DOI:
10.1039/D4RA00146J
(Paper)
RSC Adv., 2024,
14, 10182-10190
Boosting oxygen evolution reaction rates with mesoporous Fe-doped MoCo-phosphide nanosheets†
Received
6th January 2024
, Accepted 13th March 2024
First published on 27th March 2024
Abstract
Transition metal-based catalysts are commonly used for water electrolysis and cost-effective hydrogen fuel production due to their exceptional electrochemical performance, particularly in enhancing the efficiency of the oxygen evolution reaction (OER) at the anode. In this study, a novel approach was developed for the preparation of catalysts with abundant active sites and defects. The MoCoFe-phosphide catalyst nanosheets were synthesized using a simple one-step hydrothermal reaction and chemical vapor deposition-based phosphorization. The resulting MoCoFe-phosphide catalyst nanosheets displayed excellent electrical conductivity and a high number of electrochemically active sites, leading to high electrocatalytic activities and efficient kinetics for the OER. The MoCoFe-phosphide catalyst nanosheets demonstrated remarkable catalytic activity, achieving a low overpotential of only 250 mV to achieve the OER at a current density of 10 mA cm−2. The catalyst also exhibited a low Tafel slope of 43.38 mV dec−1 and maintained high stability for OER in alkaline media, surpassing the performance of most other transition metal-based electrocatalysts. The outstanding OER performance can be attributed to the effects of Mo and Fe, which modulate the electronic properties and structures of CoP. The results showed a surface with abundant defects and active sites with a higher proportion of Co2+ active sites, a larger specific surface area, and improved interfacial charge transfer. X-ray photoelectron spectroscopy (XPS) analysis revealed that the catalyst's high activity originates from the presence of Mo6+/Mo4+ and Co2+/Co3+ redox couples, as well as the formation of active metal (oxy)hydroxide species on its surface.
Introduction
The need for environmentally friendly and renewable energy sources has become urgent due to the rapid depletion of conventional fossil fuels and the increasing problem of environmental pollution.1,2 Recently, some environmentally friendly renewable and clean energy technologies, such as fuel cells, metal–air batteries, and water splitting, have been crucial to mitigating environmental pollution and the energy crisis.3–5 Hydrogen energy is considered an ideal energy carrier due to its clean and pollution-free attributes, as well as its high energy density.6,7 Electrochemical water splitting is viewed as a highly efficient method for producing hydrogen energy. However, the oxygen evolution reaction (OER), which is a crucial step in this process, requires a higher overpotential and acts as a bottleneck due to its slow reaction kinetics resulting from a four-electron transfer process.8–10 This limitation significantly impacts the overall efficiency of electrocatalysis for water splitting. Commercially used OER electrocatalysts such as IrO2 and RuO2, which are precious metal oxides with high electrocatalytic activity, face significant limitations in their widespread industrial application due to their high cost and poor stability.11,12 Therefore, there is an urgent need to develop low-cost and highly efficient electrocatalysts to reduce the overpotential required for the OER.13,14 In recent years, there has been a significant rise in the development of non-noble transition metal catalysts, primarily based on metals like cobalt (Co), molybdenum (Mo), and iron (Fe), which are highly attractive due to their low cost, relatively high conductivity, and excellent OER performance.15,16 These transition metal catalysts, which encompass a variety of forms such as metal alloys, oxides, hydroxides, nitrides, sulfides, and phosphides, showed extraordinary catalytic behavior of OER under an alkaline condition.17,18 Transition metal phosphide (TMP) catalysts have attracted considerable attention, particularly those based on cobalt (Co), iron (Fe), and molybdenum (Mo), due to their notable electrical conductivity and high catalytic activity.19–21 These materials have garnered significant interest as potential catalysts in various applications. TMPs possess metal sites and abundant oxygen vacancies (VO), which could serve as catalysts for the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) in alkaline conditions.22,23 Furthermore, they offer a combination of high catalytic activity, excellent electrical conductivity, cost-effectiveness, abundance of constituent elements, stability, and versatility, making them promising electrocatalysts for efficient water electrolysis.24,25 In addition, the favorable structures, well-defined surface morphology, and ample surface area of these materials contribute to an abundance of active sites, thereby bestowing them with distinct properties for catalytic reactions.26–28 These characteristics improve the kinetics of the conversion between the OER intermediates and enhance their overall conductivity. Subsequent results also showed that the morphology and properties of the LDH precursors were influenced by the initial solvents and that the morphology and properties of phosphides and their electrochemical activity are tightly associated.29,30 The advantages of the precursor catalyst porous nanosheets, including their high surface area, efficient mass transport, enhanced charge transport, synergistic effects, structural stability, and tunability, make them highly promising materials for efficient oxygen evolution reactions.31,32 Such unique features are obtained by beginning solvents, which is highly interesting, and TMPs made from LDH precursors with such unique properties need to demonstrate superior catalytic activity against OER. Such unique features are obtained by beginning solvents, which is highly interesting, and TMPs made from LDH precursors with such unique properties need to demonstrate superior catalytic activity against OER. Furthermore, the different kinds of metal active sites and the doping effect the catalytic activity by enhancing the electronic structure. Researchers are still working to completely understand the precise catalytic mechanism of CoFe-based electrocatalysts in the OER. While some contend that Co sites are the principal catalytic active centers,33,34 others point out that Fe species are the intrinsic active substances and that Co species primarily function as a conducting network.35 On the other hand, some suggest a dual-site catalytic mechanism, meaning that the OER involves both Fe and Co sites at the same time.36,37 In our study, Co and Fe active sites beside the Mo active sites are likely to participate in the OER, and their synergistic effect can enhance the electronic structure and the distribution of charge density and, consequently, improve the electrocatalytic performance of the OER.
Herein, we introduce porous nanosheets (PNSs) based on MoCoFeP as effective electrocatalysts for the OER. We will discuss the synthesis approaches and structural characteristics of CoP, MoCoP, and MoCoFeP, as well as its electrocatalytic performance and durability in alkaline conditions. The catalysts are synthesized using a modified method using starting mixture solvents (OAM, ethanol, and water), which begins with the hydrothermal method to create layered double hydroxides (LDH). Subsequently, the LDH is subjected to calcination with NaH2PO2 in a nitrogen (N2) atmosphere, resulting in the formation of metal-based phosphide. Electrochemical analysis reveals that MoCoFeP exhibits a remarkably low overpotential of 250 mV in alkaline conditions (1 M KOH), achieving a current density of 10 mA cm−2. The MoCoFeP PNSs demonstrate excellent conductivity, a very high electrochemical surface area with abundant active sites, a low overpotential with a low Tafel slope of 43.38 mV dec−1, and long-term stability at high current densities. These findings signify the potential of MoCoFeP as a practical way to enhance the activity and stability of water electrolysis, thereby optimizing the OER performance of MoCoFe-based catalysts.
Chemicals, experimental, and physical characterization
Detailed information regarding the materials and chemicals used, experimental procedures, electrochemical measurements, and physical characterization methods and descriptions can be found in the ESI† accompanying the research paper.
Results and discussion
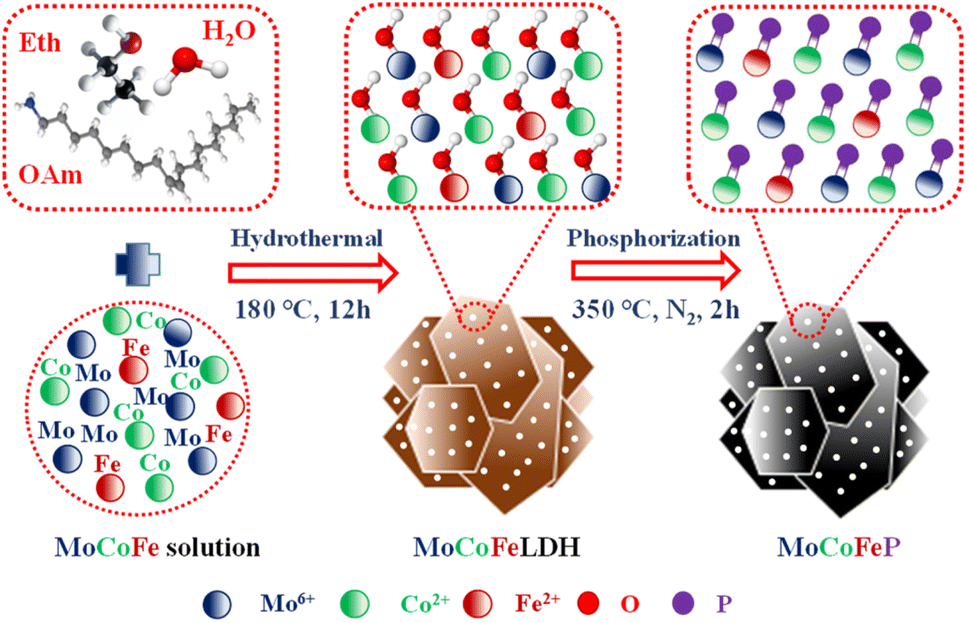
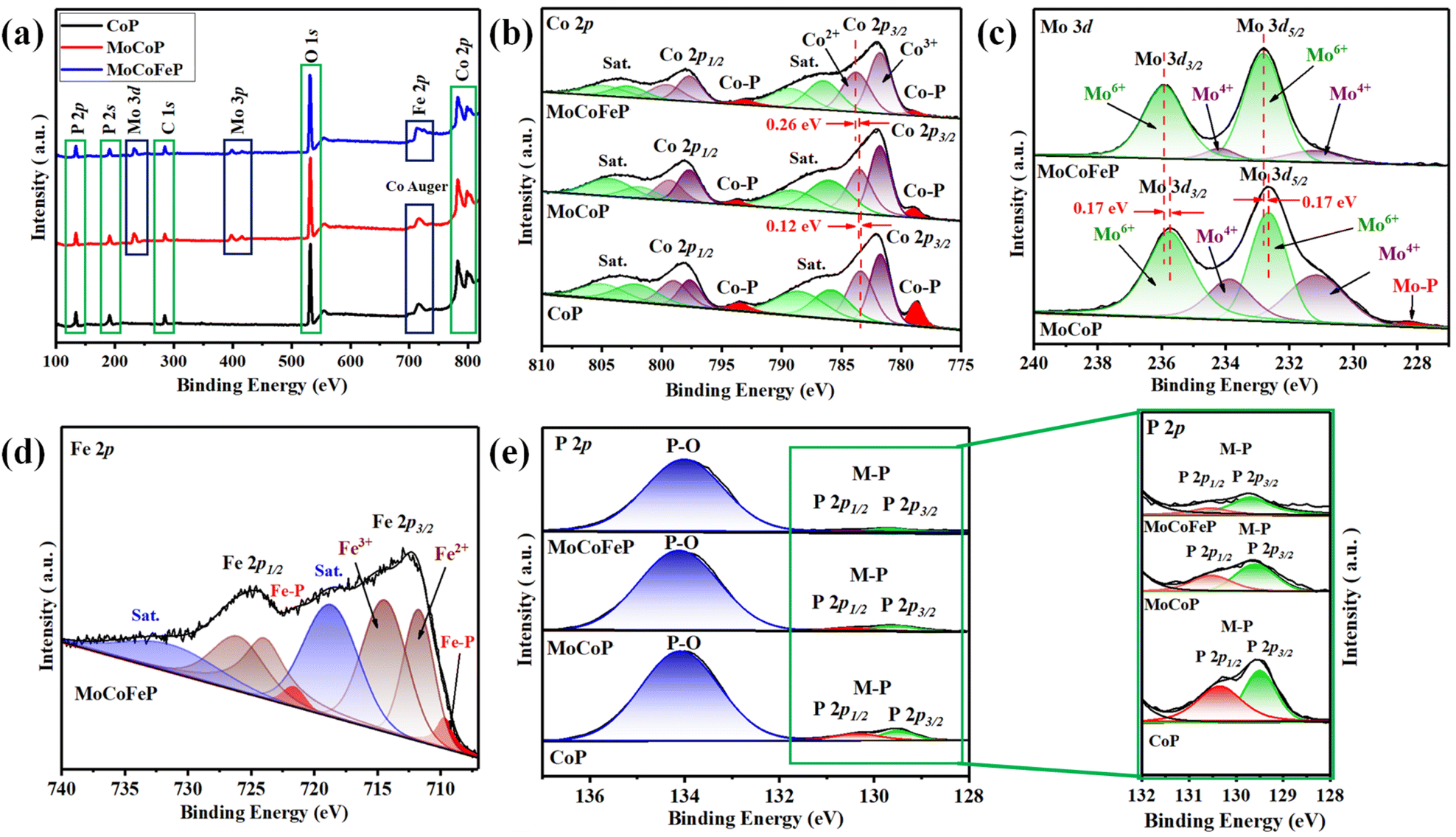
Fig. 1 illustrates the sequential steps involved in the synthesis of the MoCoFeP PNSs catalyst. Initially, MoCoFeLDH PNSs is prepared by subjecting a solution of Mo, Co, and Fe salts with a solvent mixture of water, oleylamine, and ethanol to a hydrothermal process at 180 °C for 12 h. Subsequently, the synthesized MoCoFeLDH PNSs undergo a phosphorization process in a nitrogen (N2) environment at 350 °C for 2 h, leading to the formation of MoCoFeP PNSs. We investigated the morphologies and microstructures of the obtained samples using scanning electron microscopy (SEM). Firstly, the SEM images of the CoP PNSs, MoCoP PNSs, and their precursors (Fig. S1 and S2 ESI†) clearly showed the presence of two-dimensional (2D) thin nanosheets. Secondly, the surface morphology of the MoCoFeLDH PNSs and MoCoFeP PNSs (Fig. S3, ESI†) shows a 2D morphology with thin nanosheets. The MoCoFeP PNSs possess a layered structure that is consistent with its precursor as layered double hydroxides and beneficial for creating a large surface area, which can enhance the material's reactivity and facilitate accessibility to catalytic active sites.38 In addition to SEM imaging, the surface morphology of the MoCoFeP PNSs was investigated using transmission electron microscopy (TEM) imaging. The TEM images in Fig. 2a and b confirmed a 2D morphology with ultrathin nanosheets with abundant active sites. The ultrathin nature of the nanosheets provides a high surface area and a high electrochemical surface area (ECSA), allowing for increased accessibility of active sites.39,40 Furthermore, the BET adsorption theory was used to confirm the mesoporous structure of the catalysts. The hysteresis loop in the BET curve indicated that the catalysts are a typical type IV isotherm, which reveals that the material contains a significant number of mesoporous structures. The MoCoFeP (Fig. S4, ESI†) showed a large surface area 29.4 m2 g−1 with a small pore size 3.936 nm which can enhance the accessibility of the active sites for the reactant molecules, and further improve the catalytic activity of the catalysts. Additionally, the porous structure of the nanosheets facilitates faster mass transportation during the OER process which enhancing the efficiency of the reaction. The selected area electron diffraction (SAED) image in Fig. 2c refers to a polycrystalline structure with ring patterns with low intensity diffraction spots, which indicates that the materials have a low-crystalline structure, which suggests that rich defects are present on the MoCoFeP PNSs surface.41,42 Some of these spots reveal distances of 0.282 nm, 0.247 nm, 0.196 nm, and 0.174 nm, corresponding to the crystal planes (011), (111), (112), and (103) of CoP, respectively. The high-resolution transmission electron microscopy (HRTEM) imaging of one sheet of MoCoFeP PNSs in Fig. 2d and S5 ESI† revealed that there are some lattice fringes in specific regions (areas 1 and 2). Fig. 2e and f, showed that regions (areas 1 and 2) have an interplanar spacing of 0.196 nm and 0.231 nm, respectively, corresponding to the crystal planes (112) and (201) of CoP. It's clear that the surface morphology of MoCoFeP PNSs (area 3 in Fig. S5 ESI†) has abundant lattice defects, which facilitate the development of a surface with abundant active sites.43,44 The dark-filed TEM image in Fig. 2g and elemental EDX mapping in Fig. 2h–m and S6, ESI† provide an overall distribution view of Mo, Co, Fe, P, and O elements. The results confirm that these elements are homogeneously distributed throughout the entire surface area of the MoCoFeP PNSs. Such a unique morphological characteristic makes the MoCoFeP PNSs a promising candidate for various applications, including catalysis and energy storage. The crystalline structure and chemical composition were characterized by the powder X-ray diffraction (XRD) patterns. As exhibited in Fig. S7, ESI,† the XRD pattern of the three precursors confirms the successful preparation of crystalline LDH. While the XRD pattern of the CoP in Fig. S8a, ESI,† is well matched with the standard card of CoP (JCPDS no. 29-0497), confirming the generation of crystallized CoP,.45 After the formation of bimetallic phosphide (Fig. S8b, ESI†), MoCoP showed the same XRD pattern as CoP; nevertheless, the diffraction peaks of MoCoP are weaker than those of CoP, suggesting a decreased crystallinity of MoCoP. After doping with Fe (Fig. S8c, ESI†), the diffraction peaks of the MoCoFeP become weaker and very wide, suggesting the introduction of Mo and Fe to CoP can influence the crystallization behavior of phosphide and make it has low crystalline structure.46,47 Beside the SAED and HRTEM, the XRD confirms the low crystalline structure of MoCoFeP PNSs. The decrease in the crystallinity of CoP structure after doping refers to the abundance of defects and active sites on the surface that promote catalytic activity. To determine the elemental composition of the prepared catalysts-based phosphide, they were analyzed using inductively coupled plasma-optical emission spectroscopy (ICP-OES). Based on the metal ion ratio obtained from the ICP-OES analysis, the prepared catalysts can be identified as Co5P, Mo2.18Co2.9P, and Mo2.09Co3.11Fe1.01P PNSs, respectively, as indicated in Table S1. ESI.† The results showed that the metal composition ratio is very close to their feeding ratio, which confirms the successful preparation of catalysts. The electron structure of the synthesized catalysts was analyzed using X-ray photoelectron spectroscopy (XPS) measurements. The XPS survey spectra in (Fig. 3a and Table S2. ESI†) confirmed the successful preparation of the three catalysts, as they exhibited the presence of Co, P, O, and C for all of them. Additionally, the presence of peaks corresponding to P 2p and P 2s in the samples indicated the successful formation of phosphide-based catalysts. Additionally, MoCoP and MoCoFeP have prominent Mo 2p and Mo 3d peaks, and MoCoFeP has a peak of Fe 2p. No unexpected peaks were observed in the survey spectra, indicating that the synthesized catalysts were pure and free from impurities or contaminants. The Co 2p XPS spectrum of the three catalysts in Fig. 3b, showed a two-spin orbit doublet of Co 2p1/2 and Co 2p3/2, along with satellite humps. The satellite peak and an oxidized peak refer to formation of cobalt phosphate (Co–POx). The presence of both Co3+ and Co2+ species further suggested that the Co ions were partially oxidized and could participate in redox reactions during the electrocatalytic process.48,49 The observation of two small peaks of Co–P, suggesting the involvement of phosphorus in the catalyst's formation to form cobalt phosphide. The slightly higher binding energy observed in the Co 2p XPS spectrum of MoCoFeP PNSs compared to the other catalysts indicated a beneficial for more Co2+ active species and promote the electrocatalytic process. The Mo 3d XPS spectrum displayed in Fig. 3c demonstrates the presence of two Mo 2p5/2 peaks as well as two Mo 2p3/2 peaks partially ionized to Mo4+ and Mo6+. The higher intensity of Mo6+ than Mo4+ and the slightly higher binding energy observed in the Mo 3d XPS spectrum of MoCoFeP PNSs compared to MoCoP PNSs indicated a higher valence state of Mo, which was beneficial for promoting the four-electron proton coupling transfer steps during the OER.50,51 The Fe 2p spectrum displayed in Fig. 3d demonstrates the presence of two Fe 2p3/2 peaks as well as two Fe 2p1/2 peaks. Additionally, there are two shakeup satellite peaks, indicating the presence of both Fe2+ and Fe3+ species. In the Fe 2p XPS spectrum, there are small peaks at 709.7 and 721.6 eV, which are attributed to the presence of Fe–P bonds. These peaks, along with the presence of Fe2+ and Fe3+ species and satellite peaks, suggest that the synthesized catalysts have a complex, diverse, and intricate electronic structure.52,53 The P 2p XPS spectrum (Fig. 3e) displays three peaks, which correspond to phosphate and phosphide peaks (P–O and M–P3/2, M–P1/2), respectively. M–P3/2, and M–P1/2, peaks indicate the presence of metal phosphide in the catalysts that were synthesized. The detection of P–O (phosphates PO43− or PO3− or P2O5) peaks suggests that the metal phosphide has undergone oxidation, leading to a higher degree of oxidation probably caused by the exposure of phosphide species
to air.54,55 Whether in catalysts, the peak strength of P–O is stronger than that of M–P, indicating that the P element is mainly formed by phosphates (PO43−, or PO3−, or P2O5). The intensity of the M–P peak for Co 2p, Mo 3d, and P 2p is decreased in order to CoP > MoCoP > MoCoFeP which is consistent with the change in relative atomic ratio about the P element (Table S2†). This change indicates that the doping process helps to form phosphate species, improves the ability of charge transfer, and ultimately promotes the OER reaction process. Meanwhile, the formation of phosphate can act as a ligand to facilitate the conversion from low-valent to high-valent Co in the redox reaction and promote the four-electron proton coupling transfer steps during the OER, thereby enhancing the activity of the OER.56
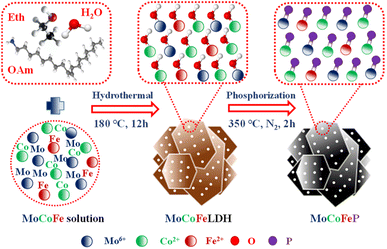 |
| | Fig. 1 Schematic diagram illustrating the synthesis process of MoCoFeP. | |
 |
| | Fig. 2 Surface morphology of MoCoFeP PNSs (a and b) TEM; (c) SAED; (d) HRTEM; (e) lattice distance of the selected area 1 of (d); (f) lattice distance of the selected area 2 of (d); (g) HADAF-STEM; (h–m) EDX mapping elements. | |
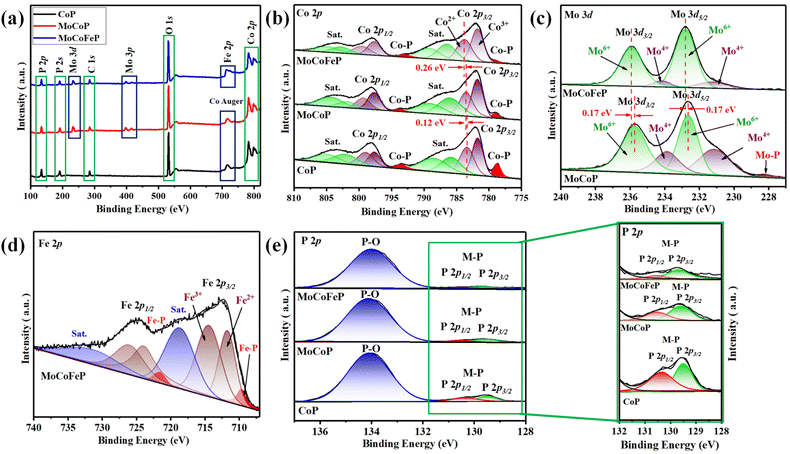 |
| | Fig. 3 XPS spectra of MoCoFeP PNSs and other samples: (a) survey spectra; (b) high-resolution Co 2p; (c) high-resolution Mo 3d; (d) high-resolution Fe 2p; and (e) high-resolution P 2p. | |
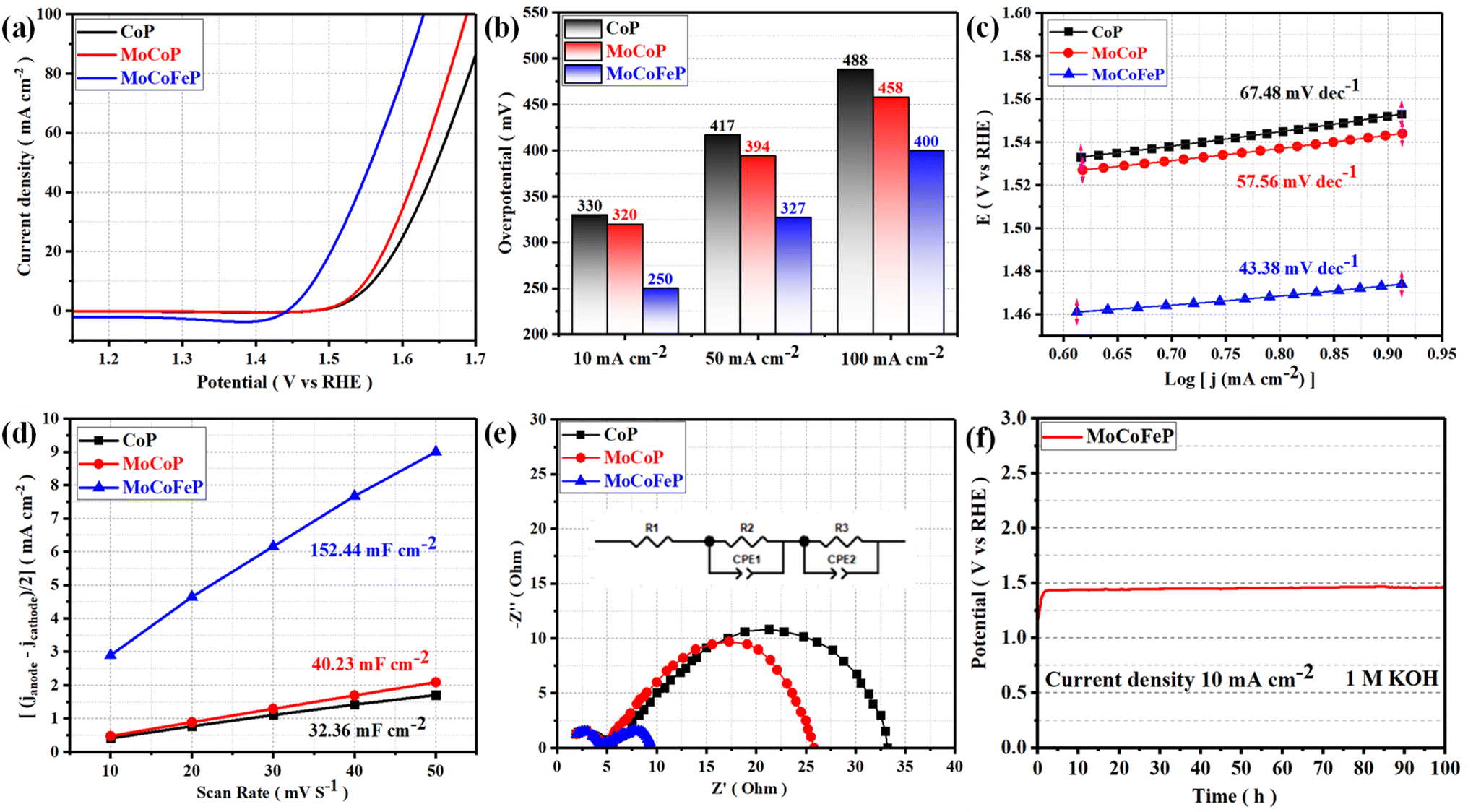
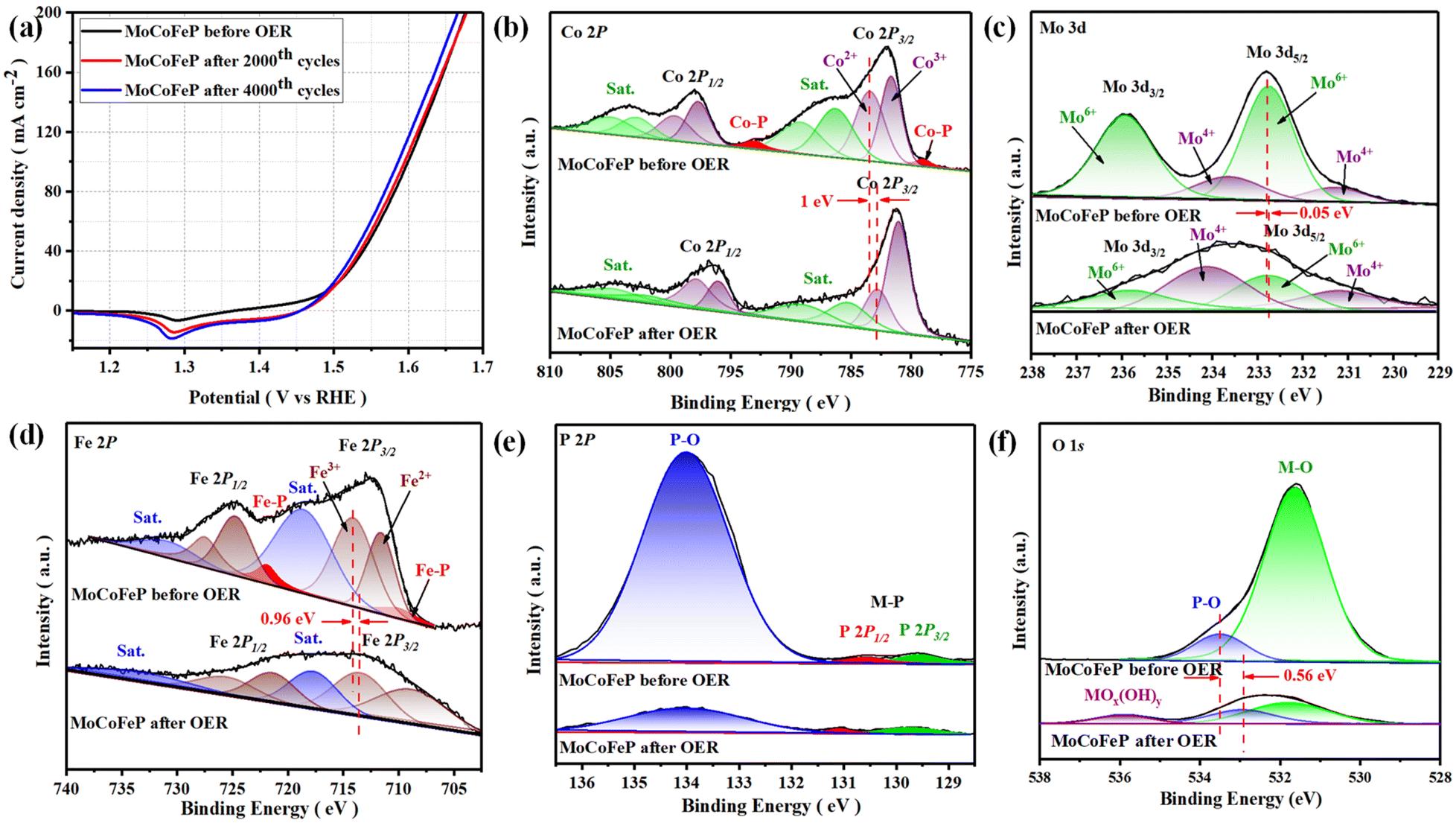
The electrocatalytic performances of the as-prepared samples were investigated in a 1 M KOH solution with typical three-electrode systems. Linear sweep voltammetry (LSV) is used to investigate the electrocatalytic activity of materials for OER. In this technique, a potential sweep was performed at a scan rate of 5 mV s−1, and a working electrode was connected to the rotating disk electrode system and rotated at 1600 rpm. The temperature of the entire system was maintained at 30 °C. Fig. 4a exhibits the LSV curves of the samples. Among these samples, MoCoFeP exhibited a comparatively lower overpotential of 250 mV at 10 mA cm−2 (η10 = 250 mV) which is better than CoP (η10 = 330 mV) and MoCoP (η10 = 320 mV). This result indicates that the introduction of a proper amount of Mo and Fe can effectively improve the electrocatalytic OER activities of the catalysts, which may be caused by forming, exposing more active sites, abundant vacancies, and defects that accelerate the charge transfer between the intermediates and the active sites and required less energy to drive the reaction compared to the other prepared catalysts. The overpotential plot of the synthesized samples at current densities of 10, 50, and 100 mA cm−2 shows that the electrocatalytic behavior of MoCoFeP is significantly better than that of the other samples (Fig. 4b). The OER reaction kinetics was evaluated by the Tafel slope derived from the LSV curves. As shown in Fig. 4c, MoCoFeP presents a Tafel slope of 43.38 mV dec−1, which is much smaller than that of MoCoP (57.56 mV dec−1), and CoP (67.48 mV dec−1), indicating that the doping of MoCoP by Fe to form MoCoFeP can improve the OER reaction kinetics and accelerate the rate of the OER. The intrinsic properties of the electrocatalytic surface of the prepared samples were investigated using electrochemical double-layer capacitance (Cdl). As shown in Fig. 4d, the Cdl value decreases in the order of MoCoFeP (152.44 mF cm−2), MoCoP (40.23 mF cm−2), and CoP (32.36 mF cm−2). The results suggest that the MoCoFeP PNSs have a superior electrochemically active surface area that is higher than that of MoCoP and CoP, which is closely related to the density of active sites on the surface, which reflects the reason for the high catalytic activity of MoCoFeP. Electrochemical impedance spectroscopy (EIS) was employed to study the charge transfer dynamics between the working electrode, catalyst, and electrolyte at a potential of 1.52 V (vs. the reversible hydrogen electrode (RHE)). The Nyquist plots of all the samples were fitted to determine the charge transfer resistance (Rct). The semicircle diameter obtained from the EIS analysis is related to the charge transfer resistance (Rct) between the catalyst and the working electrode. The results shown in Fig. 4e indicate that the MoCoFeP PNSs have the smallest Nyquist fitting semicircle diameter, which is indicative of the lowest Rct value of 5 Ω, compared to the MoCoP and CoP PNSs, which showed Rct values of 21 Ω, and 28 Ω, respectively. MoCoFeP PNSs has low impedance during charge transfer within the electrode. This is due to the high ECSA and high density of OER active sites on the surface of the MoCoFeP PNSs, which can accelerate the rate of the OER reaction and decrease the overpotential compared to the other prepared catalysts. (Fig. S9a, ESI†) exhibits the catalytic activity of the LDH precursors. Among these samples, MoCoFeLDH exhibited a comparatively lower overpotential (η10 = 288 mV), which is better than CoLDH (η10 = 310 mV) and MoCoLDH (η10 = 327 mV). This result indicates that CoLDH has better catalytic activity than MoCoLDH due to oxidization of Co(OH)2 during the hydrothermal process to form structure of Co(OH)2/Co3O4, which can generate some oxygen vacancies (VO) and improve the electronic structure. The introduction of a proper amount of Mo and Fe can effectively improve the electronic structure of the catalysts, which may be caused by forming more active sites and defects, and accelerating the charge transfer between the intermediates and the active sites. The overpotential plot of the synthesized samples at current densities of 10, 50, and 100 mA cm−2 shows that the electrocatalytic behavior of MoCoFeLDH is significantly better than that of the other samples (Fig. S9b, ESI†). As shown in Fig. S9c, ESI† MoCoFeLDH presents a Tafel slope of 43.46 mV dec−1, which is much smaller than that of MoCoLDH (56.05 mV dec−1), and CoLDH (50.07 mV dec−1), indicating that the introduction of Mo and Fe can improve the OER reaction kinetics and accelerate the rate of OER and required less energy to drive the reaction compared to the other prepared catalysts. As shown in Fig. S9d, ESI,† the Cdl value decreases in the order of MoCoFeLDH (51.28 mF cm−2), CoLDH (43.62 mF cm−2), and MoCoLDH (23.42 mF cm−2). The results suggest that the MoCoFeLDH PNSs have ECSA which is higher than CoLDH, and MoCoLDH, which is closely related to the density of active sites on the surface which reflect the reason for the high catalytic activity of MoCoFeLDH. The results shown in Fig. S9e, ESI,† indicate that the MoCoFeLDH PNSs have the lowest Rct value of 6 Ω, compared to the CoLDH and MoCoLDH PNSs, which showed Rct values of 16 Ω, and 18 Ω, respectively. MoCoFeLDH PNSs has a low Rct due to the high ECSA and high density of OER active sites on the surface of the MoCoFeLDH PNSs, which can accelerate the rate of the OER reaction and decrease the overpotential compared to the other prepared catalysts. The MoCoFeP PNSs maintained the same advantage as its precursor MoCoFeLDH surface morphology, high surface area. Meanwhile, the formation of phosphate can act as a ligand to facilitate the conversion from low-valent to high-valent Co in the redox reaction, and promotes the four-electron proton coupling transfer steps during the OER, thereby enhancing the activity of the OER. To study the effect of Fe concentration on the catalytic activity and the overpotential we prepared MoCoFeP with different Fe concentration (0.05, 0.1, 0.15, and 0.2 mmol of iron(II) nitrate nonahydrate Fe(NO3)2·9H2O) (Fig. S10, ESI†). The results shown that the MoCoFeP with Fe doping 0.1 mmol has the best catalytic activity with overpotential 250 mV at current density 10 mA cm−2. The long-term electrochemical durability of the MoCoFeP PNSs catalyst was studied by a chronopotentiometric test. As shown in Fig. 4f, the potential almost remains stable and just shows a slight increase during the 100 h OER measurement, demonstrating the good electrochemical stability of the MoCoFeP PNSs catalyst in 1 M KOH. To further approve the electrocatalytic durability of the MoCoFeP PNSs catalyst, the LSV curves of the MoCoFeP PNSs catalyst before and after 4000 continuous CV cycles are also compared (Fig. 5a). The LSV curve after the cyclic test stays almost unchanged, further validating the excellent catalytic stability. This suggests that the MoCoFeP PNSs catalyst can maintain their electrocatalytic activity and stability even under high current densities, making them a promising candidate for practical applications in various electrochemical processes. Such outstanding electrochemical stability of the MoCoFeP PNSs catalyst could come from its structural and chemical stability during the OER process. The XRD pattern of the post-OER sample (Fig. S11a, ESI†) reveals that the catalyst remains low crystalline structure without the appearance of unexpected peaks. The SEM image (Fig. S11b and c ESI†) revealed that the post-OER surface morphology of the MoCoFeP PNSs maintained nanosheets after the cyclic test. The XPS spectrum of the initial and the post-OER of the MoCoFeP PNSs catalyst was also studied to investigate the change in the electronic structure after the cyclic durability test of the OER (Fig. S12, ESI†). The XPS spectrum shows the same peaks, and there are no unexpected peaks. The Co 2p XPS profile (Fig. 5b) shows a decreasing intensity of the Co2+ peak and an increasing intensity of the Co3+ peak with a negative shift of binding energy estimated by 1 eV, which increases the high oxidation state Co3+ and decreases the lower oxidation state Co2+. Also, the two peaks that refer to Co–P disappeared after OER, which indicates that the Co–P was changed chemically during the OER to cobalt phosphate or any OER intermediates.57,58 The Mo 3d XPS profile (Fig. 5c) shows a decreasing intensity of the Mo6+ peak and an increasing intensity of the Mo4+ peak with a small
shift of binding energy by 0.05 eV, which increases the lower oxidation state Mo4+ and decreases the higher oxidation state Mo6+. The Fe 2p XPS profile (Fig. 5d) shows a negative change in binding energy equal to 0.96 eV, increasing the lower oxidation state Fe2+ and decreasing the high oxidation state Fe3+ and disappearing the Fe–P peaks. The P 2p XPS profile (Fig. 5e) showed a decrease in the peaks intensity, which refer to the fact that firstly, the metal phosphides are usually electrochemically oxidized into metal (oxy)hydroxides (CoOx(OH)y) or oxides during the OER process and P-containing species (phosphates PO43−, or PO3−, or P2O5), which is also the main mode of P–O binding. Secondly, the P-containing species (phosphates PO43−, or PO3−, or P2O5) electrochemically oxidized and facilitate the generation of CoOx(OH)y and the electron transfer during the OER reaction.59,60 The O 1s (Fig. 5f) also showed a decrease in the intensity of M–O and P–O peaks, and a new peak of CoOx(OH)y appears for MoCoFeP after OER with a negative shift of 0.56 eV for the P–O peak. The results show that during the OER process, the Co–P electrochemically oxidized to polyphosphates on the surface of MoCoFeP PNSs which dissolve in alkali and form active catalytic sites with the structure of CoOx(OH)y, as previously reported.61 Regarding the oxidation state, the XPS analysis shows that the transition from Co2+ to Co3+ is responsible for the enhanced catalytic activity of Co-based OER catalysts. The reason is that the higher oxidation state of Co3+ can facilitate the formation of more oxygen vacancies on the catalyst surface, which can then act as active sites for the OER process. The higher oxidation states can lead to the formation of more stable surface species, which can help improve the catalyst's stability and durability. To know the surface reconstruction the CV curves in the potential range from 0.9 to 1.8 V, (Fig. S13, ESI†) show two pairs of redox peaks around 1.17 and 1.49 V, which relate to the Co2+/Co3+ redox couple and Co3+/Co4+ redox couple, respectively.62 CV curves clearly show that the MoCoFeP have stronger Co2+/Co3+ and Co3+/Co4+ electrochemical response than the MoCoFeLDH, indicating that the MoCoFeP contain more electroactive sites in comparison to the MoCoFeLDH due to the abundant defect atoms at the edge of pores. The MoCoFeP PNSs show superior electrocatalytic performance compared to other MoCoFe-based OER catalysts that have been recently reported. This is demonstrated by their performance in the evaluation of both overpotential and Tafel slope, as shown in Fig. S9f and Table S3 ESI.† The exceptional electrocatalytic activity of the MoCoFeP PNSs can be attributed to their unique characteristics, including their porous nanosheets with abundant defects with a large surface area and high electrochemically active surface area (ECSA), a high concentration of active sites, and low resistance to charge transfer within the electrode.
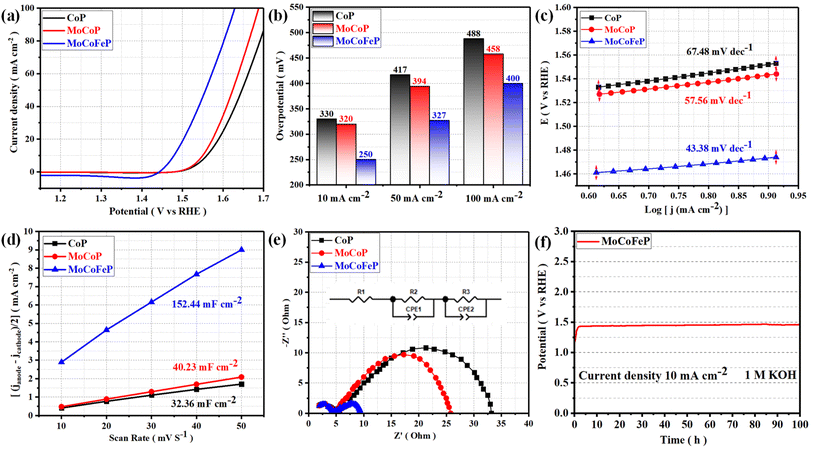 |
| | Fig. 4 Electrochemical measurement results of CoP, MoCoP, and MoCoFeP PNSs: (a) LSV curves of OER; (b) overpotential histogram at 10, 50, and 100 mA cm−2; (c) Tafel plots; (d) the capacitive current at 1.22 V vs. RHE as a function of the scan rate; (e) Nyquist plots measured at 0.6 V versus Hg/HgO (inset equivalent circuit diagram) (f) chronopotentiometry of the MoCoFeP at a current density of 10 A cm−2 for OER. | |
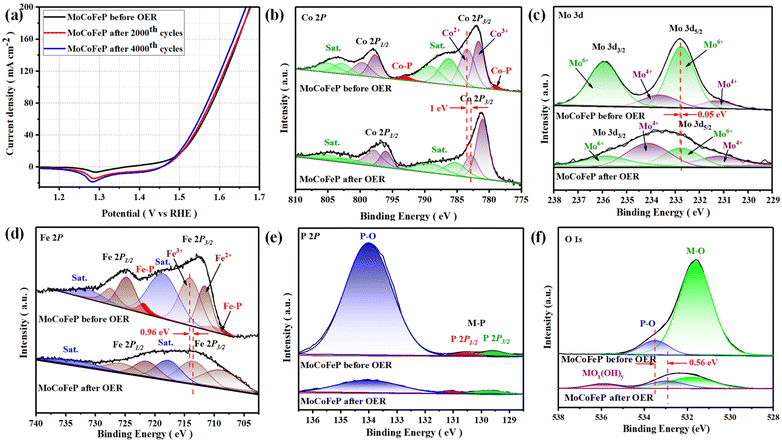 |
| | Fig. 5 (a) LSV curves of MoCoFeP PNSs before and after multiple cycles stability; XPS spectra of MoCoFeP PNSs before and after cyclic stability; (b) Co 2p; (c) Mo 3d; (d) Fe 2p; (e) P 2p; (f) O 1s. | |
Conclusion
In summary, MoCoFeP has been prepared by a simple one-step hydrothermal reaction using a mixture solvent of oleylamine, ethanol and water and chemical vapor deposition-based phosphorization. SEM and TEM revealed a low crystalline surface morphology with abundant active sites. The low crystallinity of MoCoFeP reveals exceptional electrocatalytic activity, which exhibits an overpotential of only 250 mV at 10 mA cm−2, a small Tafel slope of 43.38 mV dec−1, and excellent electrochemical durability in alkaline. It is found that the introduction of Mo and Fe can effectively affect the crystallinity of cobalt phosphide, the ECSA of the electrocatalysts, and expose more catalytic active sites. Meanwhile, the enhanced electrical conductivity promotes electron transfer, which boosts the electrocatalytic performance. X-ray photoelectron spectroscopy (XPS) reveals that the doping process for the CoP can enhance the electronic structure and produce a lot of active sites and defects. These advantages ensure that the MoCoFeP electrocatalysts have good application prospects in practical electrochemical water splitting. This work provides a new path for optimizing metal phosphide electrocatalysts.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of Hubei Province (2021CFB144) and the Large-Scale Instrument and Equipment Sharing Foundation of Wuhan University.
References
- J. L. Holechek, H. M. Geli, M. N. Sawalhah and R. Valdez, Sustainability, 2022, 14, 4792 CrossRef.
- N. Abas, A. Kalair and N. Khan, Futures, 2015, 69, 31–49 CrossRef.
- D. Likius, A. Rahman, C. Zivayi and V. Uahengo, Catal. Lett., 2020, 150, 1942–1956 CrossRef CAS.
- N. Javed, T. Noor, N. Iqbal and S. R. Naqvi, RSC Adv., 2023, 13, 1137–1161 RSC.
- R. Anand, B. Ram, M. Umer, M. Zafari, S. Umer, G. Lee and K. S. Kim, J. Mater. Chem. A, 2022, 10, 22500–22511 RSC.
- X. Zhang, P. Liu and Y. Zhang, Inorg. Chim. Acta, 2023, 557, 121683 CrossRef CAS.
- Y. Chen, M. Wu, J. Li, Q. Zhao, S. Zhang, Y. Guo, F. Feng and Z. Li, Colloids Surf., A, 2023, 678, 132514 CrossRef CAS.
- A. Rajput, A. Kundu and B. Chakraborty, ChemElectroChem, 2021, 8, 1698–1722 CrossRef CAS.
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843–3876 RSC.
- R. Y. Fan, J. Y. Xie, N. Yu, Y. M. Chai and B. Dong, Int. J. Hydrogen Energy, 2022, 47, 10547–10572 CrossRef CAS.
- N. T. T. Thao, J. U. Jang, A. K. Nayak and H. Han, Small Sci., 2024, 4, 2300109 CrossRef CAS.
- L. Liu, W. An, F. Gu, L. Cui, X. He and M. Fan, Green Chem., 2023, 25, 6149–6169 RSC.
- J. Zhao, J. J. Zhang, Z. Y. Li and X. H. Bu, Small, 2020, 16, 2003916 CrossRef CAS PubMed.
- L. Wu, H. Qin, Z. Ji, H. Zhou, X. Shen, G. Zhu and A. Yuan, Small, 2023, 20, 2305965 CrossRef PubMed.
- C. Hou, Z. Cui, S. Zhang, W. Yang, H. Gao and X. Luo, RSC Adv., 2021, 11, 37624–37630 RSC.
- L. Li, X. Cao, J. Huo, J. Qu, W. Chen, C. Liu, Y. Zhao, H. Liu and G. Wang, J. Energy Chem., 2023, 76, 195–213 CrossRef CAS.
- S. H. Li, M. Y. Qi, Z. R. Tang and Y. J. Xu, Chem. Soc. Rev., 2021, 50, 7539–7586 RSC.
- S. Gopi, V. Selvamani and K. Yun, Inorg. Chem., 2021, 60, 10772–10780 CrossRef CAS PubMed.
- Y. Li, R. Li, D. Wang, H. Xu, F. Meng, D. Dong, J. Jiang, J. Zhang, M. An and P. Yang, Int. J. Hydrogen Energy, 2021, 46, 5131–5149 CrossRef CAS.
- J. Li, Z. Wang, P. Wang, Z. Zheng, Y. Liu, H. Cheng and B. Huang, RSC Adv., 2022, 12, 13639–13644 RSC.
- L. M. Cao, J. Zhang, L. W. Ding, Z. Y. Du and C. T. He, J. Energy Chem., 2022, 68, 494–520 CrossRef CAS.
- Y. Liu, M. Ding, X. Deng, Y. Zhang and G. Zhao, RSC Adv., 2022, 12, 9051–9057 RSC.
- S. S. Liu, L. J. Ma and J. S. Li, J. Colloid Interface Sci., 2023, 631, 147–153 CrossRef CAS PubMed.
- C. D. Nguyen, V. H. Nguyen, L. M. T. Pham and T. Y. Vu, Int. J. Hydrogen Energy, 2020, 45, 15063–15075 CrossRef CAS.
- F. Tang, Y. W. Zhao, Y. Ge, Y. G. Sun, Y. Zhang, X. L. Yang, A. M. Cao, J. H. Qiu and X. J. Lin, J. Colloid Interface Sci., 2022, 628, 524–533 CrossRef CAS PubMed.
- W. Li, D. Xiong, X. Gao and L. Liu, Chem. Commun., 2019, 55, 8744–8763 RSC.
- M. Jiang, J. Li, J. Li, Y. Zhao, L. Pan, Q. Cao, D. Wang and Y. Du, Nanoscale, 2019, 11, 9654–9660 RSC.
- Z. Li, H. Feng, M. Song, C. He, W. Zhuang and L. Tian, Mater. Today Energy, 2021, 20, 100698 CrossRef CAS.
- J. Yu, Z. Li, T. Liu, S. Zhao, D. Guan, D. Chen, Z. Shao and M. Ni, Chem. Eng. J., 2023, 460, 141674 CrossRef CAS.
- M. Liu, Q. He, S. Huang, W. Zou, J. Cong, X. Xiao, P. Li, J. Cai and L. Hou, ACS Appl. Mater. Interfaces, 2021, 13, 9932–9941 CrossRef CAS PubMed.
- S. Xu, Y. Qi, Y. Lu, S. Sun, Y. Liu and D. Jiang, Int. J. Hydrogen Energy, 2021, 46, 26391–26401 CrossRef CAS.
- J. Hei, G. Xu, B. Wei, L. Zhang, H. Ding and D. Liu, Appl. Surf. Sci., 2021, 549, 149297 CrossRef CAS.
- G. A. Gebreslase, M. V. Martínez-Huerta and M. J. Lázaro, J. Energy Chem., 2022, 67, 101–137 CrossRef CAS.
- Q. Li, R. Cao, J. Cho and G. Wu, Adv. Energy Mater., 2014, 4, 1301415 CrossRef.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- D. Zhang, W. Chen, Z. Li, Y. Chen, L. Zheng, Y. Gong, Q. Li, R. Shen, Y. Han, W. C. Cheong and L. Gu, Chem. Commun., 2018, 54, 4274–4277 RSC.
- L. Bai, C. S. Hsu, D. T. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS PubMed.
- L. Lv, Z. Yang, K. Chen, C. Wang and Y. Xiong, Adv. Energy Mater., 2019, 9, 1803358 CrossRef.
- Y. Yang, M. Yuan, H. Li, G. Sun and S. Ma, Electrochim. Acta, 2018, 281, 198–207 CrossRef CAS.
- B. Chen, Z. Zhang, S. Kim, S. Lee, J. Lee, W. Kim and K. Yong, ACS Appl. Mater. Interfaces, 2018, 10, 44518–44526 CrossRef CAS PubMed.
- P. Han, S. Hua, J. Ji, Y. Wu, L. Ma, H. Xu, X. Sun, S. Yu, S. Chen, J. Xiao and Y. Dang, J. Alloys Compd., 2023, 932, 167538 CrossRef CAS.
- W. Zhang, Y. Li, L. Zhou, Q. Zheng, F. Xie, K. H. Lam and D. Lin, Electrochim. Acta, 2019, 323, 134595 CrossRef CAS.
- H. Huang, A. Cho, S. Kim, H. Jun, A. Lee, J. W. Han and J. Lee, Adv. Funct. Mater., 2020, 30, 2003889 CrossRef CAS.
- P. Viswanathan and K. Kim, ACS Appl. Mater. Interfaces, 2023, 15, 16571–16583 CrossRef CAS PubMed.
- G. Yuan, J. Bai, L. Zhang, X. Chen and L. Ren, Appl. Catal., B, 2021, 284, 119693 CrossRef CAS.
- T. Wang, C. Wang, Y. Jin, A. Sviripa, J. Liang, J. Han, Y. Huang, Q. Li and G. Wu, J. Mater. Chem. A, 2017, 5, 25378–25384 RSC.
- H. Huang, A. Cho, S. Kim, H. Jun, A. Lee, J. W. Han and J. Lee, Adv. Funct. Mater., 2020, 30, 2003889 CrossRef CAS.
- M. Hao, Z. Xu, X. Liu, J. Ma, L. Wang, C. Li and W. Wang, Int. J. Hydrogen Energy, 2023, 48, 147–159 CrossRef CAS.
- J. Guo, Z. Zhan, T. Lei and P. Yin, Int. J. Hydrogen Energy, 2022, 47, 5974–5989 CrossRef CAS.
- L. Feng, P. Yang, W. Ling, S. Wang, J. Shi and F. Wang, J. Phys. Chem. Solids, 2021, 148, 109697 CrossRef CAS.
- L. Zhao, S. Chen, Y. Zhao, Q. Kuang, Q. Fan and Y. Dong, Solid State Ionics, 2020, 348, 115280 CrossRef CAS.
- M. Gao, P. Gao, T. Lei, C. Ouyang, X. Wu, A. Wu and Y. Du, J. Mater. Chem. A, 2022, 10, 15569–15579 RSC.
- X. Ren, Y. Tian, F. Shaik, J. Yang, R. Liu, K. Guo and B. Jiang, Adv. Sustainable Syst., 2022, 6, 2100436 CrossRef CAS.
- X. Li, L. Xiao, L. Zhou, Q. Xu, J. Weng, J. Xu and B. Liu, Angew. Chem., 2020, 132, 21292–21299 CrossRef.
- Y. Zhang, L. Gao, E. J. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- P. Kumar, F. Dinsmore and W. Miao, ACS Appl. Energy Mater., 2022, 5, 12602–12613 CrossRef CAS.
- L. Chen, J. T. Ren and Z. Y. Yuan, Sci. China Mater., 2022, 65, 2433–2444 CrossRef CAS.
- L. Yan, B. Zhang, J. Zhu, Y. Li, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2020, 265, 118555 CrossRef CAS.
- H. Xiao, X. Du, M. Zhao, Y. Li, T. Hu, H. Wu, J. Jia and N. Yang, Nanoscale, 2021, 13, 7381–7388 RSC.
- H. Han, F. Yi, S. Choi, J. Kim, J. Kwon, K. Park and T. Song, J. Alloys Compd., 2020, 846, 156350 CrossRef CAS.
- R. Beltrán-Suito, P. W. Menezes and M. Driess, J. Mater. Chem. A, 2019, 7, 15749–15756 RSC.
- Y. Li, F.-M. Li, X.-Y. Meng, S.-N. Li, J.-H. Zeng and Y. Chen, ACS Catal., 2018, 8, 1913–1920 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Gongzhen Cheng
c,
Gongzhen Cheng