01; H, 2.92; N, 7.79.
4.1.1.24 3-(5-(4-Bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-5-chloro-1-ethylindolin-2-one 6e. Off-white powder, m.p. 275–277 °C, yield (0.44 g, 76%). 1H NMR (400 MHz, DMSO-d6): δ = 1.10 (t, 3H, J = 7.2 Hz, CH3), 3.68 (q, 2H, J = 6.8 Hz, CH2), 6.30 (s, 1H, CH-indole), 7.20–7.22 (m, 2H, H–Ar). 7.31–7.35 (m, 1H, H–Ar), 7.37–7.39 (m, 1H, H–Ar), 7.44–7.48 (m, 2H, H–Ar), 7.57–7.66 (m, 3H, H–Ar), 7.72 (d, 2H, J = 8.8 Hz, H–Ar), 7.82 (dd, 1H, J = 1.2 and 8.0 Hz, H–Ar), 12.97 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 12.65, 35.17, 60.27, 69.51, 95.35, 106.89, 111.32, 115.41, 116.77, 123.88, 124.08, 124.87, 125.45, 127.09, 127.30, 127.79, 130.38, 131.82, 132.46, 133.68, 142.40, 145.61, 148.15, 152.48, 160.04, 163.56, 170.94. Anal. calcd for C28H19BrClN3O4 (576.82): C, 58.30; H, 3.32; N, 7.28. Found: C, 58.48; H, 3.50; N, 7.44.
4.1.1.25 1-Benzyl-3-(5-(4-bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-5-chloroindolin-2-one 6f. Violet crystals, m.p. 222–223 °C, yield (0.42 g, 66%). 1H NMR (400 MHz, DMSO-d6): δ = 4.91 (d, 1H, J = 16.0 Hz, CHa-benzyl), 5.03 (d, 1H, J = 16.00 Hz, CHb-benzyl), 6.49 (s, 1H, CH-indole), 7.02 (d, 1H, J = 8.4 Hz, H–Ar), 7.29 (s, 1H, H–Ar), 7.34–7.45 (m, 8H, H–Ar), 7.53–7.54 (m, 1H, H–Ar), 7.67–7.74 (m, 3H, H–Ar), 7.79–7.81 (m, 2H, H–Ar), 7.88 (dd, 1H, J = 1.2 and 7.6 Hz, H–Ar), 12.96 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.49, 60.21, 95.35, 106.89, 111.90, 115.47, 116.83, 123.97, 124.06, 125.00, 125.54, 127.02, 127.44, 127.64, 127.85, 128.06, 129.18, 129.68, 130.35, 131.90, 132.59, 133.75, 135.82, 142.50, 146.04, 148.46, 152.56, 160.08, 163.65, 171.71. Anal. calcd for C33H21BrClN3O4 (637.04): C, 62.04; H, 3.31; N, 6.58. Found: C, 62.22; H, 3.51; N, 6.77.
4.1.1.26 5-Bromo-3-(5-(4-bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl) indolin-2-one 6g. Off-white powder, m.p. 298–299 °C, yield (0.36 g, 60%). 1H NMR (400 MHz, DMSO-d6): δ = 6.27 (s, 1H, CH-indole), 6.97 (d, 1H, J = 8.4 Hz, H–Ar), 7.25 (s, 1H, H–Ar), 7.36–7.44 (m, 4H, H–Ar), 7.66–7.70 (m, 3H, H–Ar), 7.77 (d, 2H, J = 8.4 Hz, H–Ar), 7.87 (dd, 1H, J = 1.2 and 8.0 Hz, H–Ar), 10.99 (s, 1H, NH), 13.09 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 60.74, 95.39, 106.75, 112.19, 115.48, 116.81, 123.89, 124.12, 124.91, 125.59, 126.70, 127.67, 127.89, 130.38, 131.83, 132.53, 133.72, 142.10, 145.75, 148.15, 152.54, 160.08, 163.62, 173.05. Anal. calcd. For C26H15Br2N3O4 (593.22): C, 52.64; H, 2.55; N, 7.08. Found: C, 52.75; H, 2.69; N, 7.22.
4.1.1.27 5-Bromo-3-(5-(4-bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-1-ethylindolin-2-one 6h. Buff powder, m.p. 276–278 °C, yield (0.41 g, 66%). 1H NMR (400 MHz, DMSO-d6): δ = 1.14 (t, 3H, J = 7.2 Hz, CH3), 3.66 (q, 2H, J = 4.8 Hz, CH2), 6.34 (s, 1H, CH-indole), 7.16–7.29 (m, 1H, H–Ar). 7.39–7.44 (m, 3H, H–Ar), 7.54–7.59 (m, 2H, H–Ar), 7.62–7.70 (m, 3H, H–Ar), 7.87 (d, 2H, J = 8.4 Hz, H–Ar), 7.98 (dd, 1H, J = 1.2 and 7.6 Hz, H–Ar), 12.97 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 12.60, 34.83, 95.17, 102.50, 116.45, 116.75, 120.77, 124.13, 124.70, 125.98, 127.13, 127.48, 127.92, 128.04, 129.52, 129.57, 129.64, 131.65, 133.26, 133.89, 140.20, 142.69, 148.02, 152.58, 156.48, 160.51, 164.54, 171.13. Anal. calcd for C28H19Br2N3O4 (618.97): C, 54.13; H, 3.08; N, 6.76. Found: C, 54.29; H, 3.22; N, 6.92.
4.1.1.28 1-Benzyl-5-bromo-3-(5-(4-bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)indolin-2-one 6i. Violet crystals, m.p. 218–220 °C, yield (0.44 g, 64%). 1H NMR (400 MHz, DMSO-d6): δ = 4.73 (d, 1H, J = 16.0 Hz, CH-benzyl), 4.86 (d, 1H, J = 16.0 Hz, CH-benzyl), 6.32 (s, 1H, CH-indole), 6.80 (d, 1H, J = 8.4 Hz, H–Ar), 7.12 (s, 1H, H–Ar), 7.14–7.27 (m, 7H, H–Ar), 7.38 (d, 1H, J = 8.4 Hz, H–Ar), 7.48–7.54 (m, 4H, H–Ar), 7.62–7.64 (m, 2H, H–Ar), 7.71 (d, 1H, J = 6.8 Hz, H–Ar), 12.81 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 43.46, 60.11, 95.35, 106.93, 112.38, 115.32, 115.46, 116.82, 124.05, 124.99, 127.34, 127.43, 127.49, 128.06, 128.19, 129.18, 129.65, 131.55, 131.68, 133.21, 133.74, 135.22, 135.78, 142.92, 145.98, 148.44, 152.55, 160.08, 163.64, 171.61, Anal. calcd for C33H21Br2N3O4 (683.34): C, 58.00; H, 3.10; N, 6.15. Found: C, 58.17; H, 3.25; N, 6.33.
4.1.1.29 3-(5-(4-Bromophenyl)-3-(4-hydroxy-2-oxo-2H-chromen-3-yl)-1H-pyrazol-1-yl)-5-(trifluoromethoxy)indolin-2-one 6j. Yellow crystals, m.p. 298–300 °C, yield (0.46 g, 77%). 1H NMR (400 MHz, DMSO-d6): δ = 7.06 (d, 1H, J = 8.4 Hz, H–Ar), 7.25–7.28 (m, 3H, H–Ar), 7.46–7.49 (m, 1H, H–Ar), 7.59–7.69 (m, 4H, H–Ar), 7.78–7.82 (m, 3H, H–Ar), 7.84 (dd, 1H, J = 1.6 and 8.0 Hz, H–Ar), 10.89 (s, 1H, NH), 13.03 (s, 1H, OH). 13C NMR (101 MHz, DMSO-d6): δ = 69.95, 89.80, 92.82, 112.45, 116.68, 119.29, 120.03, 120.69, 121.83, 124.09, 125.86, 125.98, 126.14, 129.63, 130.90, 132.44, 132.59, 134.40, 141.34, 144.23, 153.38, 156.54, 163.38, 174.13. Anal. calcd for C27H15BrF3N3O5 (598.32): C, 54.20; H, 2.53; N, 7.02. Found: C, 54.39; H, 2.71; N, 7.17.
4.2. Biology
IC50 of normal cells/IC50 of tumor cells.62000 cells were collected and the distribution of cell cycle phases was analyzed using the ACEA Novo Express™ software (ACEA Biosciences Inc., San Diego, USA).644.3. Molecular modeling
The molecular docking studies were conducted using the Molecular Operating Environment (MOE 2022.02)67 package license purchased from Chemical Computing Group Inc. and a Sherbrooke St, Montreal, QC, Canada. Triangle matcher was used as a placement method. The London ΔG scoring method was applied to estimate the free energy of binding (kcal mol−1). Validation of docking was done by computing (GBVI/WSA) dG, which is a force field-based scoring function to estimate the free energy of binding (kcal mol−1). Redocking of the co-crystalized ligand SB-5902885 was done as a type of validation as well. The root mean square deviation (RMSD) of the docked ligands was computed to measure the deviation from the co-crystalized ligand. The crystal structures of BRAF (PBD = 2FB8) and VEGFR-2 (PBD = 3VO3) were downloaded from the Protein Data Bank (https://www.rcsb.org/).4.4. ADME prediction
ADME prediction of all pharmacokinetic properties was performed and validated using two platforms, i.e. ADMETLab 2.068 and pKCSM.69
Ethical approval
This study was conducted according to the appropriate ethical standards as required by guidance from The Medical Research Ethics Committee (MREC), Egypt, Ethical Clearance Certificate No. 9446072023.Conflicts of interest
There are no conflicts of interest to declare.References
- G. C. Leonardi, L. Falzone, R. Salemi, A. Zanghì, D. A. Spandidos, J. A. Mccubrey, S. Candido and M. Libra, Cutaneous melanoma: from pathogenesis to therapy (Review), Int. J. Oncol., 2018, 52, 1071–1080 CAS
.
- B. A. Shoo and M. Kashani-Sabet, Melanoma arising in African-, Asian-, Latino- and Native-American populations, Semin. Cutaneous Med. Surg., 2009, 28, 96–102 CrossRef CAS PubMed
- V. Lai, W. Cranwell and R. Sinclair, Epidemiology of skin cancer in the mature patient, Clin. Dermatol., 2018, 36, 167–176 CrossRef PubMed
- T. Amaral, T. Sinnberg, F. Meier, C. Krepler, M. Levesque, H. Niessner and C. Garbe, The mitogen-activated protein kinase pathway in melanoma part I - Activation and primary resistance mechanisms to BRAF inhibition, Eur. J. Cancer, 2017, 73, 85–92 CrossRef CAS PubMed
- G. Maurer, B. Tarkowski and M. Baccarini, Raf kinases in cancer-roles and therapeutic opportunities, Oncogene, 2011, 30, 3477–3488 CrossRef CAS PubMed
- D. T. Leicht, V. Balan, A. Kaplun, V. Singh-Gupta, L. Kaplun, M. Dobson and G. Tzivion, Raf kinases: function, regulation and role in human cancer, Biochim. Biophys. Acta, 2007, 1773, 1196–1212 CrossRef CAS PubMed
- M. A. Rahman, A. Salajegheh, R. A. Smith and A. K. Lam, B-Raf mutation: a key player in molecular biology of cancer, Exp. Mol. Pathol., 2013, 95, 336–342 CrossRef CAS PubMed
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins and S. Clegg, et al., Mutations of the BRAF gene in human cancer, Nature, 2002, 417, 949–954 CrossRef CAS PubMed
- M. Dankner, A. A. N. Rose, S. Rajkumar, P. M. Siegel and I. R. Watson, Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations, Oncogene, 2018, 37, 3183–3199 CrossRef CAS PubMed
- Y. S. Jo, S. Li, J. H. Song, K. H. Kwon, J. C. Lee, S. Y. Rha, H. J. Lee, J. Y. Sul, G. R. Kweon, H. K. Ro, J. M. Kim and M. Shong, Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer, J. Clin. Endocrinol. Metab., 2006, 91, 3667–3670 CrossRef CAS PubMed
- P. A. Ascierto, J. M. Kirkwood, J. J. Grob, E. Simeone, A. M. Grimaldi, M. Maio, G. Palmieri, A. Testori, F. M. Marincola and N. Mozzillo, The role of BRAF V600 mutation in melanoma, J. Transl. Med., 2012, 10, 85 CrossRef CAS PubMed
- S. C. Lalla, A. B. Kumar, J. S. Lehman, C. M. Lohse and J. D. Brewer, Association of BRAF V600E Status of Incident Melanoma and Risk for a Second Primary Malignancy: A Population-Based Study, Cutis, 2022, 110, 150–154 Search PubMed
- A. Vultur, J. Villanueva and M. Herlyn, Targeting BRAF in advanced melanoma: a first step toward manageable disease, Clin. Cancer Res., 2011, 17, 1658–1663 CrossRef CAS PubMed
- K. Singh, P. Sonawane, A. Kumar, H. Singh, V. Naumovich, P. Pathak, M. Grishina, H. Khalilullah, M. Jaremko, A. H. Emwas, A. Verma and P. Kumar, Challenges and Opportunities in the Crusade of BRAF Inhibitors: From 2002 to 2022, ACS Omega, 2023, 8, 27819–27844 CrossRef PubMed
- M. Holderfield, T. E. Nagel and D. D. Stuart, Mechanism and consequences of RAF kinase activation by small-molecule inhibitors, Br. J. Cancer, 2014, 111, 640–645 CrossRef CAS PubMed
- Y. Liu and N. S. Gray, Rational design of inhibitors that bind to inactive kinase
conformations, Nat. Chem. Biol., 2006, 2, 358–364 CrossRef CAS PubMed
- Y. Wang, S. Wan, Z. Li, Y. Fu, G. Wang, J. Zhang and X. Wu, Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrazolo[3,4-d] pyrimidine derivatives as BRAFV600E and VEGFR-2 dual inhibitors, Eur. J. Med. Chem., 2018, 155, 210–228 CrossRef CAS PubMed
- I. H. Ali, H. T. Abdel-Mohsen, M. M. Mounier, M. T. Abo-Elfadl, A. M. El Kerdawy and I. A. Y. Ghannam, Design, synthesis and anticancer activity of novel 2-arylbenzimidazole/2-thiopyrimidines and 2-thioquinazolin-4(3H)-ones conjugates as targeted RAF and VEGFR-2 kinases inhibitors, Bioorg. Chem., 2022, 126, 105883 CrossRef CAS PubMed
- A. Kim and M. S. Cohen, The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma, Expert Opin. Drug Discovery, 2016, 11, 907–916 CrossRef CAS PubMed
- R. Kainthla, K. B. Kim and G. S. Falchook, Dabrafenib. Recent Results Cancer Res., 2014, 201, 227–240 CAS
- P. Koelblinger, O. Thuerigen and R. Dummer, Development of encorafenib for BRAF-mutated advanced melanoma, Curr. Opin. Oncol., 2018, 30, 125–133 CrossRef CAS PubMed
- R. M. Anforth, T. C. Blumetti, R. F. Kefford, R. Sharma, R. A. Scolyer, S. Kossard, G. V. Long and P. Fernandez-Peñas, Cutaneous manifestations of dabrafenib (GSK2118436): a selective inhibitor of mutant BRAF in patients with metastatic melanoma, Br. J. Dermatol., 2012, 167, 1153–1160 CrossRef CAS PubMed
- G. S. Falchook, G. V. Long, R. Kurzrock, K. B. Kim, T. H. Arkenau, M. P. Brown, O. Hamid, J. R. Infante, M. Millward, A. C. Pavlick, S. J. O'Day, S. C. Blackman, C. M. Curtis, P. Lebowitz, B. Ma, D. Ouellet and R. F. Kefford, Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial, Lancet, 2012, 379, 1893–1901 CrossRef CAS PubMed
- K. T. Flaherty, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. J. O'Dwyer, R. J. Lee, J. F. Grippo, K. Nolop and P. B. Chapman, Inhibition of mutated, activated BRAF in metastatic melanoma, N. Engl. J. Med., 2010, 363, 809–819 CrossRef CAS PubMed
- J. Blanc, R. Geney and C. Menet, Type II kinase inhibitors: an opportunity in cancer for rational design, Anti Canc, Agents Med. Chem., 2013, 13, 731–747 CrossRef CAS PubMed
- M. Moore, H. W. Hirte, L. Siu, A. Oza, S. J. Hotte, O. Petrenciuc, F. Cihon, C. Lathia and B. Schwartz, Phase I study to determine the safety and pharmacokinetics of the novel Raf kinase and VEGFR inhibitor BAY 43-9006, administered for 28 days on/7 days off in patients with advanced, refractory solid tumors, Ann. Oncol., 2005, 16, 1688–1694 CrossRef CAS PubMed
- B. Izar, W. Sharfman, F. S. Hodi, D. Lawrence, K. T. Flaherty, R. Amaravadi, K. B. Kim, I. Puzanov, J. Sosman, R. Dummer, S. M. Goldinger, L. Lam, S. Kakar, Z. Tang, O. Krieter, D. F. McDermott and M. B. Atkins, A first-in-human phase I, multicenter, open-label, dose-escalation study of the oral RAF/VEGFR-2 inhibitor (RAF265) in locally advanced or metastatic melanoma independent from BRAF mutation status, Cancer Med., 2017, 6, 1904–1914 CrossRef CAS PubMed
- S. M. Wilhelm, C. Carter, L. Tang, D. Wilkie, A. McNabola, H. Rong, C. Chen, X. Zhang, P. Vincent, M. McHugh, Y. Cao, J. Shujath, S. Gawlak, D. Eveleigh, B. Rowley, L. Liu, L. Adnane, M. Lynch, D. Auclair, I. Taylor, R. Gedrich, A. Voznesensky, B. Riedl, L. E. Post, G. Bollag and P. A. Trail, BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis, Cancer Res., 2004, 64, 7099–7109 CrossRef CAS PubMed
- S. M. Wilhelm, J. Dumas, L. Adnane, M. Lynch, C. A. Carter, G. Schütz, K. H. Thierauch and D. Zopf, Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity, Int. J. Cancer, 2011, 129, 245–255 CrossRef CAS PubMed
- T. E. Williams, S. Subramanian, J. Verhagen, C. M. McBride, A. Costales, L. Sung, W. Antonios-McCrea, M. McKenna, A. K. Louie, S. Ramurthy, B. Levine, C. M. Shafer, T. Machajewski, P. A. Renhowe, B. A. Appleton, P. Amiri, J. Chou, D. Stuart, K. Aardalen and D. Poon, Discovery of RAF265: A Potent mut-B-RAF Inhibitor for the Treatment of Metastatic Melanoma, ACS Med. Chem. Lett., 2015, 6, 961–965 CrossRef CAS PubMed
- R. Z. Batran, E. Y. Ahmed, H. M. Awad, K. A. Ali and N. A. Abdel Latif, EGFR and PI3K/m-TOR inhibitors: design, microwave assisted synthesis and anticancer activity of thiazole-coumarin hybrids, RSC Adv., 2023, 13, 29070–29085 RSC
- Y. J. Jeon, J. Y. Jang, J. H. Shim, P. K. Myung and J. I. Chae, Esculetin, a Coumarin Derivative, Exhibits Anti-proliferative and Pro-apoptotic Activity in G361 Human Malignant Melanoma, J. Cancer Prev., 2015, 20, 106–112 CrossRef PubMed
- R. D. Thornes, L. Daly, G. Lynch, B. Breslin, H. Browne, H. Y. Browne, T. Corrigan, P. Daly, G. Edwards and E. Gaffney, et al., Treatment with coumarin to prevent or delay recurrence of malignant melanoma, J. Cancer Res. Clin. Oncol., 1994, 120(Suppl), S32–S34 CrossRef PubMed
- T. Aoki, I. Hyohdoh, N. Furuichi, S. Ozawa, F. Watanabe, M. Matsushita, M. Sakaitani, K. Ori, K. Takanashi, N. Harada, Y. Tomii, M. Tabo, K. Yoshinari, Y. Aoki, N. Shimma and H. Iikura, The sulfamide moiety affords higher inhibitory activity and oral bioavailability to a series of coumarin dual selective RAF/MEK inhibitors, Bioorg. Med. Chem. Lett., 2013, 23, 6223–6227 CrossRef CAS PubMed
- S. Han, V. Zhou, S. Pan, Y. Liu, M. Hornsby, D. McMullan, H. E. Klock, J. Haugen, S. A. Lesley, N. Gray, J. Caldwell and X. J. Gu, Identification of coumarin derivatives as a novel class of allosteric MEK1 inhibitors, Bioorg. Med. Chem. Lett., 2005, 15, 5467–5473 CrossRef CAS PubMed
- T. K. Mohamed, R. Z. Batran, S. A. Elseginy, M. M. Ali and A. E. Mahmoud, Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis, Bioorg. Chem., 2019, 85, 253–273 CrossRef CAS PubMed
- R. Z. Batran, D. H. Dawood, S. A. El-Seginy, M. M. Ali, T. J. Maher, K. S. Gugnani and A. N. Rondon-Ortiz, New Coumarin Derivatives as Anti-Breast and Anti-Cervical Cancer Agents Targeting VEGFR-2 and p38α MAPK, Arch. Pharm., 2017, 350, e1700064 CrossRef PubMed
- M. A. Dickson, M. S. Gordon, G. Edelman, J. C. Bendell, R. R. Kudchadkar, P. M. LoRusso, S. H. Johnston, D. O. Clary and G. K. Schwartz, Phase I study of XL281 (BMS-908662), a potent oral RAF kinase inhibitor, in patients with advanced solid tumors, Invest. New Drugs, 2015, 33, 349–356 CrossRef CAS PubMed
- J. Zhang, T. Liu, M. Chen, F. Liu, X. Liu, J. Zhang, J. Lin and Y. Jin, Synthesis and Biological Evaluation of Indole-2-carbohydrazide Derivatives as Anticancer Agents with Anti-angiogenic and Antiproliferative Activities, ChemMedChem, 2018, 13, 1181–1192 CrossRef CAS PubMed
- M. S. Nafie and A. T. A. Boraei, Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer, Bioorg. Chem., 2022, 122, 105708 CrossRef CAS PubMed
- M. H. Kim, M. Kim, H. Yu, H. Kim, K. H. Yoo, T. Sim and J. M. Hah, Structure based design and syntheses of amino-1H-pyrazole amide derivatives as selective Raf kinase inhibitors in melanoma cells, Bioorg. Med. Chem., 2011, 19, 1915–1923 CrossRef CAS PubMed
- S. F. Wang, Y. L. Zhu, P. T. Zhu, J. A. Makawana, Y. L. Zhang, M. Y. Zhao, P. C. Lv and H. L. Zhu, Design, synthesis and biological evaluation of novel 5-phenyl-1H-pyrazole derivatives as potential BRAF(V600E) inhibitors, Bioorg. Med. Chem., 2014, 22, 6201–6208 CrossRef CAS PubMed
- C. R. Wang, Z. F. Wang, L. Shi, Z. C. Wang and H. L. Zhu, Design, synthesis, and biological evaluation of pyrazole derivatives containing acetamide bond as potential BRAFV600E inhibitors, Bioorg. Med. Chem. Lett., 2018, 28, 2382–2390 CrossRef CAS PubMed
- H. L. Li, M. M. Su, Y. J. Xu, C. Xu, Y. S. Yang and H. L. Zhu, Design and biological evaluation of novel triaryl pyrazoline derivatives with dioxane moiety for selective BRAFV600E inhibition, Eur. J. Med. Chem., 2018, 155, 725–735 CrossRef CAS PubMed
- Y. S. Yang, B. Yang, Y. Zou, G. Li and H. L. Zhu, Design, biological evaluation and 3D QSAR studies of novel dioxin-containing triaryl pyrazoline derivatives as potential B-Raf inhibitors, Bioorg. Med. Chem., 2016, 24, 3052–3061 CrossRef CAS PubMed
- Y. S. Yang, Q. S. Li, S. Sun, Y. B. Zhang, X. L. Wang, F. Zhang, J. F. Tang and H. L. Zhu, Design, modification and 3D QSAR studies of novel 2,3-dihydrobenzo[b][1,4]dioxin-containing 4,5-dihydro-1H-pyrazole derivatives as inhibitors of B-Raf kinase, Bioorg. Med. Chem., 2012, 20, 6048–6058 CrossRef CAS PubMed
- R. Z. Batran, D. H. Dawood, S. A. El-Seginy, M. M. Ali, T. J. Maher, K. S. Gugnani and A. N. Rondon-Ortiz, New Coumarin Derivatives as Anti-Breast and Anti-Cervical Cancer Agents Targeting VEGFR-2 and p38α MAPK, Arch. Pharm., 2017, 350, e1700064 CrossRef PubMed
- O. M. Hafez, M. I. Nassar, S. M. El-Kousy, A. F. Abdel-Razik, M. M. Sherien and M. M. El-Ghonemy, Synthesis of some new carbonitriles and pyrazole coumarin derivatives with potent antitumor and antimicrobial activities, Acta Pol. Pharm., 2014, 71, 594–601 Search PubMed
- H. S. Anbar, M. I. El-Gamal, H. Tarazi, B. S. Lee, H. R. Jeon, D. Kwon and C. H. Oh, Imidazothiazole-based potent inhibitors of V600E-B-RAF kinase with promising anti-melanoma activity: biological and computational studies, J. Enzyme Inhib. Med. Chem., 2020, 35, 1712–1726 CrossRef CAS PubMed
- M. Koley, J. Han, V. A. Soloshonok, S. Mojumder, R. Javahershenas and A. Makarem, Latest developments in coumarin-based anticancer agents: mechanism of action and structure–activity relationship studies, RSC Med. Chem., 2024, 15, 10–54 RSC
- T. Neamatallah, A. M. Malebari, A. J. Alamoudi, S. Nazreen, M. M. Alam, H. H. Bin-Melaih, O. A. Abuzinadah, S. M. Badr-Eldin, G. Alhassani, L. Makki and M. Z. Nasrullah, Andrographolide nanophytosomes exhibit enhanced cellular delivery and pro-apoptotic activities in HepG2 liver cancer cells, Drug Delivery, 2023, 30, 2174209 CrossRef PubMed
- L. Arce-Ramos, J. C. Castillo and D. Becerra, Synthesis and Biological Studies of Benzo[b]furan Derivatives: A Review from 2011 to 2022, Pharmaceuticals, 2023, 16, 1265 CrossRef CAS PubMed
- A. A. Bahman, M. S. I. Abaza, S. I. Khoushiash and R. J. Al-Attiyah, Sequence-dependent effect of sorafenib in combination with natural phenolic compounds on hepatic cancer cells and the possible mechanism of action, Int. J. Mol. Med., 2018, 42, 1695–1715 CAS
- J. O. Pinho, M. Matias, A. Godinho-Santos, J. D. Amaral, E. Mendes, M. J. Perry, A. P. Francisco, C. M. P. Rodrigues and M. M. Gaspar, A step forward on the in vitro and in vivo assessment of a novel nanomedicine against melanoma, Int. J. Pharm., 2023, 640, 123011 CrossRef CAS PubMed
- C. Rosen, T. Mayes, C. Overholt and B. Lucke-Wold, Treatment of Melanoma Metastasis: Surgical, Chemotherapy, and Innovation, Med. Discovery, 2023, 2, 1032 Search PubMed
- K. Palm, P. Stenberg, K. Luthman and P. Artursson, Polar molecular surface properties predict the intestinal absorption of drugs in humans, Pharm. Res., 1997, 14, 568–571 CrossRef CAS PubMed
- J. Kelder, P. D. Grootenhuis, D. M. Bayada, L. P. Delbressine and J. P. Ploemen, Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs, Pharm. Res., 1999, 16, 1514–1519 CrossRef CAS PubMed
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed
- M. F. Abo-Ashour, W. M. Eldehna, A. Nocentini, A. Bonardi, S. Bua, H. S. Ibrahim, M. M. Elaasser, V. Kryštof, R. Jorda, P. Gratteri, S. M. Abou-Seri and C. T. Supuran, 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: synthesis, in vitro biological evaluation and in silico insights, Eur. J. Med. Chem., 2019, 184, 111768 CrossRef CAS PubMed
- W. M. Eldehna, S. T. Al-Rashood, T. Al-Warhi, R. O. Eskandrani, A. Alharbi and A. M. El Kerdawy, Novel oxindole/benzofuran hybrids as potential dual CDK2/GSK-3β inhibitors targeting breast cancer: design, synthesis, biological evaluation, and in silico studies, J. Enzyme Inhib. Med. Chem., 2021, 36, 270–285 CAS
- D. A. Abosedera, S. A. Emara, O. A. S. Tamam, O. M. Badr, S. A. M. Khalifa, H. R. El-Seedi and M. S. Refaey, Metabolomic profile and in vitro evaluation of the cytotoxic activity of Asphodelus microcarpus against human malignant melanoma cells A375, Arabian J. Chem., 2022, 15, 104174 CrossRef CAS
- A. A. Hassabo, M. Abdelraof and R. M. Allam, L-arginase from Streptomyces diastaticus MAM5 as a potential therapeutic agent in breast cancer: purification, characterization, G1 phase arrest and autophagy induction, Int. J. Biol. Macromol., 2023, 224, 634–645 CrossRef CAS PubMed
- T. Atakul, Serum Levels of Angiogenic Factors Distinguish Between Women with Preeclampsia and Normotensive Pregnant Women But Not Severity of Preeclampsia in an Obstetric Center in Turkey, Med. Sci. Monit., 2019, 25, 6935–6942 CrossRef PubMed
- O. E. Abdel-Sattar, R. M. Allam, A. M. Al-Abd, B. Avula, K. Katragunta, I. A. Khan, A. M. El-Desoky, S. O. Mohamed, A. El-Halawany, E. Abdel-Sattar and M. R. Meselhy, Cytotoxic and chemomodulatory effects of Phyllanthus niruri in MCF-7 and MCF-7ADR breast cancer cells, Sci. Rep., 2023, 13, 2683 CrossRef CAS PubMed
- M. M. F. Ismail, T. Z. Shawer, R. S. Ibrahim, R. M. Allam and Y. A. Ammar, Novel quinoxaline-based VEGFR-2 inhibitors to halt angiogenesis, Bioorg. Chem., 2023, 139, 106735 CrossRef CAS PubMed
- K. Shari, R. A. El Gedaily, R. M. Allam, K. M. Meselhy, A. E. Khaleel and E. Abdel-Sattar, Jatrophone: a cytotoxic macrocylic diterpene targeting PI3K/AKT/NF-κB pathway, inducing apoptosis
and autophagy in resistant breast cancer cells, BMC Complementary Med. Ther., 2023, 23, 293 CrossRef CAS PubMed
- C.C.G. Inc., Molecular Operating Environment (MOE), 2022.02, Chemical Computing Group Inc., Sherbooke St. West, Suite# 910, Montreal, QC, Canada Search PubMed
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed
- D. E. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00157e |
| This journal is © The Royal Society of Chemistry 2024 |