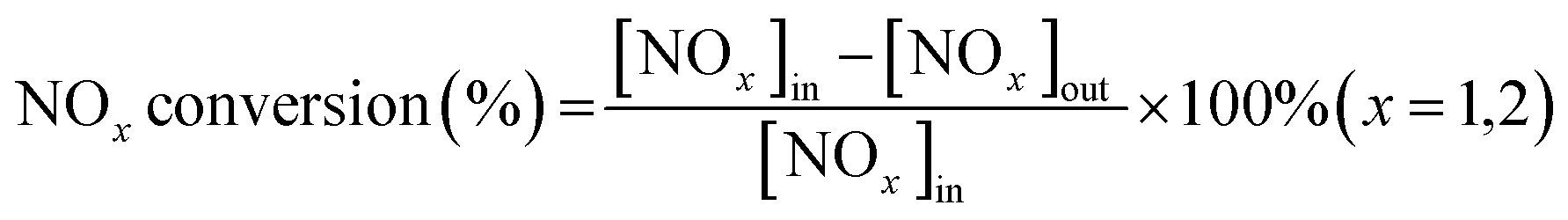DOI:
10.1039/D3RA08782D
(Paper)
RSC Adv., 2024,
14, 7206-7214
Mechanism of SO2/H2O enhanced rare earth tailings catalysts in NH3-SCR at medium and high temperature
Received
23rd December 2023
, Accepted 12th January 2024
First published on 28th February 2024
Abstract
Rare earth tailings (RET) NH3-SCR catalysts were prepared by mechanical and microwave activation of a large amount of rare earth tailings after beneficiation of Bayan Ebo rare earth ore. The effects of SO2/H2O on the denitrification performance of the RET catalysts were evaluated by conducting denitrification activity tests, SO2/H2O tolerance tests and in situ DRIFTs mechanistic analysis. The results showed that the denitrification activity was significantly increased in the presence of SO2/H2O. And in situ DRIFTs analysis showed that in the presence of SO2/H2O, SO2 could be adsorbed as SO32− groups by the hydroxyl groups on the catalyst surface and react with SO42− to form S2O72− species. And in the presence of NH3, S2O72− would decompose into unstable SO42− species and SO32− and continue to react cyclically to form S2O72− species, providing the RET catalyst provides more acid sites, facilitating the SCR reaction.
1 Introduction
NOx is considered to be one of the serious air pollutants and its main source is industrial combustion emissions from fossil fuels in thermal power plants. NH3-SCR technology is an effective method for NOx removal.1–3 V2O5–WO3(MoO3)/TiO2 has been used industrially for many years,4,5 but its applicable denitrification temperature range is narrow. To broaden or reduce the applicable temperature range, researchers have carried out work on the preparation of NH3-SCR catalysts using transition metal elements,6–13 which mainly focused on Mn-based, Fe-based and rare earth-based catalysts. It was found that Mn-based catalysts have a good De-NOX activity in the low temperature band but poor SO2/H2O tolerance.6–8 Fe-based catalysts have some activity (lower than Mn-based catalysts) in the medium to high temperature band (150–450 °C) and have better SO2 tolerance.9–13 Single metal oxide catalysts usually have certain defects, so they are usually loaded with other elements to form composite metal oxides for the preparation of the NH3-SCR catalysts to compensate for each other's defects.
Bayan Ebo has a large amount of rare earth tailings after beneficiation, which contains a rich variety of NH3-SCR active elements (Fe, Ce, Mn),14 of which rare earth elements are excellent NH3-SCR catalyst active components.15 In addition, there are congenial relationships such as adjacency, leaching and encapsulation of various active minerals in rare earth tailings, and the interactions that exist between these congenial minerals can also facilitate the SCR reaction process,16,17 so rare earth tailings are naturally excellent materials that can be used to prepare NH3-SCR catalysts.
SO2/H2O poisoning phenomenon seriously affects the activity of SCR catalysts. Due to the increase of HSO4− and SO42− on the catalyst surface, it often leads to accumulation of ammonium sulfate on the catalyst surface and irreversible sulfation of the active components of the catalyst leading to decreased activity. So improving the SO2/H2O tolerance of catalysts is directly related to the practical application of catalysts and is an unavoidable indicator in the evaluation of NH3-SCR catalysts. Composite oxide catalysts doped with rare earth elements exhibit better SO2/H2O tolerance and denitrification efficiency compared to Fe-based and Mn-based denitrification catalysts.18–21 In contrast, rare earth tailings with multiple active components (Fe, Ce, Mn) are natural composite oxides, and rare earth tailings catalysts have better SO2/H2O tolerance performance than most composite oxide catalysts.
Therefore, it is necessary to investigate the SO2/H2O tolerance characteristics of the Bayan Ebo rare earth tailings catalysts (RET) in the NH3-SCR denitrification process and to investigate the SO2/H2O tolerance mechanism in the denitrification process. This paper explores the process and tolerance mechanism of SO2/H2O on the denitrification performance of the RET catalysts and expands the research field of NH3-SCR denitrification with rare earth tailings catalysts from Bayan Ebo.
2 Materials and methods
2.1 Catalysts preparation
The rare earth tailings catalyst was prepared by ball mills. And the rare earth tailings come from Bayan Ebo (Baotou, China). The beneficiated rare earth tailings material was fixed in a canister on a planetary ball mill and ball milled at 300 rpm for 2 h. The obtained material was passed through a 200 mesh sieve and microwave roasted at 1100 w, 250 °C for 20 min to obtain the catalyst powder. The elemental content of the catalyst was quantified by XRF. As shown in Table 1, the results of the XRF analysis, in which Fe, Ca, Si, Mg, Ce and Al were the major elements with a total content of 50.4%, and the remaining 49.6% are Mn and other trace elements, and the content of each trace element is <1%. The Bayan Ebo rare earth tailings catalyst is indicated as RET.
Table 1 Rare earth tailings catalyst (RET) XRF results
| Element |
Fe |
Ca |
Si |
Mg |
Ce |
Al |
Mn |
Other |
| Percentage (%) |
17.394 |
17.798 |
8.804 |
3.104 |
1.783 |
1.485 |
0.926 |
48.706 |
2.2 Catalytic performance experiment
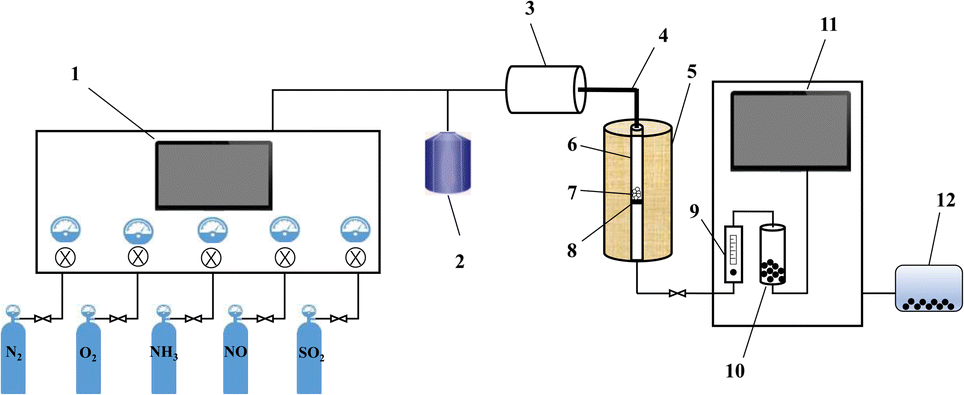
The NH3-SCR performance experiments with the RET catalysts were carried out in a SCR denitrification evaluation unit, as shown in Fig. 1. The denitrification evaluation unit is consisted of gas distribution system (1–4), reaction system (5–8) and analysis system (9–11), and is equipped with a tail gas treatment unit (12). The sample is fixed in a 9 mm inner diameter quartz fixed bed reaction tube. The reaction gas mixture consists of NO (500 ppm), NH3 (500 ppm), H2O (6 vol% when in use), SO2 (500 ppm when in use), O2 (6 vol%) and N2 are controlled by the gas distribution system at a flow rate (100 mL min−1), and the GHSV are set to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The gas mixture were measured by on-line multi-component flue gas analyzer (HP-OMGA, China), the measurement accuracy is ≤± 2% FS, and the NOX conversion is obtained from the formula (1):
000 h−1. The gas mixture were measured by on-line multi-component flue gas analyzer (HP-OMGA, China), the measurement accuracy is ≤± 2% FS, and the NOX conversion is obtained from the formula (1):| |
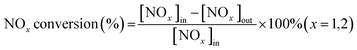 | (1) |
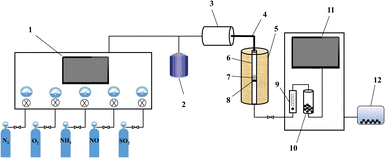 |
| | Fig. 1 Schematic diagram of the NH3-SCR evaluation unit ((1) gas flow control system; (2) heating water tank; (3) gas distribution tank; (4) heating pipes; (5) heating furnace; (6) quartz tubes; (7) RET catalyst; (8) quartz wool; (9) rotameter; (10) condensation drying device; (11) flue gas online detection device; (12) tail gas treatment device). | |
2.3 Catalysts characterization
The RET catalysts were characterization by the X-ray diffraction (XRD), temperature programmed reduction by H2 and NH3 (H2-TPR and NH3-TPD), and in situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS).
3 Results and discussion
The RET catalysts after denitrification under different operating conditions were characterised as sample 0, 1, 2 and 3. The total flow rate for all operating condition cases is 100 mL min−1, as shown in Table 2.
Table 2 Working conditions for the preparation of the four RET catalysts
| Catalysts |
NH3/ppm |
NO/ppm |
O2/% |
H2O/% |
SO2/ppm |
| Sample 0 |
500 |
500 |
6 |
— |
— |
| Sample 1 |
500 |
500 |
6 |
6 |
— |
| Sample 2 |
500 |
500 |
6 |
— |
500 |
| Sample 3 |
500 |
500 |
6 |
6 |
500 |
3.1 Structural changes of catalysts
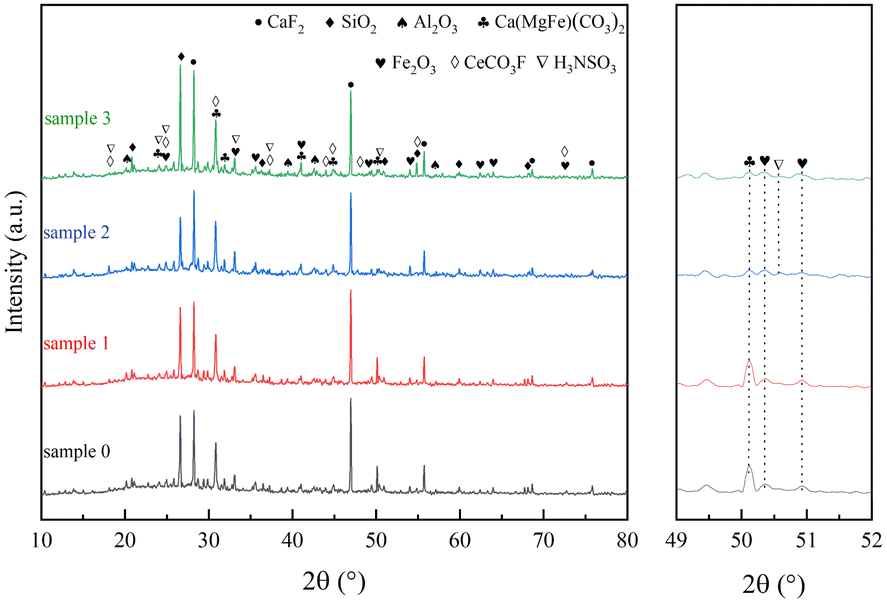
As shown in Fig. 2, the diffraction peaks of fluorite (CaF2), quartz (SiO2), aluminium trioxide (Al2O3), iron dolomite {Ca(MgFe)[(CO3)2]}, hematite (Fe2O3) and cerium fluoride (CeCO3F) were mainly detected on the surface of sample 0, where the Ca(MgFe)[(CO3)2], Fe2O3 and CeCO3F diffraction peaks are overlap, it is due to the natural action of these reactive minerals to form a solid solution structure.18,19 In comparison to sample 0, no significant crystalline phase changes were observed for sample 1 when the catalyst was reacted under H2O conditions for 24 hours. However, when the catalyst was reacted under SO2 or SO2 and H2O conditions for 24 hours, a decrease in the intensity of the Ca(MgFe)[(CO3)2] and Fe2O3 diffraction peak intensities was found for samples 2 and 3, but no characteristic metal sulphate peaks were detected, indicating that the SO2 sulphide phase was present in an amorphous form and could not be observed by XRD. However, in the presence of SO2 or SO2/H2O, new diffraction peaks were observed for samples 2, 3 at 18.43°, 24.17°, 24.84°, 33.14°, 37.02° and 50.68°, which can be attributed to sulphonate (NH3SO3) (PDF#08-0483). And these diffraction peaks overlap with or are similar to the active mineral diffraction peaks of Ca(MgFe)[(CO3)2], Fe2O3 and CeCO3F. It can be inferred that the newly formed crystalline phase NH3SO3 is produced because the acid groups formed on the catalyst surface after SO2/H2O is involved in the denitrification process and during the drop to low temperature interact with the NH3 adsorbed species and a reduction reaction occurs to reduce the active metal centres in the active minerals to their original state.22–24
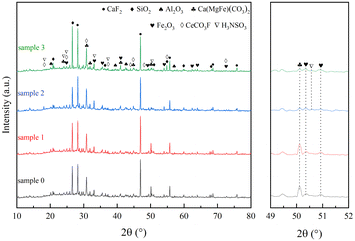 |
| | Fig. 2 Rare earth tailings catalyst (RET) XRD results. | |
3.2 Catalyst performance change
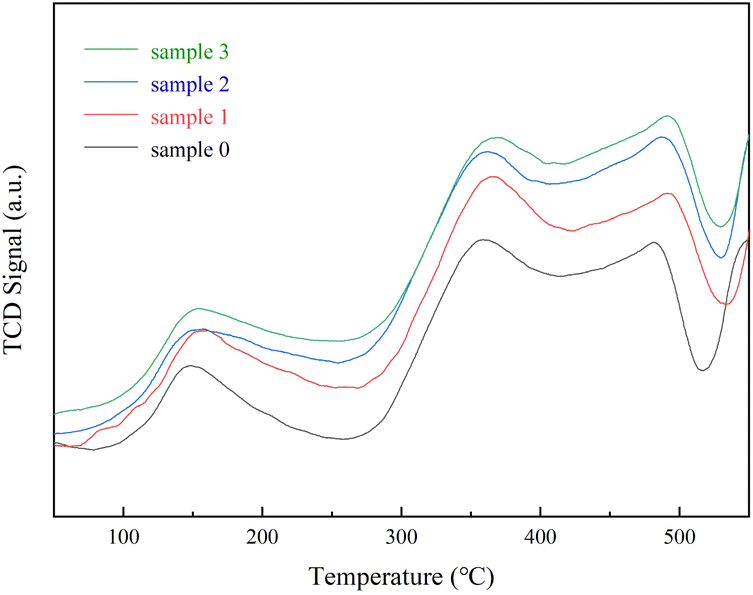
To examine the effect of SO2/H2O on the NH3 adsorption of the RET catalyst as shown in Fig. 3. The peak temperatures of the desorption peaks of sample 0 were 147 °C (weak acid centers), 356 °C (medium centers) and 480 °C (strong centers).25 The NH3 adsorption capacity of samples 1, 2 and 3 was significantly higher than sample 0, while the NH3 adsorption capacity of samples 2 and 3 was slightly higher than sample 1. Sample 0 showed a desorption peak at 147 °C corresponding to the weak acid site, a desorption peak at 356 °C corresponding to at the medium to strong acidic site, and a desorption peak at 480 °C corresponding to the strong acidic site. At 480 °C corresponds to the desorption peak from the strongly acidic site. Samples 1, 2 and 3 also showed three peaks in the low and medium-high temperature sections, each acidic site being shifted back and enhanced compared to sample 0, indicating that the presence of a suitable amount of SO2/H2O can strengthen the acidic sites on the RET catalyst. The peak at 150 °C for sample 3, which was influenced by the combined presence of SO2/H2O, was attributed to the weakly acidic sites, with a significantly greater ability to adsorb NH3 on the weakly acidic sites relative to the RET catalysts influenced by the presence of SO2 or H2O alone with larger desorption peaks at 365 °C and 489 °C attributed to the moderately strong and strong acidic sites. Significantly larger peak areas in the mid to high temperature fractions compared to samples 1 and 2, which were affected by SO2/H2O alone. It means that co-modification of SO2 and H2O can also substantially enhance the ability of medium to strong acidic sites to adsorb NH3. Overall, the RET catalyst with the influence of SO2/H2O both increased the acidic sites and improved NH3 adsorption capacity. Combined with XRD analysis, it was found that the increase in acidic sites after the catalyst was affected by SO2/H2O can be attributed to the formation of NH3SO3, which is readily decomposed by heat, thus providing more acidic sites.
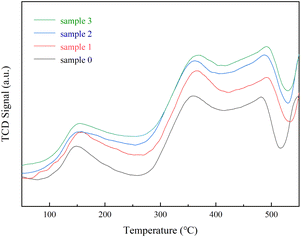 |
| | Fig. 3 Rare earth tailings catalyst (RET) NH3-TPD results. | |
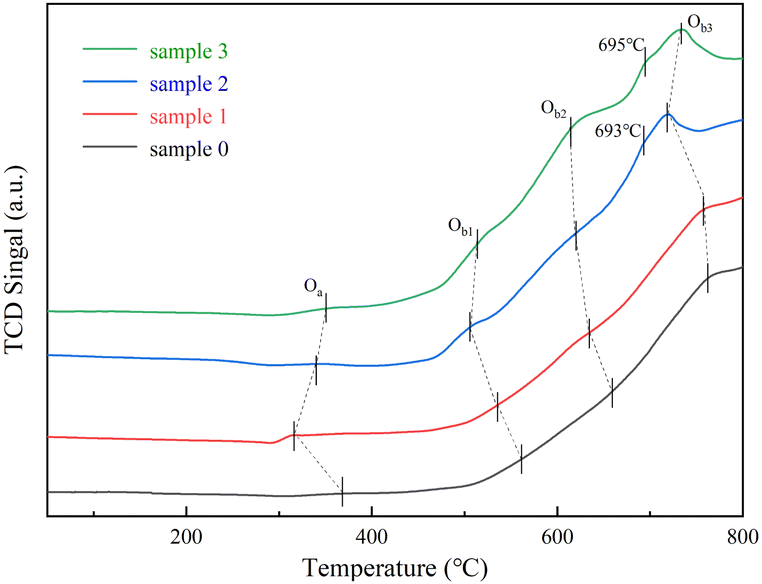
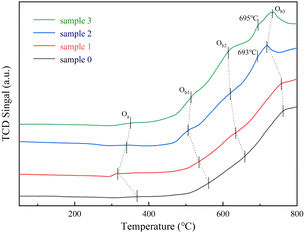
H2-TPR tests were using to investigate the effect of the SO2/H2O on the redox ability of the RET catalyst. In Fig. 4, the Oa peak located below 400 °C is the reduction peak of oxygen species adsorbed on the catalyst surface. The Ob1 peak located between 400 and 600 °C is the peak of H2 consumption (Fe2O3 to Fe3O4 and the reduction of the CeO2).26,27 The Ob2 peak between 600 and 700 °C is the peak of H2 consumption (Fe3O4 to FeO).26 The Ob3 peak above 700 °C is the peak of H2 consumption (FeO to Fe).26 The reduction peaks of the RET catalysts with SO2 or SO2/H2O are shifted towards a lower temperature region and the peak area is slightly increased compared to the RET catalysts without the influence of SO2 or H2O. This means that SO2 and H2O can influence the RET catalysts to induce a beneficial change in redox capacity towards a more beneficial catalytic reduction reaction occurring, with new shoulder peaks are due to the reduction of the CeO2 at 693 °C and 695 °C,27 and the introduction of SO2/H2O leads to a more pronounced shoulder peak shape at 695 °C. Which combined with XRD analysis, is presumed to be due to the influence of SO2, which induces the reduction of RET catalysts with the natural co-occurrence of active minerals associated properties of the RET catalyst resulted from the joint participation of multiple substances in the redox reaction.28 From the H2-TPR results, electron transfer paths can be derived (Fe3+ ↔ Fe2+ and Ce4+ ↔ Ce3+).
 |
| | Fig. 4 Rare earth tailings catalyst (RET) H2-TPR results. | |
The surface of the rare earth tailings catalyst (Fig. 5a) showed an uneven and pitted morphology, and the surface of the rare earth tailings catalyst (Fig. 5b) did not change much after the H2O tolerance test. After being affected by SO2 alone and SO2/H2O tolerance, the catalysts (Fig. 5c and d) showed some mobility, which was in the form of stacked lamellar fragmentation with a few fine particles attached to the surface, after being affected by SO2/H2O tolerance, there was a large volume of lamellar particles stacked on the surface of the catalysts, and after the catalysts were affected by the simultaneous presence of SO2 and H2O, the lamellar particles were attached to the surface of the catalysts.
 |
| | Fig. 5 SEM and EDS surface sweeps before and after the effect of SO2/H2O on RET catalysts ((a) fresh RET catalyst; (b) after H2O; (c) after SO2; (d) after SO2/H2O). | |
3.3 In situ DRIFTs analysis
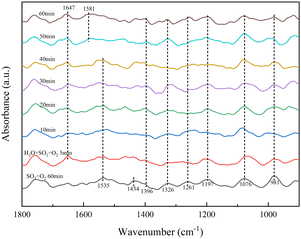
3.3.1 Effect of H2O on the adsorption of SO2 on the catalyst surface. The adsorption of SO2 in the presence of H2O at 350 °C was investigated by in situ DRIFTS, as shown in Fig. 6.
 |
| | Fig. 6 Effect of H2O on the adsorption of SO2 by the RET catalyst. | |
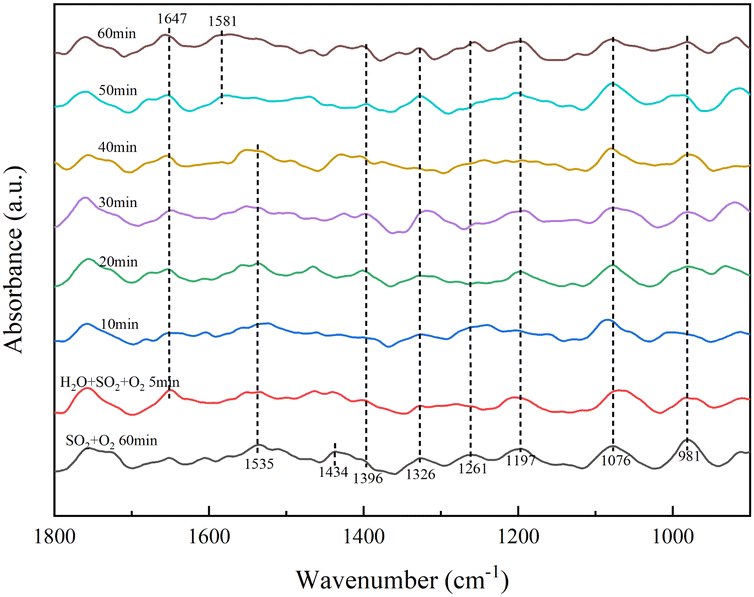
As shown in Fig. 6, the RET catalyst was exposed to SO2/O2 for 60 min and treated in a pure N2 gas stream (100 mL min−1) at 350 °C for a further 30 min. 5 min after the addition of H2O, an absorption peak of molecular water (1647 cm−1) appeared, with little change in peak intensity with increasing time, meaning that the H2O tolerance of the RET catalyst was good. The absorption peak of sulfite (1434 cm−1) disappeared with increasing time and the absorption peak of pyrosulfate (1396 cm−1) gradually became stronger. The absorption peaks of bridged tri-dentate sulphate (1326 cm−1), double-dentate sulphate (1076 cm−1), bridged double-dentate sulphate (1197 cm−1) and single-dentate sulphate (1261 cm−1) showed little change.31,32,35,36 The crystalline water variable angle vibrational peak shifted from 1535 to 1581 cm−1, it is due to the chemisorption of gaseous SO2 onto the catalyst surface via hydroxyl groups.37 The increase in persulphate is due to the presence of H2O, which causes the formation of hydroxyl groups on the RET catalyst surface, and adsorb more SO2 to form SO32−, which in turn reacts with the catalyst surface sulphate species to form S2O72−, so H2O promotes the formation of S2O72− species on the RET catalyst surface.
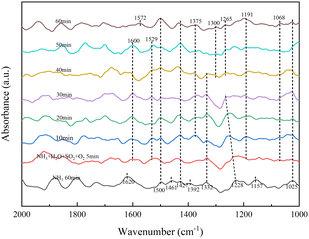
3.3.2 Effect of SO2/H2O on surface acidity. Fig. 7 shows that NH3 was first adsorbed without SO2 for 60 min at 350 °C and then treated in a pure N2 gas stream (100 mL min−1) for a further 30 min.
 |
| | Fig. 7 Effect of SO2/H2O on catalyst NH3 adsorption. | |
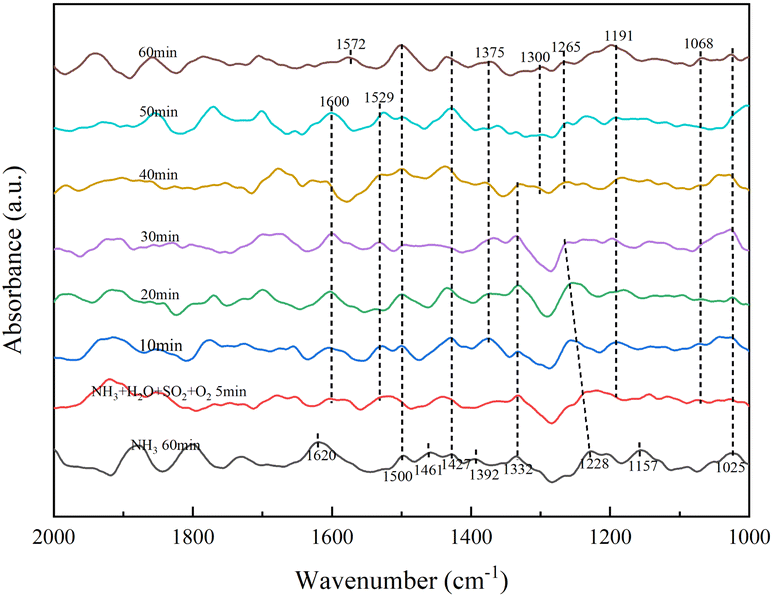
Fig. 7 shows the absorption peaks of NH4+ adsorption at the Brønsted acid site (1392, 1427, 1461, 1500 cm−1), NH3 adsorption at the Lewis acid site (1025, 1157, 1228, 1620 cm−1) and it causes NH3 form –NH2 (1332 cm−1).29,30 After continuing the ammonia stream and 5 min of SO2, H2O and O2, the absorption peaks of NH4+ adsorption (1392, 1461 cm−1) and NH3 adsorption at the Lewis acid site (1157, 1228, 1620 cm−1) disappeared. The intermediate product –NH2 (1529 cm−1)29,30 and NH3 adsorption (1600 cm−1)29,30 with increasing peak intensity with time, meaning that the presence of SO2/H2O promoted NH3 adsorption. Bidentate sulfate (1068 cm−1), bridged bidentate sulfate (1191 cm−1), and monodentate sulfate (1265 cm−1) absorption peaks were also present after 40 min.31,35,36 The intensity of the bidentate and bridged bidentate sulfate absorption peaks frequently changes but no pyrosulfate species were found at 1391 cm−1. In combination with XRD analysis, was attributed to the formation of NH3SO3 from NH3 adsorbed species bound to the catalyst surface. After 40 min of addition of SO2, H2O and O2, the intermediate product –NH2 (1300, 1572 cm−1) appeared, resulting from the creation of a new Lewis acid site, which is consistent with NH3-TPD results. SO2/H2O caused the formation of an unstable sulphate species, which adsorbed on the active metal sites are strongly acidic, so this unstable sulphate species provides more acidic sites for the NH3-SCR process and is less likely to form ammonium sulphate species with NH3. The combination of NH3 with catalyst surface species to form NH3SO3 resulted in the disappearance of persulphate species from the RET catalyst surface, indicating that the presence of NH3 promoted the decomposition of S2O72−.
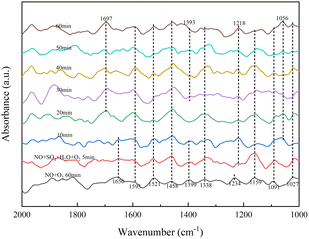
3.3.3 Effect of SO2 on the adsorbed NO species. As in step with Fig. 8, the in situ DRIFTS spectra showed the effect of SO2/H2O on NO adsorption on the RET catalyst at 350 °C.
 |
| | Fig. 8 Effect of SO2/H2O on NO adsorption by RET catalysts. | |
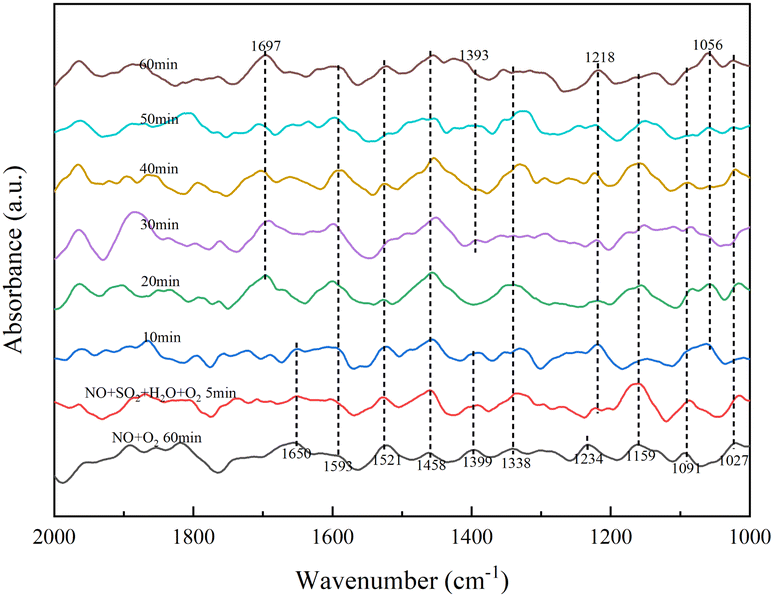
In Fig. 8, the addition of NO and O2 gas streams followed by a pure N2 gas stream shows monodentate nitrite (1027, 1091, 1159 cm−1), bridged nitrite (1234 cm−1), intermediate product-NO2 (1338, 1399, 1458 cm−1), intermediate product-NO3 (1521 cm−1) and adsorbed NO2 (1593, 1650 cm−1).33,34 After simultaneous introduction of NO, O2, H2O and SO2 for 5 min, the absorption peak of bridged nitrite shifted from 1234 to 1218 cm−1 with little change in peak intensity with increasing time, with bridged nitrite still occupying the active site. After 10 min of addition of H2O and SO2, the absorption peaks of –NO2 (1399 cm−1) and adsorbed NO2 (1650 cm−1) disappeared and absorption peaks of bidentate sulfate (1056 cm−1), pyrosulfate (1393 cm−1) and NO2 (1697 cm−1) generated by the reaction of adsorbed NO on the surface appeared.33,34,38 In the presence of SO2/H2O, the absorption peak of S2O72− species gradually increased with time and the –NO2 disappeared, due to the competition between SO2 and NO adsorption on this active site to produce S2O72− species. It occupies part of the NO adsorption site, and the increase in surface active oxygen species due to SO2 acidification of the RET catalyst, converting more NO to NO2 and hence the absorption peak of NO2.
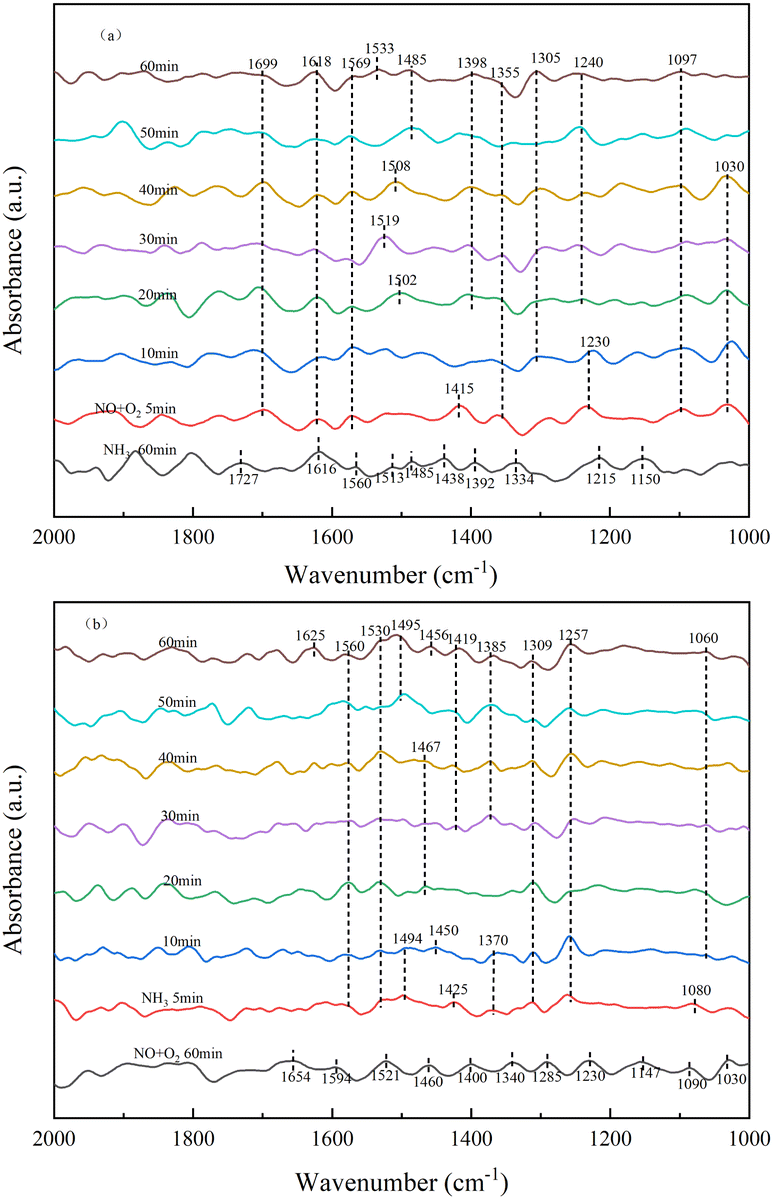
3.3.4 Transient reactions of NH3 and NO on the catalyst surface. As shown in Fig. 9a, the in situ DRIFTs spectra of the reaction of NO with pre-adsorbed NH3 species on the RET catalyst surface under the influence of SO2/H2O and Fig. 9b shows the in situ DRIFTs spectra of the reaction of NH3 with pre-adsorbed NO species on the RET catalyst surface under the influence of SO2/H2O.
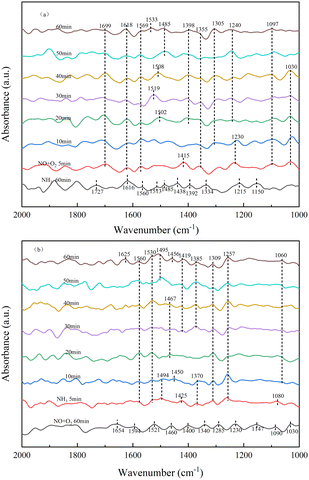 |
| | Fig. 9 RET Catalytic transient reaction in the presence of SO2/H2O ((a) NH3/NO; (b) NO/NH3). | |
As shown in Fig. 9a, the RET catalysts were firstly exposed to NH3 gas flow for 60 min in the addition of SO2/H2O, and then purged with N2 at 350 °C for 20 min.
Fig. 9a shows the absorption peaks of NH4+ adsorption (1392, 1438, 1485, 1513, 1727 cm−1), NH3 adsorption (1150, 1215, 1616 cm−1) and –NH2 (1334, 1560 cm−1).29,30 After 5 min of the addition of NO and O2, the NH3 adsorbed species disappeared and peaks appeared for monodentate nitrite (1030, 1097 cm−1), bridged nitrite (1230 cm−1), intermediate product –NO2 (1355, 1415, 1618 cm−1), bidentate nitrate (1569 cm−1) and NO2 (1699 cm−1).33,34 The peak of –NO2 (1415 cm−1) disappeared rapidly with increasing time. The peak pattern of bridged nitrite shifted from 1230 to 1240 cm−1 with little change in peak intensity. Monodentate nitrite (1097 cm−1) and bidentate nitrate (1569 cm−1) showed little change. Absorption peaks for intermediate product-NO2 (1305, 1398 cm−1) and intermediate product-NO3 (1502 cm−1) appeared after 20 min of the addition of NO/O2. Absorption peaks of –NO3 at 1519, 1508, 1485 and 1533 cm−1 appeared, with significant changes in peak pattern with increasing time. The absorption peaks of –NO2 did not change much after 20 min of NO/O2 addition, and the peak intensity increased. This is due to the fact that when NO/O2 were first added, the NH3 adsorbed species mainly combined with –NO2 to form the intermediate product, and after 20 min as NO was oxidized to more NO2, the –NO3 group was generated to participate in the reaction. The results show that –NO2 is easier to combine with NH4+ than –NO3 to form the intermediate product NH4NO2. And when NH3 is present, the NO species mainly form the –NO2 group directly to participate in the reaction, without oxidation between the catalyst and NO2 to form the –NO3 group, shortening the reaction process in favor of the NH3-SCR reaction.
In Fig. 9b, the samples were first exposed to NO and O2 for 60 min in the presence of SO2/H2O, and then treated in a pure N2 gas stream (100 mL min−1) at 350 °C for a further 30 min. The appearance of monodentate nitrite (1030, 1090, 1147 cm−1) was observed in Fig. 9b, with bridging nitrite (1230 cm−1), –NO2 (1285, 1340, 1400, 1460 cm−1), –NO3 (1521 cm−1) and adsorbed NO2 (1594, 1654 cm−1) absorption peaks.33,34 After 5 min of the addition of NH3, the peaks of the nitrate-like adsorbed species, adsorbed state NO2, intermediate products –NO2 and –NO3 disappeared, along with the peaks of adsorbed NH4+ (1370, 1425, 1494 cm−1), adsorbed NH3 (1080, 1257 cm−1) and –NH2 (1309, 1530, 1560 cm−1),29,30 where the absorption peak of –NH2 increased in intensity with time. The peak of adsorbed NH3 was less variable, and the peak of adsorbed NH4+ disappeared quickly. Absorption peaks for NH4+ (1385, 1419, 1456, 1467, 1495 cm−1) and –NH2 (1625 cm−1) adsorbed at the Brønsted acid site appeared after 20 min of the addition of NH3.29,30 When NH3 was present, the –NO2 and –NO3 groups as well as the adsorbed NO2 disappeared from the catalyst surface and NH4+ and –NH2 groups were generated at subsequent time periods of the reaction, indicating that intermediate products NH4NO3, NH4NO2 and NH2NO2 were generated on the RET catalyst surface to remove NO adsorption products, suggesting that both the E–R mechanism and the L–H mechanism, which acted together to remove NOX through both pathways.
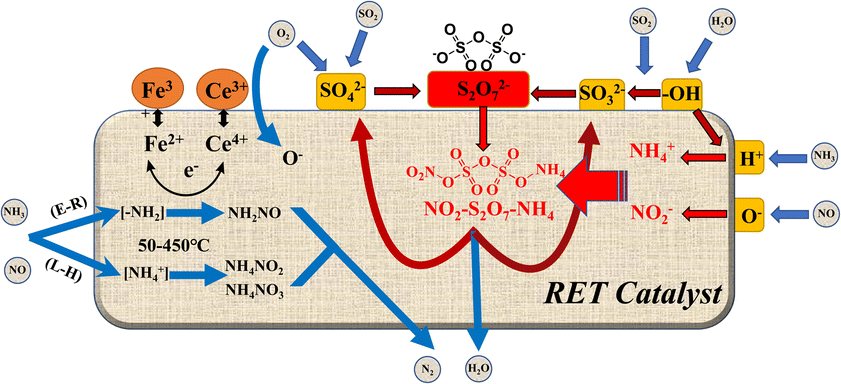
4 Denitrification mechanism of SO2/H2O enhanced rare earth tailings catalyst
Combining the results of in situ DRIFTs analysis and SO2 characterization,22,23,38,39 We can surmise the following reaction mechanism (Fig. 10) as well as reaction equations (eqn (2)–(16)) can be derived for the effect of SO2/H2O on the RET catalysts. As shown in Fig. 10, combined with the analysis of the H2-TPR results, it is found that the conversion between Fe3+ ↔ Fe2+ and Ce4+ ↔ Ce3+ on the RET catalyst surface provides the active sites for SCR catalytic denitrification, and the conversion between Ce3+ and Ce4+ mainly acts as oxygen storage and release (eqn (2)–(5)). When SO2/H2O is added, SO2 is adsorbed by the hydroxyl groups formed by H2O and reacts with O2 to form SO32− groups, while combining with SO42− groups to form S2O72− (eqn (7)–(9)). When NH3 and NO are present, they are converted to NH4+ and NO2− respectively, which are then reduced to N2 and H2O (eqn (10)–(15)). The adsorption of H2O by the catalyst surface increases the –OH sites on the catalyst surface, leading to easier adsorption and conversion of SO2 to form SO32−, while the S2O72− group is converted to NH3SO3 and the unstable SO42− group by the reduction of NH3 (eqn (16)), allowing the catalyst reactive sites on the surface are restored, inhibiting the continued sulphation of the catalyst surface. NH3 also adsorbs to the Brønsted and Lewis acidic sites on the RET catalyst surface as –NH2 and NH4+, and perform NH3-SCR reaction.| | |
Ce3+ + O2(g) ↔ Ce4+ + 2O−
| (4) |
| | |
H2O(g) + △ → △ − OH + H+
| (6) |
| | |
△ − OH + SO2(g) → △ − SO2−3 + H2O
| (7) |
| | |
2SO2(g) + 3/2O2(g) + H2O → 2SO42− + 2H +
| (8) |
| | |
SO42− + △ − SO32− → △ − S2O72−
| (9) |
| | |
△ − S2O72− + H+ → △ − S2O72–H+
| (10) |
| | |
△ − S2O72−H+ + NH3(g) → △ − S2O72−NH4+
| (11) |
| | |
△ − S2O72−NH4+ + NO(g) + 1/2O2(g) → △–NO2−S2O72–NH4+
| (12) |
| | |
△ − NO2−S2O72–NH4+ → △ − S2O72− + N2(g) + 2H2O
| (13) |
| | |
△ − S2O72− + NH3(g) → △ − H2NS2O72−H+
| (14) |
| | |
△ − H2NS2O72−H+ + NO(g) → △S2O72−H+ + N2(g) + H2O
| (15) |
| | |
△ − S2O72−H+ + NH3(g) + H+ → NH3SO3 + SO42− + △
| (16) |
where △ represents the surface oxygen vacancy in the active metal center. The symbol (g) represents NO, NH3, SO2, NO2, N2, and O2 in the gaseous state. The symbol (a) represents the adsorbed state of NH3, NH4+ and SO3 on the catalyst surface.
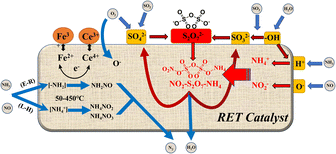 |
| | Fig. 10 Mechanism of SO2/H2O influence on the surface of rare earth tailings (RET) catalysts (△ represents the active metal sites on the RET catalyst surface; blue arrows represent the gas absorption and release process; black arrows represent the internal electron transfer process of the catalyst; red arrows represent the reaction cycle process of the S2O72− group). | |
5 Catalyst performance
5.1 Effect of H2O and SO2 on catalytic NOX reduction
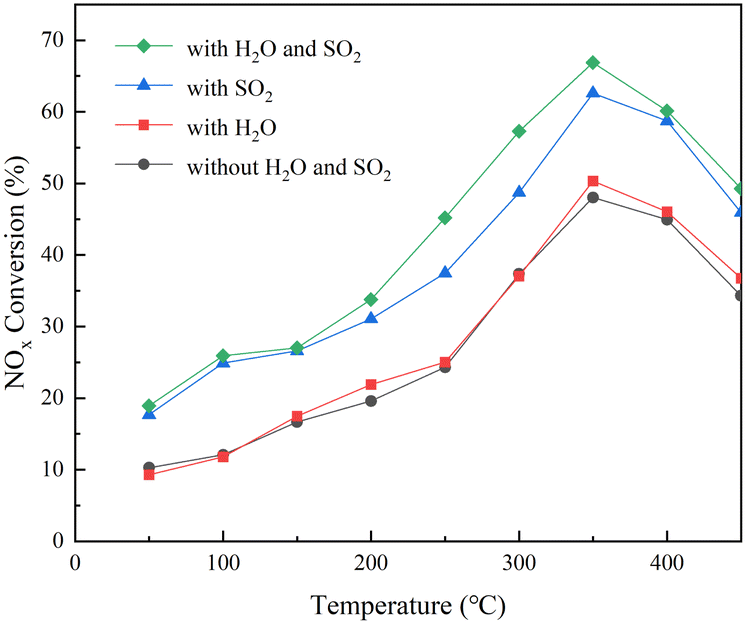
The effect of SO2/H2O on the catalytic denitrification activity of the RET catalyst was studied in the temperature range of 50–450 °C with φNO, φNH3 and φSO2 of 500 × 10−6, φO2 of 6 × 10−2 and φH2O of 6 × 10−2. After the SCR reactor outlet φNO was stabilized for 10 min, the NH3 was started to be introduced and the denitrification rate for each temperature section was calculated as the average of the NOX outlet concentration values within 180 min of stabilization were calculated and the results, as shown in Fig. 11.
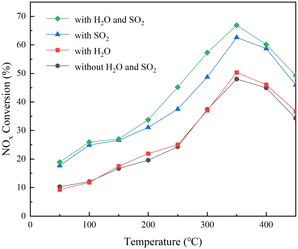 |
| | Fig. 11 RET catalyst NOX removal rate. | |
Fig. 11 shows that the NOX conversion of the RET catalyst in the absence of SO2/H2O and SO2 is much lower than that in the presence of SO2 and H2O, and H2O has almost no effect on the NOX conversion of the RET catalyst. At relatively low temperatures (50 to 200 °C), the enhancement of catalytic activity in the presence of SO2 and H2O was similar, about 10 ± 1%. However, when the reaction temperature is in the medium to high temperature range (250–450 °C), the activity enhancement under SO2/H2O can reach 16 ± 1%, and the activity enhancement of the RET catalyst in SO2 only condition is only 10 ± 1%. Furthermore, at 350 °C, SO2/H2O in the gas supply leads to a significant increase in SCR activity of about 18 ± 1%, with a NOX conversion of 66 ± 1% for the RET catalyst. This enhancement is due to SO2 gas phase acidification of the active metal oxides under SO2.37 It suggests that the appropriate conditions of SO2/H2O presence are favorable to improve the catalytic activity of the RET catalysts.
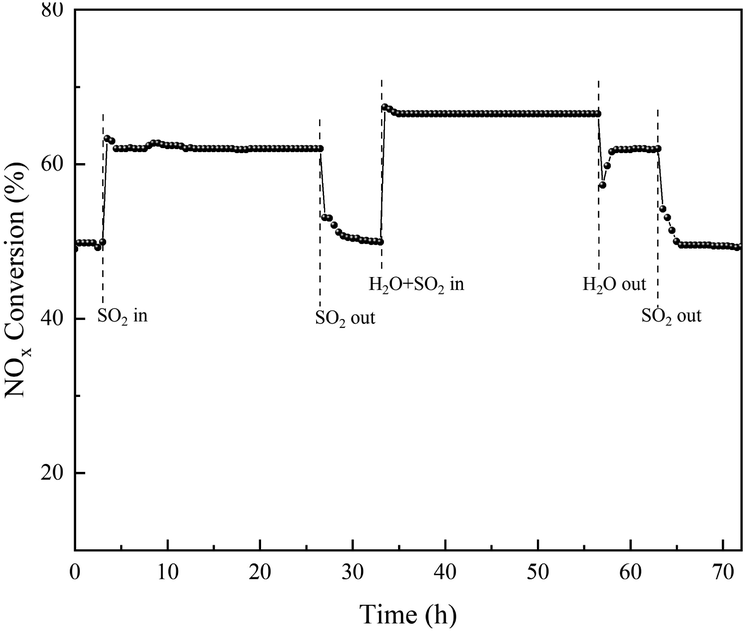
With the same steps, the stability of the effect of SO2/H2O on the NOX conversion in the temperature range of 350 °C of the RET catalyst over time is shown in Fig. 12 (the data points in the figure are the average values of NOX conversion over 30 min).
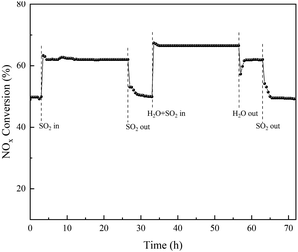 |
| | Fig. 12 Stability of RET catalyst NOX removal rates under the influence of SO2/H2O. | |
Fig. 12 showed stable catalytic activity at 350 °C during the 72 h test. When SO2 was added to the gas stream, the NOX conversion increased from a stable 49 ± 1% in the first 3 h to obtain a stable NOX conversion of 62 ± 1% in 24 h, indicating that SO2 has a significant effect on the catalyst activity. The addition of SO2/H2O increases the catalytic activity from 49 ± 1% to 66 ± 1%. However, when H2O is removed from the gas, the activity can be restored, implying that the enhancement effect of H2O in promoting SO2 influence on the activity is reversible. The NOX conversion of the RET catalyst can be recovered when there is no SO2 or H2O in the reaction gas mixture, which proves that the RET catalyst has a good effect on SO2/H2O, and the SCR activity of the RET catalyst can be obtained at 350 °C with a 17 ± 1% enhancement under the conditions of both SO2 and H2O.
6 Conclusion
In summary, this paper prepared rare earth tailings (RET) catalysts from Bayan Ebo rare earth tailings by ball milling and microwave roasting, and analyzed the SO2/H2O tolerance mechanism of the RET catalysts by activity testing, characterization and in situ DRIFTs experiments. And revealed the mechanism of SO2/H2O to enhance the denitrification performance of catalysts by promoting the cyclic reaction of S2O72− groups on the RET catalysts surface. Although RET catalysts are less active, they can be used as a catalyst active carrier through their excellent SO2 tolerance properties.
(1) The effect of SO2/H2O on the RET catalyst was facilitated, and the NOX conversion of the RET catalyst increased from 48% to 66 ± 1% at 350 °C under SO2/H2O, which was higher than that of SO2 or H2O alone.
(2) Rare earth tailings catalyst NH3-SCR denitrification followed the combined L–H and E–R mechanism. The Fen+–Cen+ coactivation of the RET catalyst provides Brønsted and Lewis acid sites.
(3) The effect of SO2 on the RET catalyst is mainly the formation of SO42− and SO32− species, while H2O promotes the formation of hydroxyl groups and facilitates SO2 adsorption to form SO32−, which promotes the reaction of SO32− with SO42− to form S2O72− groups, which provide acid sites for the RET catalyst and improve the adsorption of NH3. At the same time, the S2O72− group decomposes into SO32− and unstable SO42− groups, thus continuing the cyclic reaction to form S2O72−groups, thereby increasing the denitrification activity of the RET catalyst activity and inhibit the continued sulphation of the catalyst surface.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research was financially supported by the Natural Science Foundation of Inner Mongolia (Grant No. 2019ZD13).
References
- K. Skalska, J. S. Miller and S. Ledakowicz, Trends in NOx abatement: A review, Sci. Total Environ., 2010, 408(19), 3976–3989 CrossRef CAS PubMed.
- G. L. Manney, L. Froidevaux, J. W. Waters, R. W. Zurek, W. G. Read, L. S. Elson, J. B. Kumer, J. L. Mergenthaler, A. E. Roche, A. O'Neill, R. S. Harwood, I. Mackenzie and R. Swinbank, Chemical depletion of ozone in the Arctic lower stratosphere during winter 1992-93, Nature, 1994, 370(6489), 429–434 CrossRef CAS.
- D. Maric and J. P. Burrows, Formation of N2O in the photolysis/photoexcitation of NO, NO2 and air, J. Photochem. Photobiol., A, 1992, 66(3), 291–312 CrossRef CAS.
- H. Bosch and F. Janssen, Catalytic reduction of nitrogen oxides-A review on the fundamental and technology, Catal. Today, 1988, 2(4), 369–532 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: A review, Appl. Catal., B, 1998, 18(1–2), 1–36 CrossRef CAS.
- F. Kapteijn, L. Singoredjo and A. Andreini, Activity and selectivity of pure manganese oxides in the selective catalytic reduction of nitric oxide with ammonia, Appl. Catal., B, 1994, 3(1), 173–189 CrossRef CAS.
- T. S. Park, S. K. Jeong, S. H. Hong and S. C. Hong, Selective catalytic reduction of nitrogen oxides with NH3 over natural manganese ore at low temperature, Ind. Eng. Chem. Res., 2001, 40(21), 4491–4495 CrossRef CAS.
- L. Singoredjo, R. Korver and F. Kapteijin andJ.Moulijn, Alumina supported manganese oxides for the low-temperature selective catalytic reduction of nitric oxide with ammonia, Appl. Catal., B, 1992, 1(4), 297–316 CrossRef CAS.
- C. Liu, S. Yang, L. Ma, Y. Peng, A. Hamidreza, H. Chang and J. Li, Comparison on the Performance of alpha-Fe2O3 and gamma-Fe2O3 for Selective Catalytic Reduction of Nitrogen Oxides with Ammonia, Catal. Lett., 2013, 143(7), 697–704 CrossRef CAS.
- A. Kato, S. Matsuda, T. Kamo, F. Nakajima, H. Kuroda and T. Narita, Reaction between NOX and NH3 on iron oxide-titanium oxide catalyst, J. Phys. Chem., 1981, 85(26), 276–287 CrossRef.
- F. Liu, H. He and C. Zhang, Novel iron titanate catalyst for the selective catalytic reduction of NO with NH3 in the medium temperature range, Chem. Commun., 2008, 164(17), 2043–2045 RSC.
- F. Liu, H. He, C. Zhang, Z. Feng, L. Zheng, Y. Xie and T. Hu, Selective catalytic reduction of NO with NH3 over iron titanate catalyst: Catalytic performance and characterization, Appl. Catal., B, 2010, 96(3), 408–420 CrossRef CAS.
- F. Liu, K. Asakura, H. He, Y. Liu, W. Shan, X. Shi and C. Zhang, Influence of calcination temperature on iron titanate catalyst for the selective catalytic reduction of NOX with NH3, Catal. Today, 2011, 164(1), 520–527 CrossRef CAS.
- L. Hou, S. Fu, X. Yan, C. Qiao and W. Wu, Effects of sulfate on denitration performance of rare earth tailings for selective catalytic reduction of NO with NH3, Mol. Cryst. Liq. Cryst., 2023, 755(1), 91–106 CrossRef CAS.
- P. Zhang, Study on the process mineralogy of large-sized Bayan Obo Fe-REE-Nb deposit, J. Chin. Rare Earth Soc., 1991, 9(4), 350–353 Search PubMed.
- J. Wang, C. Zhu, B. Li, Z. Gong, Z. Meng, G. Xu and W. Wu, Prepare a catalyst consist of rare earth minerals to denitrate via NH3-SCR, Green Process. Synth., 2019, 9(1), 191–202 Search PubMed.
- X. Bai, J. Lin, Z. Chen, L. Hou and W. Wu, A Study on the Effect of Different Ball Milling Methods on the NH3-SCR Activity of Aluminum-Laden Bayan Obo Tailings, Catalysts, 2021, 11(5), 568 CrossRef CAS.
- G. Qi and R. Yang, Low-temperature selective catalytic reduction of NO with NH3 over iron and manganese oxides supported on titania, Appl. Catal., B, 2003, 44(3), 217–225 CrossRef CAS.
- Z. Chen, F. Wang, H. Li, Q. Yang, L. Wang and X. Li, Low-Temperature Selective Catalytic Reduction of NOX with NH3 over Fe-Mn Mixed-Oxide Catalysts Containing Fe3Mn30s Phase, Ind. Eng. Chem. Res., 2012, 51(1), 202–212 CrossRef CAS.
- B. Shen, T. Liu, N. Zhao, X. Yang and L. Deng, Iron-doped Mn-Ce/TiO2 catalyst for low temperature selective catalytic reduction of NO with NH3, J. Environ. Sci., 2010, 22(9), 1447–1454 CrossRef CAS PubMed.
- F. Wang, B. Shen, S. Zhu and Z. Wang, Promotion of Fe and Co doped Mn-Ce/TiO2 catalysts for low temperature NH3-SCR with SO2 tolerance, Fuel, 2019, 249, 54–60 CrossRef CAS.
- E. Hartley and M. J. Matteson, Sulfur dioxide reactions with ammonia in humid air, Ind. Eng. Chem. Fundam., 1975, 14(1), 67–72 CrossRef CAS.
- Z. Gao, W. Yang, X. Ding, G. Lv and W. Yang, Support effects in single atom iron catalysts on adsorption characteristics of toxic gases (NO2, NH3, SO3 and H2S), Appl. Surf. Sci., 2018, 436, 585–595 CrossRef CAS.
- L. Liu and Z. Yuan, Ordered Mesoporous Carbon Materials Synthesized by Organic-Organic Self-Assembly, Prog. Chem., 2014, 26(05), 756–771 CAS.
- X. Zhu, Y. Wang, Y. Huang and Y. Cai, Selective catalytic reduction of NO with NH3 over Ce-W-Ti oxide catalysts prepared by solvent combustion method, Appl. Sci., 2018, 8(12), 2430 CrossRef CAS.
- F. Liu and H. He, Structure-Activity Relationship of Iron Titanate Catalysts in the Selective Catalytic Reduction of NOX with NH3, J. Phys. Chem. C, 2010, 114(40), 16929–16936 CrossRef CAS.
- X. Huang, P. Wang, J. Tao and Z. Xi, CeO2-modified Mn-Fe-O composites and their catalytic performance for NH3-SCR denitrification, J. Inorg. Mater., 2020, 35(05), 573–580 CrossRef.
- Q. Fu, J. Wen, L. Li, Y. Xiao and K. Gao, The Study of REE Occurrence State in Bayan Obo Tailings, Chin. Rare Earths, 2017, 38(05), 103–110 Search PubMed.
- J. Sun, Y. Lu, L. Zhang, C. Ge, C. Tang, H. Wan and L. Dong, Comparative Study of Different Doped Metal Cations on the Reduction, Acidity, and Activity of Fe9M1OX (M= Ti4+, Ce4+/3+, Al3+) Catalysts for NH3-SCR Reaction, Ind. Eng. Chem. Res., 2017, 56(42), 12101–12110 CrossRef CAS.
- G. Ramis and M. A. Larrubia, An FT-IR study of the adsorption and oxidation of N-containing compounds over Fe2O3/Al2O3 SCR catalysts, J. Mol. Catal. A: Chem., 2004, 215(1–2), 161–167 CrossRef CAS.
- A. A. Ioffe, V. P. Bulatov, V. A. Lozovsky, M. Y. Goldenberg, O. M. Sarkisov and S. Ya, Umansky. On the reaction of the NH2 radical with SO2 at 298–363 K, Chem. Phys. Lett., 1989, 156(5), 425–432 CrossRef CAS.
- D. Ye, R. Qu, C. Zheng, K. Cen and X. Gao, Mechanistic investigation of enhanced reactivity of NH4HSO4 and NO on Nb- and Sb- doped V-W/Ti SCR catalysts, Appl. Catal., A, 2018, 549, 310–319 CrossRef CAS.
- F. Liu and H. He, Selective catalytic reduction of NO with NH3 over manganese substituted iron titanate catalyst: Reaction mechanism and H2O/SO2 inhibition mechanism study, Catal. Today, 2010, 153(3–4), 70–76 CrossRef CAS.
- L. Chen, X. Yao, J. Cao, F. Yang, C. Tang and L. Dong, Effect of Ti4+ and Sn4+ co-incorporation on the catalytic performance of CeO2-MnOX catalyst for low temperature NH3-SCR, Appl. Surf. Sci., 2019, 476, 283–292 CrossRef CAS.
- F. Gao, X. Tang, H. Yi, S. Zhao, J. Wang and T. Gu, Improvement of activity, selectivity and H2O&SO2-tolerance of micro-mesoporous CrMn2O4 spinel catalyst for low-temperature NH3-SCR of NOX, Appl. Surf. Sci., 2019, 466, 411–424 CrossRef CAS.
- S. Pan, H. Luo, L. Li, Z. Wei and B. Huang, H2O and SO2 deactivation mechanism of MnOX/MWCNTs for low-temperature SCR of NOX with NH3, J. Mol. Catal. A: Chem., 2013, 377, 154–161 CrossRef CAS.
- T. Gu, L. Yue, H. Wang and Z. Wu, The enhanced performance of ceria with surface sulfation for selective catalytic reduction of NO by NH3, Catal. Commun., 2010, 12(4), 310–313 CrossRef CAS.
- W. S. A. El-Yazeed, M. Eladl, A. I. Ahmed and A. A. Ibrahim, Sulfamic: acid incorporated tin oxide: Acidity and activity relationship, J. Alloys Compd., 2021, 858, 158–192 Search PubMed.
- E. Hayon, A. Treinin and J. Wilf, Electronic spectra, photochemistry, and autoxidation mechanism of the sulfite bisulfite pyrosulfite systems. SO2-, SO3-, SO4-, and SO5-radicals, J. Am. Chem. Soc., 1972, 94(1), 47–57 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jingran Zhanga and
Wenfei Wu
a,
Jingran Zhanga and
Wenfei Wu