
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem manganese catalysis for the chemo-, regio-, and stereoselective hydroboration of terminal alkynes: in situ precatalyst activation as a key to enhanced chemoselectivity†
Victor Duran Arroyo and
Rebeca Arevalo*
and
Rebeca Arevalo*
Department of Chemistry and Biochemistry, University of California, 5200 North Lake Road, 95343, Merced, California, USA. E-mail: rebecaarevalo@ucmerced.edu
First published on 13th February 2024
Abstract
The manganese(II) complex [Mn(iPrPNP)Cl2] (iPrPNP = 2,6-bis(diisopropylphosphinomethyl)pyridine) was found to catalyze the stereo- and regioselective hydroboration of terminal alkynes employing HBPin (pinacolborane). In the absence of in situ activators, mixtures of alkynylboronate and E-alkenylboronate esters were formed, whereas when NaHBEt3 was employed as the in situ activator, E-alkenylboronate esters were exclusively accessed. Mechanistic studies revealed a tandem C–H borylation/semihydrogenation pathway accounting for the formation of the products. Stoichiometric reactions hint toward reaction of a Mn–H active species with the terminal alkyne as the catalyst entry pathway to the cycle, whereas reaction with HBPin led to catalyst deactivation.
Introduction
The discovery of chemoselective catalysts for the functionalization of small molecules is key to access versatile synthons with applications in fine chemistry. The functionalization of alkynes with boronate esters (HB(OR)2) is a widely-used transformation, providing access to synthetically valuable alkynyl-, alkenyl- or alkyl-boronate esters.1 However, when the alkyne is terminal, chemoselectivity issues arise due to the competition of the functionalization of the triple and of the C(sp)–H bonds, and the control of the chemoselectivity of the reaction employing a catalyst is key. In transition metal-catalyzed reactions of terminal alkynes with boronate esters, the preferred reactivity, either hydroboration2 or C–H borylation,3 is determined by the identity of the metal complex, which dictates whether C–H activation or alkyne insertion steps are preferred. To avoid this competition, some catalysts have been reported to be efficient only for internal alkynes,4 and there are few insights into the keys leading to chemoselective hydroboration of terminal alkynes,2c,d including how the other components of the catalytic system (such as in situ activators, solvent identity or temperature) impact the chemoselectivity.2dManganese catalysis is a rising field, with Mn(I) catalysts containing CO ligands being well-established, particularly for reduction processes.5 In contrast, low-oxidation state (0, +I or +II) Mn complexes lacking CO ligands have been comparatively less explored for small molecule functionalization.6 We have recently reported that when the manganese complex [Mn(SiNSi)Cl2]6a (Mn1) was employed as precatalyst for the functionalization of terminal alkynes with HBPin, alkynylboronate esters were exclusively formed by C–H borylation (Scheme 1a, top).7 However, this precatalyst was found inefficient for the functionalization of either internal or terminal alkynes when it was in situ activated with NaHBEt3. In contrast, Rueping and coworkers reported that in situ activation with NaHBEt3 triggered the catalytic activity of the [Mn(EtPDI)Cl2] complex in the hydroboration of symmetrical internal alkynes to yield alkenylboronate esters from the syn addition of HBPin (Scheme 1a, bottom).8 However, the substrate scope was limited to symmetric internal alkynes, where competition of C(sp)–H activation was not a concern. Whereas Earth-abundant transition metal catalysts, mainly of Fe and Co,2b–d,f–o for the hydroboration of terminal alkynes are known, most of them being Z-stereoselective, a manganese catalyst for this transformation has not been described. Due to manganese being the third most abundant transition-metal in the Earth crust, a manganese catalyst for this process would contribute to the development of sustainable processes in synthetic chemistry. The main challenge to address to discover this catalyst is to understand which components of the catalytic system play a role in controlling the chemo-, stereo- and regioselectivity of the reaction for the functionalization of terminal alkynes.
 | ||
| Scheme 1 (a) Manganese-catalyzed dehydrogenative borylation of terminal alkynes (top) and hydroboration of internal alkynes (bottom) with two different Mn complexes; (b) this work: stereo- and regioselective hydroboration of terminal alkynes. | ||
Interested in elucidating the role that the ligand plays on the chemoselectivity of the manganese-catalyzed reaction between a terminal alkyne and HBPin, we hypothesized that a Mn complex containing a less electron-donating and sterically hindered pincer ligand than the SiNSi9 in Mn1, may render access to a catalyst capable of insertion steps affording alkenylboronate esters. In this work we report that the Mn(II) complex, [Mn(iPrPNP)Cl2], is the first catalyst for the hydroboration of unactivated terminal alkynes showing an excellent stereo- and regio-selectivity for the E-alkene upon in situ activation with NaHBEt3. The role of NaHBEt3 as activator is key to enhance the chemoselectivity of the catalyst for hydroboration. Insights into the reaction pathway as well as on the precatalyst entry to the cycle and deactivation pathways are provided.
Results and discussion
Catalytic competency of [Mn(iPrPNP)Cl2] for the functionalization of terminal alkynes with HBPin
Our research commenced with the synthesis of the manganese precatalyst [Mn(iPrPNP)Cl2]10 (Mn2, iPrPNP = 2,6-bis(diisopropylphosphinomethyl)pyridine) and the assessment of its efficiency for the functionalization of phenylacetylene (1) with HBPin (Pin = pinacolate) in the absence of strong hydride or alkyl sources as in situ activators. The initial conditions employed for the reaction were those identified as optimal in our lab for the C–H borylation of terminal alkynes catalyzed by [Mn(SiNSi)Cl2] (Mn1)7 (5 mol% catalyst loading, 2.5 equiv. HBPin, 80 °C, 24 h, 0.5 M solution in THF). Under these conditions, Mn2 led to a low conversion of the starting material (23%) affording the E-alkenylboronate ester 1b as the major product in a 14% yield, with the alkynylboronate ester 1a formed in <5% yield (crude yields and conversion determined by GC). A mixture of the alkynyl (15% yield) and the E-alkenylboronate ester (7% yield) was obtained for 4-fluorophenylacetylene with the reaction also proceeding to low conversion (24%, see page S5 in the ESI†). However, when 2-fluoro- or 3-fluorophenylacetylene were employed as substrates, the reactions proceeded to 51% and 52% conversions respectively affording the corresponding alkynylboronate esters exclusively (45% and 19% yields respectively, see page S5 in the ESI†). These results suggest that Mn2 is less efficient than Mn1 for the functionalization of terminal alkynes with HBPin and lacks chemoselectivity, as the identity of the substrate was capable of determining whether hydroboration or C–H borylation was preferred. Employing the Mn(II) dihalide complexes containing a tBuPNP (2,6-bis(di-tert-butylphosphinomethyl)pyridine, Mn3) or a EtPDI ligand (EtPDI = (2,6-diethylphenyl)pyridyldiimine, Mn4,8 see entries 3 and 4 in Scheme 2a) as precatalysts, resulted in low conversion of the starting material with formation of the E-alkenylboronate ester as the major product, although in low to moderate yields (13% for Mn3 and 43% for Mn4). It is worth noting that all the precatalysts employed (Mn2-Mn4) were chemoselective for hydroboration when phenylacetylene was employed as the substrate, suggesting that the stronger-electron donating and sterically demanding SiNSi ligand in Mn1 is key to favor C–H borylation over hydroboration.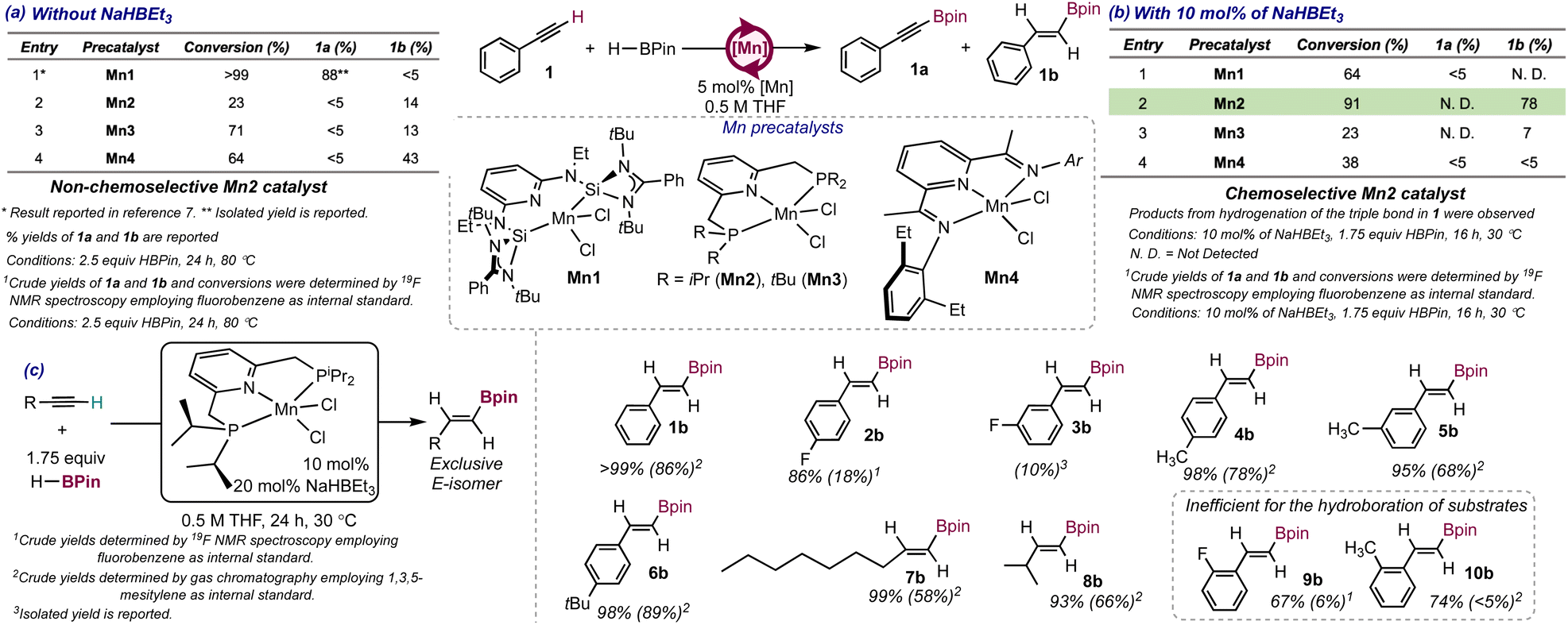 | ||
| Scheme 2 Catalyst screening for the functionalization of phenylacetylene with HBPin (a) without and (b) with NaHBEt3 as in situ activator and (c) substrate scope for the hydroboration of terminal alkynes employing Mn2/NaHBEt3 as precatalyst. | ||
Aiming to understand the factors that contributed to the loss of the chemoselectivity, the functionalization of 4-fluorophenylacetylelene (2) with HBPin catalyzed by Mn2 was monitored for 4 hours by 1H, 19F, 11B and 31P NMR spectroscopy in THF-d8 at 80 °C in a J. Young NMR tube where the headspace was evacuated (see pages S15–S19 in the ESI†). The results support that both products, the alkynyl- (2a, at −108.4 ppm in the 19F NMR spectrum) and the E-alkenylboronate (2b, at −111.9 ppm in the 19F NMR spectrum) esters were formed in a 0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 2a:2b ratio after 1 h (determined by integration of the 19F NMR spectrum). In contrast, when Mn1 was employed as precatalyst, product 2b was only formed at high substrate conversion,7 again highlighting the key role of the SiNSi ligand in the chemoselectivity for C–H borylation. The 2a:2b ratio changed over the course of 4 hours (1.4:1 after 2 h, 2.3:1 after 3 h and 3.4:1 after 4 h), hinting a faster increase in the amount of alkynylboronate ester than that of E-alkenylboronate ester, and suggesting two independent catalytic cycles operative in solution, one for C–H borylation and one for hydroboration. Further supporting this hypothesis, the 31P NMR spectra showed two signals at 38.0 and 38.5 ppm, consistent with the presence of two diamagnetic catalyst-resting states, one for each of the cycles operative (see Fig. S5 in the ESI†).
:1 2a:2b ratio after 1 h (determined by integration of the 19F NMR spectrum). In contrast, when Mn1 was employed as precatalyst, product 2b was only formed at high substrate conversion,7 again highlighting the key role of the SiNSi ligand in the chemoselectivity for C–H borylation. The 2a:2b ratio changed over the course of 4 hours (1.4:1 after 2 h, 2.3:1 after 3 h and 3.4:1 after 4 h), hinting a faster increase in the amount of alkynylboronate ester than that of E-alkenylboronate ester, and suggesting two independent catalytic cycles operative in solution, one for C–H borylation and one for hydroboration. Further supporting this hypothesis, the 31P NMR spectra showed two signals at 38.0 and 38.5 ppm, consistent with the presence of two diamagnetic catalyst-resting states, one for each of the cycles operative (see Fig. S5 in the ESI†).
Hydroboration of terminal alkynes with HBPin catalyzed by [Mn(iPrPNP)Cl2] in the presence of NaHBEt3 as in situ activator
Aiming to determine the impact that the presence of a strong hydride or alkyl source as in situ activator had on the chemoselectivity of the catalytic system, the efficiency of Mn2 for the functionalization of 1 with HBPin was assessed in the presence of strong hydride and alkyl sources. Employing 10 mol% of RLi (R = Me, Ph) or KOtBu as in situ activators of 5 mol% of Mn2 resulted in poor conversion of 1 and yield of the E-alkenylboronate ester 1b (see Table S1 in the ESI†). In contrast, when 10 mol% of NaHBEt3 were employed as the activator, full conversion of the starting material was observed, with 1b being formed as the major product in a 78% yield (see Scheme 2b, entry 2 in table). The E-isomer was exclusively obtained, and neither the Z isomer, α-alkenylboronate esters nor product 1a were detected by GC in the reaction crude, supporting Mn2/NaHBEt3 as the first stereo-, regio- and chemoselective catalytic system for the hydroboration of 1. Styrene was detected in <5% yield in the crude reaction mixture by GC, as well as trace amounts of 1,2,4 and 1,3,5-triphenylbenzene from alkyne cyclotrimerization.11The presence of styrene in the reaction medium can be attributed to the semihydrogenation of 1 catalyzed by Mn2/NaHBEt3 and is consistent with the report of Mn2 as an efficient catalyst for the Z-selective semihydrogenation of internal and terminal alkynes employing NH3BH3 as the H2 source.10
Whereas the use of NaHBEt3 as in situ activator in the reaction of 1 with HBPin catalyzed by Mn1 led to a mixture of unidentified products (see Scheme 2b, entry 1 in table), the results presented above support that Mn2 was capable of generating an active species by reaction of NaHBEt3 that catalyzed the hydroboration of 1. The presence of NaHBEt3 as the activator increased the catalyst efficiency but, more importantly, was key to drive the chemoselectivity of the reaction toward hydroboration. Employing [Mn(tBuPNP)Cl2] (Mn3) as the precatalyst, containing tBu groups at the P-donors, resulted in a lower conversion of the starting material (23%) and the formation of 1b in low yield (7%) (Scheme 2b, entry 3 in table). Even though Mn4 has been previously reported as an efficient catalyst for the hydroboration of internal alkynes,8 it showed poor catalytic activity for the hydroboration of terminal alkynes under the conditions in Scheme 2b (see entry 4 in table).
Control experiments for the reaction of 1 and HBPin in THF at 30 °C for 24 h were run (a) in the presence of 10 mol% of NaHBEt3, (b) in the presence of 5 mol% of MnCl2 and 10 mol% of NaHBEt3, (c) in the presence of 5 mol% of iPrPNP and 10 mol% of NaHBEt3 and (d) without any added species (see pages S10–S12 in the ESI†). In all cases, the reactions proceeded to <35% conversion of the starting material yielding a mixture of products that did not include 1b except in (c) and (d) where 1b was formed in <5% yield (determined by GC). These results support the need of all the reagents to access 1b in a synthetically useful yield and rule out trace borane as the species responsible for the catalytic activity (experiment (a)).12
Encouraged by the chemoselectivity shown by the Mn2/NaHBEt3 system for the reaction of 1 with HBPin, we set out to investigate the factors that impacted the yield of the product. The efficiency of other borylating agents, mainly HBDan and B2Pin2 for the functionalization of 1 was assessed (see Table S2 in the ESI†). In both cases, the yields for 1b were lower than when HBPin was employed (<5% for HBDan and <5% for B2Pin2). Employing 5 mol% of MnCl2, 5 mol% of iPrPNP and 10 mol% of NaHBEt3 as the precatalyst instead of in situ activated Mn2, led to a catalytic activity comparable to that of Mn2/NaHBEt3 (74% yield for 1b), suggesting that the active species can be in situ generated from the previous mixture. In contrast, when MnBr2, Mn(OAc)2 or Mn(OTf)2 were employed as the Mn source, the yield of 1b decreased significantly (18% yield of 1b for MnBr2 and <5% yield for Mn(OAc)2 and Mn(OTf)2, see Table S3 in the ESI†), highlighting the relevance of the Mn source in the formation of the active species. The order of addition of the components in the catalytic reaction also impacted the yield of 1b (see Table S4 in the ESI†). When (a) NaHBEt3 was added to a mixture of Mn2 and 1 followed by the addition of HBPin, the yield of 1b decreased to 44%, whereas when (b) NaHBEt3 was added to a mixture of Mn2 and HBPin followed by addition of 1, product 1b was formed only in trace amount. These observations suggest that the in situ activator should be added last, presumably to minimize decomposition of the catalytically active species as well as the semihydrogenation of 1 in the absence of HBPin in case (a) (see below for a mechanistic rationale).
Aiming to increase the yield of 1b, the reaction conditions were optimized (see Table S5 in the ESI†). The results showed that conducting the reaction with 10 mol% of catalyst loading, 20 mol% of NaHBEt3 at 30 °C, in a 0.5 M solution of THF and 1.75 equiv. of HBPin for 24 h afforded 1b in an 86% yield with the reaction proceeding to >99% conversion (catalyst TON = 9 and TOF = 1.3 × 10−4 s−1). Increasing the temperature of the reaction above 30 °C resulted in lower yields for 1b, presumably due to competition of the semihydrogenation of 1, as evidenced by the increased amounts of styrene formed as byproduct. Employing other solvents in the reaction such as toluene, acetonitrile or methanol led to decreased yields of 1b (acetonitrile) or to complete loss of the catalytic activity (toluene and methanol). Preactivation of Mn2 with 2 equiv. NaHBEt3 also led to a diminished yield for product 1b (34%), suggesting that the catalytically active species generated upon reaction of Mn2 with NaHBEt3 might decompose in the absence of the substrates.
The substrate scope for the hydroboration of terminal alkynes catalyzed by Mn2 was explored under the optimized conditions (see Scheme 2c). Mn2 was an efficient catalyst for the hydroboration of a moderate number of aromatic and aliphatic alkynes. The yields for the E-alkenylboronate esters from hydroboration of phenylacetylene (1, 86%, product 1b) and aromatic alkynes containing electron-donating substituents in the 4-position of the phenyl ring (e.g. products 4b and 6b with CH3 and tBu substituents formed in 78% and 89% yields respectively) were high with an excellent chemoselectivity for hydroboration. In contrast, the yield for the E-alkenylboronate ester 2b, obtained when 4-fluorophenylacetylene (2) was the substrate, was low (18%), suggesting that electron-withdrawing substituents drastically decrease the efficiency of the catalyst. The position of the substituent in the phenyl ring impacted the efficiency of the catalyst when electron-donating or electron-withdrawing groups were present at the phenyl ring. For example, when the F or CH3 groups were at the 3-position (substrates 3 and 5 respectively), the yields of products 3b and 5b decreased to 10% and 68% respectively, and when these groups were at the 2-position (substrates 9 for and 10 respectively), the E-alkenylboronate esters were formed in low yields (6% and <5% for 9b and 10b respectively). These results stand in contrast with those observed in the C–H borylation of terminal alkynes employing Mn1, where the position of the substituent in the phenyl ring did not impact the efficiency of the catalytic system.7 The decreased yields as the substituents are closer to the triple bond may be attributed to higher barriers for the insertion steps involved to access E-alkenylboronate esters, due to increased steric hindrance between the substrates and the catalytically active species. In line with this hypothesis, the aliphatic alkynes 7 and 8, containing more flexible alkyl chains, afforded products 7b and 8b in higher yields (58% and 66% respectively) than 2-substituted aryl acetylenes.
Mechanistic insights into the hydroboration of terminal alkynes with HBPin catalyzed by Mn(iPrPNP)Cl2/NaHBEt3
To gain insights into the reaction pathway as well as into the identity of the Mn active species, the catalytic hydroboration of 4-fluorophenylacetylene (2) with HBPin in the presence of 10 mol% of Mn2 and 20 mol% of NaHBEt3 employing 1,4-difluorobenzene as internal standard was monitored by 1H, 11B, 31P and 19F NMR spectroscopy in THF-d8 at 30 °C for 19 h. An induction period was not observed, and the 1H and 19F NMR spectra showed the immediate formation of several species at the expense of the starting material (see Scheme 3a for the 19F NMR spectra). The major species was identified as the E-alkenylboronate ester 2b whereas the minor species present were (a) the alkynylboronate ester 2a, (b) 4-fluorophenylethylene (2c) and (c) 4-fluorophenylethane (2d).
 | ||
| Scheme 3 Mechanistic experiments to gain insights into the reaction pathway, (a) 19F NMR monitoring of the catalytic reaction, (b) proposed reaction pathway and quantitative monitoring of the species during the catalytic reaction and (c) attempted hydroboration of an internal alkyne. | ||
Minor upfield-shifted unidentified signals were also present in the 19F NMR spectra, which can be tentatively attributed to the presence of products from alkyne cyclotrimerization (detected in the crude GC for the hydroboration of 1) and/or Mn-alkenyl or Mn-alkynyl complexes. The fact that 2a is present in the reaction medium hints that Mn2 is efficient for the C(sp)-H borylation of 2. Consistent with this hypothesis, H2 was observed (singlet at 4.55 ppm) in the 1H NMR spectra. The presence of 2c in the reaction medium supports that Mn2 is also efficient for the semihydrogenation of 2, which is consistent with previous reports describing Mn2 as an efficient catalyst for the Z-stereoselective transfer semihydrogenation of internal alkynes.10 The starting material was consumed in 10 hours, however, the 2a:2b ratio changed after full conversion (Scheme 3b). The changes in the yields of 2a and 2b over time supported that 2b formed at the expense of 2a, with this conversion being complete in 19 h, time after which the amounts of all the species in solution remained constant. The 1H NMR spectra showed the formation of H2 followed by its gradual consumption as 2b was forming at the expense of 2a (see Fig. S8 in the ESI†). These observations point toward a reaction pathway involving tandem C–H borylation/semihydrogenation to access 2b with the C–H borylation cycle being the regioselectivity-determining and the semihydrogenation cycle the stereoselectivity-determining (Scheme 3b). Because both, 2a and 2b, are present at the initial stages of the reaction, exclusive formation of 2b by stereo- and regioselective hydroboration of 2 may also be operative. Interestingly, Mn2/NaHBEt3 is E-stereoselective for the semihydrogenation of 2a, which stands in contrast to the reported Z stereoselectivity for the semihydrogenation of internal alkynes with NH3BH3 catalyzed by Mn2,10 suggesting that the identity of the substrate (internal alkyl or aryl alkyne vs. alkynylboronate ester) and/or the identity of the activator (NH3BH3 vs. NaHBEt3) play a relevant role in the stereoselectivity of the semihydrogenation reaction.
Because 4-fluorophenylstyrene (2c) is also formed as a byproduct, at least three catalytic cycles are operative in the reaction medium: (a) one for the semihydrogenation of 2, (b) one for the C–H borylation of 2, and (c) one for the semihydrogenation of 2a, with cycles (b) and (c) being responsible for the formation of 2b. The 31P NMR spectra showed signals for 3 P-containing species (at 0.60 (major), −0.50 (minor) and −2.18 ppm (minor), see bottom spectrum in Scheme 4e) which did not correspond to free iPrPNP ligand, neither to putative products from the reaction of the free iPrPNP ligand with HBPin and can be tentatively assigned to catalyst resting-states. The signal at −0.50 ppm disappeared after 2 h of reaction, whereas the those at 0.60 and −2.18 ppm remained intact during catalytic turnover, pointing toward these species as the catalyst resting-states for the C–H borylation and semihydrogenation cycles responsible for the formation of 2b. Furthermore, both signals appeared at different chemical shifts than those for the catalyst resting-states when Mn2 was employed in the absence of NaHBEt3 (38.0 and 38.5 ppm, see Fig. S5 in the ESI†), supporting different catalyst resting-states when the reaction is conducted in the presence of NaHBEt3. Signals attributable to diamagnetic Mn species were not identified in the 1H NMR spectra of the reaction, presumably obscured by the signals of 2, 2a, 2b and 2c.
 | ||
| Scheme 4 (a) Product yields in the catalytic reaction after 16 h. Mechanistic experiments to gain insights into precatalyst entry pathway to the cycle: (b) addition of NaHBEt3 to a mixture of Mn2 and excess 2; (c) addition of NaHBEt3 to a mixture of Mn2 and excess 2 followed by addition of excess HBPin and (d) addition of NaHBEt3 to a mixture of Mn2 and excess HBPin followed by addition of excess 2. (e) Stacked 31P NMR spectra in THF-d8 for the catalytic reaction (a) and reactions (c) and (d); (f) proposed catalyst entry to the cycle and deactivation pathway. Chemical shifts are reported in ppm close to each signal. * denotes an unidentified signal at −3.13 ppm. | ||
Further supporting a tandem C–H borylation/semihydrogenation pathway operative in catalysis, the hydroboration of the internal alkyne 1-phenyl-1-propyne catalyzed by Mn2/NaHBEt3 proceeded to low conversion with the corresponding E-alkenylboronate ester and alkene detected in trace amounts in the reaction crude (by 1H NMR spectroscopy and GC, see Scheme 3c). Because this substrate lacks C(sp)–H groups, a C–H borylation/semihydrogenation pathway is not accessible, and only a hydroboration cycle involving alkyne insertion and C(sp2)–B formation steps can be operative to yield the alkenylboronate ester. However, this cycle may be inaccessible due to high energy barriers resulting in no catalytic activity for this process.
Although catalysts capable of performing tandem C–H borylation/hydroboration are known,13 the potential of manganese catalysts in tandem processes, which is still the realm of precious metals,14 is underexploited. Therefore, the results reported here constitute a first step toward the design of manganese catalysts efficient for tandem transformations.
To identify whether the putative Mn–H enters the cycle by reaction with the alkyne, the reaction of Mn2 with 2 equiv. of NaHBEt3 in the presence of 10 equivalents of 2 was conducted in THF-d8 in a J. Young NMR tube at room temperature in the presence of 1,4-difluorobenzene as internal standard and the 1H, 19F and 31P NMR spectra were registered (see second 19F and 1H NMR spectra from the bottom in Fig. S16–S19† and bottom 31P NMR spectrum in Fig. S21 in the ESI† and Scheme 4b). Immediately after the addition of NaHBEt3 to the mixture of Mn2 and 2, the 19F NMR spectra showed signals consistent with the presence of 2c (−113.4 ppm) and 4-fluorophenylethane (−117.0 ppm) (see second from the bottom19F NMR spectrum in Fig. S16 in the ESI†), formed by hydrogenation of 2 (−109.4 ppm). Accordingly, the 1H NMR spectra hinted the presence of H2 (at 4.50 ppm, see second 1H NMR spectrum from the bottom in Fig. S18†). As in the monitoring of the catalytic reaction, minor upfield shifted signals in the 19F NMR spectra may hint the presence of Mn-alkenyl or Mn-alkynyl complexes and of products from alkyne cyclotrimerization. The 31P NMR spectra did not show any signal attributable to diamagnetic Mn complexes neither to free iPrPNP (see bottom 31P NMR spectrum in Fig. S21 in the ESI†). These results suggest that, in the absence of HBPin, Mn2 can catalyze the semihydrogenation of 2 to 2c and the full hydrogenation to 2d with the H2 generated from precatalyst activation and allow to rationalize the presence of 2c and 2d as byproducts in the hydroboration of 2.
Addition of excess HBPin to the reaction mixture resulted in the immediate formation of 2a (−108.0 ppm) and 2b (−111.5 ppm) (see Fig. S16 in the ESI†). This result supports reaction of the putative Mn–H active species with the alkyne 2 as the catalyst entry pathway to the cycle (Scheme 4f). Therefore, the active Mn–H would activate the C–H bond of 2 to yield 2a in a first C–H borylation cycle followed by insertion of the triple bond of 2a in a second semihydrogenation cycle that would yield 2b. The 31P NMR spectra showed the presence of 3 species at 0.60 (major), −0.50 (minor) and −2.18 ppm (minor), consistent with diamagnetic Mn complexes present in the reaction medium (see spectrum for reaction (c) after 16 h in Scheme 4e). More importantly, the species at 0.60 and −2.18 ppm were also present in the 31P NMR spectra of the catalytic reaction (see spectrum for reaction (a) after 16 h in Scheme 4e), which supports them as the catalyst resting-states for the C–H borylation and semihydrogenation cycles responsible for the formation of 2b, and the reaction of the Mn–H with the alkyne as the catalyst entry pathway to the C–H borylation cycle. Attempts to isolate or characterize a Mn complex from the reaction of Mn2 with 2 equiv. of NaHBEt3 in the presence of 3 equivalents of 2 failed and the crude 1H and 19F NMR spectra showed peaks consistent with the presence of 2c and 2d, suggesting that the in situ generated Mn–H catalyzed the hydrogenation of 2.
The results presented above support that, when only H2 is present in the reaction medium after precatalyst activation, the C–H bond activation pathway is unproductive and the insertion pathways yielding 2c and 2d are preferred. However, in the presence of HBPin, the tandem C–H borylation/semihydrogenation catalytic cycles are preferred, accounting for the formation of the major product 2b. Because the amount of 2c increases over time even in the presence of HBPin but to a lower extent than the amounts of 2b and 2a, the semihydrogenation of 2 is still operative in the presence of HBPin, however, it is slower than the tandem C–H borylation/semihydrogenation that yields 2b.
Aiming to identify other catalytically active species, the reaction of Mn2 with 2 equiv. of NaHBEt3 in the presence of 10 equivalents of HBPin was conducted in THF-d8 in a J. Young NMR tube and the 1H, 11B and the 31P NMR spectra were registered at room temperature (see Fig. S22–S26 in the ESI†). The 1H and 11B NMR spectra only showed signals attributable to HBPin, however, the 31P showed two signals, at 13.4 ppm (major), attributable to free iPrPNP ligand, and at −0.50 ppm (minor) (see bottom 31P NMR spectrum in Fig. S26 in the ESI†), which also appeared as a minor species at early stages of the catalytic reaction and when HBPin was added to a mixture of Mn2, 2 and NaHBEt3 (see spectrum for reaction (c) after 16 h in Scheme 4e). The presence of free iPrPNP ligand in the reaction crude points toward partial decomposition of the manganese complex formed upon reaction of Mn2 with HBPin and NaHBEt3.
Addition of excess 2 to the reaction mixture resulted in the formation of products 2a, 2b and 2c (see 19F NMR monitoring in Fig. S22 of the ESI†), suggesting catalytic turnover taking place after addition of 2. However, the reaction only proceeded to 18% conversion after 16 hours, significantly lower than the conversion for the catalytic reaction (98% after 16 hours), and than when HBPin was added to a mixture of Mn2, NaHBEt3 and 2 (64% after 16 hours). This result points to the reaction of the putative Mn–H with HBPin as a catalyst deactivation pathway (Scheme 4f), consistent with the presence of free iPrPNP ligand in the 31P NMR spectra, and allows to rationalize why the order of addition of the reagents in the catalytic reaction impacted the yield of product 1b. When NaHBEt3 was added to a mixture of HBPin and Mn2 followed by addition of 1 (see above), trace amount of 1b was observed due to deactivation of the catalytically active Mn–H by HBPin before the addition of 1. The presence of this deactivation pathway in the catalytic reaction prevents the recyclability of the catalyst and limits its lifetime to 19 h for the hydroboration of 2. Alternatively, when NaHBEt3 was added to a mixture of 1 and Mn2 followed by the addition of HBPin, the catalytic reaction proceeded as the Mn–H entered the first catalytic cycle by reaction with 1. However, in this instance 1b was formed in lower yield (44%) than when NaHBEt3 was added to a mixture of Mn2, HBPin and 1 (78%), which could be attributed to partial decomposition of the active species formed upon reaction of the Mn–H with 1 before the addition of HBPin.
Additionally, the 31P NMR spectra showed the presence of the signals at 0.60, −0.50 and −2.18 ppm (see top spectrum in Scheme 4e), consistent with the presence of catalytically active species that would be responsible for the formation of products 2a, 2b and 2c. However, because the species formed after reaction of Mn2/NaHBEt3 with HBPin decomposed to a large extent as evidenced by the presence of free iPrPNP ligand as the major product, the concentration of the catalytically active species is presumed to be low, resulting in a low conversion of the starting material and low yields of the products.
Further mechanistic studies are currently undergoing in our laboratory aiming to elucidate the identity of the catalytically active species.
Conclusions
In summary, we have discovered the first manganese catalyst for the hydroboration of terminal alkynes. The presence of NaHBEt3 as precatalyst activator was found to enhance the chemoselectivity of the process and the efficiency of the catalytic system. Aromatic alkynes with electron-donating groups at the phenyl rings and aliphatic alkynes, mainly 1-octene and 3-methyl-1-butyne, were hydroborated with an excellent chemo-, regio- and stereoselectivity. In contrast, when electron-withdrawing substituents were present in the phenyl ring the catalytic activity plummeted. In all the cases, the position of the substituent in the phenyl ring (2 vs. 3 vs. 4) impacted the efficiency of the system, with 2-substituted aryl alkynes leading to the loss of the catalytic activity. The hydroboration reaction was found to proceed by a tandem C–H borylation/E-stereoselective semihydrogenation pathway, with the C–H borylation step resulting on β-regioselectivity and the semihydrogenation step affording exclusively the E-alkenylboronate ester. Stoichiometric reactions support that the precatalyst entered the cycle by reaction with NaHBEt3 followed by reaction with the alkyne, whereas reaction with HBPin led to decomposition of the Mn complex and constituted a catalyst deactivation pathway. Further mechanistic studies are currently being carried out in our laboratory to elucidate the identity of the Mn species involved in the catalytic cycles.Author contributions
R. A. designed the experiments, supervised the experimental work and drafted the manuscript. V. D. A. designed the experiments, conducted the experimental work and drafted the ESI.† The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. A. acknowledges the NSF for a LEAPS-MPS grant (# 2316526) and UC Merced for start-up funds. R. A. and V. D. A. thank Himani Ahuja for a generous gift of HBDan.Notes and references
- (a) J. Jiao and Y. Nishihara, Alkynylboron Compounds in Organic Synthesis, J. Organomet. Chem., 2012, 3–16 CrossRef CAS; (b) S. Nandy, S. Paul, K. K. Das, P. Kumar, D. Ghorai and S. Panda, Synthesis and reactivity of alkynyl boron compounds, Org. Biomol. Chem., 2021, 19, 7276–7297 RSC; (c) J. Carreras, A. Caballero and P. J. Perez, Alkenyl Boronates: Synthesis and Applications, Chem.–Asian J., 2019, 14, 329–343 CrossRef CAS PubMed; (d) J. El-Maiss, T. M. El Dine, C.-S. Lu, I. Karame, A. Kanj, K. Polychronopoulou and J. Shaya, Recent advances in metal-catalyzed alkyl-boron (C(sp3)-C(sp2)) Suzuki-Miyaura cross-couplings, Catalysts, 2020, 10, 296 CrossRef CAS; (e) W. N. Palmer, C. Zarate and P. J. Chirik, Benzyltriboronates: Building Blocks for Diastereoselective Carbon–Carbon Bond Formation, J. Am. Chem. Soc., 2017, 139, 2589–2592 CrossRef CAS PubMed.
- (a) S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, The transition metal-catalysed hydroboration reaction, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC; (b) A. Singh, S. Shafiei-Haghighi, C. R. Smith, D. K. Unruh and M. Findlater, Hydroboration of Alkenes and Alkynes Employing Earth-Abundant Metal Catalysts, Asian J. Org. Chem., 2020, 9, 416–420 CrossRef CAS; (c) E. A. Romero, R. Jazzar and G. Bertrand, (CAAC)CuX-catalyzed hydroboration of terminal alkynes with pinacolborane directed by the X-ligand, J. Organomet. Chem., 2017, 829, 11e13 CrossRef; (d) K. Jaiswal, K. Groutchik, D. Bawari and R. Dobrovetsky, An “On-Demand”, Selective Dehydrogenative Borylation or Hydroboration of Terminal Alkynes Using Zn2+-based Catalyst, ChemCatChem, 2022, 14, e202200004 CrossRef CAS; (e) S. Weber, D. Zobernig, B. Stöger, L. F. Veiros and K. Kirchner, Hydroboration of Terminal Alkenes and trans-1,2-Diboration of Terminal Alkynes Catalyzed by a Manganese(I) Alkyl Complex, Angew. Chem., Int. Ed., 2021, 60, 24488–24492 CrossRef CAS PubMed; (f) N. Gorgas, L. G. Alves, B. Stöger, A. M. Martins, L. F. Veiros and K. Kirchner, Stable, Yet Highly Reactive Nonclassical Iron(II) Polyhydride Pincer Complexes: Z-Selective Dimerization and Hydroboration of Terminal Alkynes, J. Am. Chem. Soc., 2017, 139, 8130–8133 CrossRef CAS PubMed; (g) J. V. Obligacion, J. M. Neely, A. N. Yazdani, I. Pappas and P. J. Chirik, Cobalt Catalyzed Z-Selective Hydroboration of Terminal Alkynes and Elucidation of the Origin of Selectivity, J. Am. Chem. Soc., 2015, 137, 5855–5858 CrossRef CAS PubMed; (h) S. Mandal, S. Mandal and K. Geetharani, Zinc-Catalysed Hydroboration of Terminal and Internal Alkynes, Chem.–Asian J., 2019, 14, 4553–4556 CrossRef CAS PubMed; (i) H. Ben-Daat, C. L. Rock, M. Flores, T. L. Groy, A. C. Bowman and R. J. Trovitch, Hydroboration of alkynes and nitriles using an a-diimine cobalt hydride catalyst, Chem. Commun., 2017, 53, 7333–7336 RSC; (j) J. Chen, X. Shen and Z. Lu, Cobalt-Catalyzed Markovnikov-Type Selective Hydroboration of Terminal Alkynes, Angew. Chem., Int. Ed., 2021, 60, 690–694 CrossRef CAS PubMed; (k) C. K. Blasius, V. Vasilenko, R. Matveeva, H. Wadepohl and L. H. Gade, Reaction Pathways and Redox States in a-Selective Cobalt-Catalyzed Hydroborations of Alkynes, Angew. Chem., Int. Ed., 2020, 59, 23010–23014 CrossRef CAS PubMed; (l) K. Nakajima, T. Kato and Y. Nishibayashi, Hydroboration of Alkynes Catalyzed by Pyrrolide-Based PNP Pincer−Iron Complexes, Org. Lett., 2017, 19, 4323–4326 CrossRef CAS PubMed; (m) W. J. Jang, W. L. Lee, J. H. Moon, J. Y. Lee and J. Yun, Copper-Catalyzed trans-Hydroboration of Terminal Aryl Alkynes: Stereodivergent Synthesis of Alkenylboron Compounds, Org. Lett., 2016, 18, 1390–1393 CrossRef CAS PubMed; (n) M. J. González, F. Bauer and B. Breit, Cobalt-Catalyzed Hydroboration of Terminal and Internal Alkynes, Org. Lett., 2021, 23, 8199–8203 CrossRef PubMed; (o) S. Garhwal, N. Fridman and G. de Ruiter, Z-Selective Alkyne Functionalization Catalyzed by a trans-Dihydride N-Heterocyclic Carbene (NHC) Iron Complex, Inorg. Chem., 2020, 59, 13817–13821 CrossRef CAS PubMed; (p) H. Yoshida, Borylation of Alkynes underBase/Coinage Metal Catalysis: Some Recent Developments, ACS Catal., 2016, 6, 1799–1811 CrossRef CAS; (q) K. Yamamoto, Y. Mohara, Y. Mutoh and S. Saito, Ruthenium-Catalyzed (Z)-Selective Hydroboration of Terminal Alkynes with Naphthalene-1,8-diaminatoborane, J. Am. Chem. Soc., 2019, 141, 17042–17047 CrossRef CAS PubMed; (r) C. Gunanathan, M. Hölscher, F. Pan and W. Leitner, Ruthenium Catalyzed Hydroboration of Terminal Alkynes to Z-Vinylboronates, J. Am. Chem. Soc., 2012, 134, 14349–14352 CrossRef CAS PubMed; (s) A. L. Narro, H. D. Arman and Z. J. Tonzetich, Mechanistic Studies of Alkyne Hydroboration by a Well-Defined Iron Pincer Complex: Direct Comparison of Metal-Hydride and Metal-Boryl Reactivity, Inorg. Chem., 2022, 61, 10477–10485 CrossRef CAS PubMed.
- (a) C.-I. Lee, J. Zhou and O. V. Ozerov, Catalytic dehydrogenative borylation of terminal alkynes by a SiNN pincer complex of iridium, J. Am. Chem. Soc., 2013, 135, 3560–3566 CrossRef CAS PubMed; (b) B. J. Foley, N. Bhuvanesh, J. Zhou and O. V. Ozerov, Combined Experimental and Computational Studies of the Mechanism of Dehydrogenative Borylation of Terminal Alkynes Catalyzed by PNP Complexes of Iridium, ACS Catal., 2020, 10, 9824–9836 CrossRef CAS; (c) B. J. Foley and O. V. Ozerov, Air- and Water-Tolerant (PNP)Ir Precatalyst for the Dehydrogenative Borylation of Terminal Alkynes, Organometallics, 2020, 39, 2352–2355 CrossRef CAS; (d) E. A. Romero, R. Jazzar and G. Bertrand, Copper-catalyzed dehydrogenative borylation of terminal alkynes with pinacolborane, Chem. Sci., 2017, 8, 165–168 RSC; (e) R. J. Procter, M. Uzelac, J. Cid, P. J. Rushworth and M. J. Ingleson, Low-Coordinate NHC-Zinc Hydride Complexes Catalyze Alkyne C-H Borylation and Hydroboration Using Pinacolborane, ACS Catal., 2019, 9, 5760–5771 CrossRef CAS; (f) D. Lewandowski, T. Cytlak, R. Kempe and G. Hreczycho, Ligand-controlled Cobalt-Catalyzed formation of Carbon-Boron bonds: Hydroboration vs. C-H/B-H dehydrocoupling, J. Catal., 2022, 413, 728–734 CrossRef CAS; (g) C. J. Pell and O. V. Ozerov, Catalytic dehydrogenative borylation of terminal alkynes by POCOP-supported palladium complexes, Inorg. Chem. Front., 2015, 2, 720–724 RSC; (h) T. Tsuchimoto, H. Utsugi, T. Sugiura and S. Horio, Alkynylboranes: A Practical Approach by Zinc-Catalyzed Dehydrogenative Coupling of Terminal Alkynes with 1,8-Naphthalenediaminatoborane, Adv. Synth. Catal., 2015, 357, 77–82 CrossRef CAS; (i) D. Wei, B. Carboni, B. J.-B. Sortais and C. Darcel, Iron-Catalyzed Dehydrogenative Borylation of Terminal Alkynes, Adv. Synth. Catal., 2018, 360, 3649–3654 CrossRef CAS; (j) J.-R. Hu, L.-H. Liu, X. Hu and H.-D. Ye, Ag(I)-catalyzed C-H borylation of terminal alkynes, Tetrahedron, 2014, 70, 5815–5819 CrossRef CAS; (k) M. Luo, Y. Qin, X. Chen, Q. Xiao, B. Zhao, W. Yao and M. Ma, ZnBr2-Catalyzed Dehydrogenative Borylation of Terminal Alkynes, J. Org. Chem., 2021, 86, 16666–16674 CrossRef CAS PubMed.
- (a) M. Haberberger and S. Enthaler, Straightforward Iron-Catalyzed Synthesis of Vinylboronates by the Hydroboration of Alkynes, Chem.–Asian J., 2013, 8, 50–54 CrossRef CAS PubMed; (b) L. Ferrand, Y. Lyu, A. Rivera-Hernández, B. J. Fallon, M. Amatore, C. Aubert and M. Petit, Hydroboration and Diboration of Internal Alkynes Catalyzed by a Well-Defined Low-Valent Cobalt Catalyst, Synth, 2017, 49, 3895–3904 CrossRef CAS; (c) F. Rami, F. Bächtle and B. Plietker, Hydroboration of internal alkynes catalyzed by Fe(CO)(NO)(PPh3)2: a case of boron-source controlled regioselectivity, Catal. Sci. Technol., 2020, 10, 1492–1497 RSC; (d) G. Zhang, H. Zeng, S. Zheng, M. C. Neary and P. A. Dub, Vanadium-Catalyzed Stereo- and Regioselective Hydroboration of Alkynes to Vinyl Boronates, ACS Catal., 2022, 12, 5425–5429 CrossRef CAS.
- (a) K. Das, M. K. Barman and B. Maji, Advancements in multifunctional manganese complexes for catalytic hydrogen transfer reactions, Chem. Commun., 2021, 57, 8534–8549 RSC and references therein;; (b) K. Das, S. Waiba, A. Jana and B. Maji, Manganese-catalyzed hydrogenation, dehydrogenation, and hydroelementation reactions, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC and references therein;; (c) E. S. Gulyaeva, E. S. Osipova, R. Buhaibeh, Y. Canac, J. B. Sortais and D. A. Valyaev, Towards ligand simplification in manganese-catalyzed hydrogenation and hydrosilylation processes, Coord. Chem. Rev., 2022, 458, 214421 CrossRef CAS; (d) P. Schlichter and C. Werle, The Rise of Manganese-Catalyzed Reduction Reactions, Synth, 2022, 54, 517–534 CrossRef CAS; (e) A. Torres-Calis and J. J. García, Homogeneous Manganese-Catalyzed Hydrofunctionalizations of Alkenes and Alkynes: Catalytic and Mechanistic Tendencies, ACS Omega, 2022, 7, 37008–37038 CrossRef CAS PubMed; (f) D. M. Sharma, A. B. Shabade, R. G. Gonnade and B. Punji, Manganese(I)-Catalyzed Chemoselective Transfer Hydrogenation of the C=C Bond in Conjugated Ketones at Room Temperature, Chem.–Eur. J., 2023, 29, e202301174 CrossRef CAS PubMed; (g) R. A. Farrar-Tobar, S. Weber, Z. Csendes, A. Ammaturo, S. Fleissner, H. Hoffmann, L. F. Veiros and K. Kirchner, E-Selective Manganese-Catalyzed Semihydrogenation of Alkynes with H2 Directly Employed or In Situ-Generated, ACS Catal., 2022, 12, 2253–2260 CrossRef CAS PubMed.
- (a) Y. P. Zhou, Z. Mo, M. P. Luecke and M. Driess, Stereoselective Transfer Semi-Hydrogenation of Alkynes to E-Olefins with N-Heterocyclic Silylene-Manganese Catalysts, Chem.–Eur. J., 2018, 24, 4780–4784 CrossRef CAS PubMed; (b) R. J. Trovitch, The Emergence of Manganese-Based Carbonyl Hydrosilylation Catalysts, Acc. Chem. Res., 2017, 50, 2842–2852 CrossRef CAS PubMed; (c) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, A Highly Active Manganese Precatalyst for the Hydrosilylation of Ketones and Esters, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef CAS PubMed; (d) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, A β-diketiminate manganese catalyst for alkene hydrosilylation: substrate scope, silicone preparation, and mechanistic insight, Chem. Sci., 2018, 9, 7673–7680 RSC; (e) G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Highly Selective Hydroboration of Alkenes, Ketones and Aldehydes Catalyzed by a Well-Defined Manganese Complex, Angew. Chem., 2016, 128, 14581–14584 CrossRef; (f) L. Britton, M. Krodzki, G. S. Nichol, A. P. Dominey, P. Pawluc, J. H. Docherty and S. P. Thomas, Manganese-Catalyzed C(sp2)-H Borylation of Furan and Thiophene Derivatives, ACS Catal., 2021, 11, 6857–6864 CrossRef CAS.
- H. Ahuja, H. Kaur and R. Arevalo, Chemoselective C(sp)–H borylation of terminal alkynes catalyzed by a bis(N-heterocyclicsilylene) manganese complex, Inorg. Chem. Front., 2023, 10, 6067–6076 RSC.
- A. Brzozowska, V. Zubar, R.-C. Ganardi and M. Rueping, Chemoselective Hydroboration of Propargylic Alcohols and Amines Using a Manganese(II) Catalyst, Org. Lett., 2020, 22, 3765–3769 CrossRef CAS PubMed.
- (a) S. Kalra, D. Pividori, D. Fehn, C. Dai, S. Dong, S. Yao, J. Zhu, K. Meyer and M. Driess, A bis(silylene)pyridine pincer ligand can stabilize mononuclear manganese(0) complexes: facile access to isolable analogues of the elusive d7-Mn(CO)5 radical, Chem. Sci., 2022, 13, 8634–8641 RSC; (b) H. Ren, Y.-P. Zhou, Y. Bai, C. Cui and M. Driess, Cobalt-Catalyzed Regioselective Borylation of Arenes: N-Heterocyclic Silylene as an Electron Donor in the Metal-Mediated Activation of C−H Bonds, Chem.–Eur. J., 2017, 23, 5663–5667 CrossRef CAS PubMed; (c) R. Arevalo, T. P. Pabst and P. J. Chirik, C(sp2)-H Borylation of Heterocycles by Well-Defined Bis(silylene)pyridine Cobalt(III) Precatalysts: Pincer Modifica-tion, C(sp2)-H Activation, and Catalytically Relevant Intermedi-ates, Organometallics, 2020, 39, 2763–2773 CrossRef CAS PubMed; (d) Y. P. Zhou and M. Driess, Isolable Silylene Ligands Can Boost Efficiencies and Selectivities in Metal-Mediated Catalysis, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed; (e) D. Gallego, S. Inoue, B. Blom and M. Driess, Highly Electron-Rich Pincer-Type Iron Complexes Bearing Innocent Bis(metallylene)pyridine Ligands: Syntheses, Structures, and Catalytic Activity, Organometallics, 2014, 33, 6885–6897 CrossRef CAS.
- A. Brzozowska, L. M. Azofra, V. Zubar, I. Atodiresei, L. Cavallo, M. Rueping and O. El-Sepelgy, Highly Chemo- and Stereoselective Transfer Semihydrogenation of Alkynes Catalyzed by a Stable, Well-Defined Manganese(II) Complex, ACS Catal., 2018, 8, 4103–4109 CrossRef CAS.
- S. Arepally, P. Nandhakumar, G. A. González-Montiel, A. Dzhaparova, G. Kim, A. Ma, K. M. Nam, H. Yang, P. H.-Y. Cheong and J. K. Park, Unified Electrochemical Synthetic Strategy for [2 + 2 + 2] Cyclotrimerizations: Construction of 1,3,5- and 1,2,4-Trisubstituted Benzenes from Ni(I)-Mediated Reduction of Alkynes, ACS Catal., 2022, 12, 6874–6886 CrossRef CAS.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Hidden Boron Catalysis: Nucleophile-Promoted Decomposition of HBpin, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed.
- (a) Q. Lai and O. V. Ozerov, Dehydrogenative Diboration of Alkynes Catalyzed by Ir/CO/tBuNC System, J. Organomet. Chem., 2021, 931, 121614 CrossRef CAS; (b) Y. Gao, Z. Q. Wu and K. M. Engle, Synthesis of Stereodefined 1,1-Diborylalkenes via Copper-Catalyzed Di-boration of Terminal Alkynes, Org. Lett., 2020, 22, 5235–5239 CrossRef CAS PubMed; (c) S. Krautwald, M. J. Bezdek and P. J. Chirik, Cobalt-Catalyzed 1,1-Diboration of Terminal Alkynes: Scope, Mechanism, and Synthetic Applications, J. Am. Chem. Soc., 2017, 139, 3868–3875 CrossRef CAS PubMed; (d) C. I. Lee, W. C. Shih, J. Zhou, J. H. Reibenspies and O. V. Ozerov, Synthesis of Triborylalkenes from Terminal Alkynes by Iridium-Catalyzed Tandem C-H Borylation and Diboration, Angew. Chem., Int. Ed., 2015, 54, 14003–14007 CrossRef CAS PubMed; (e) X. Liu, W. Ming, Y. Zhang, A. Friedrich and T. B. Marder, Copper-Catalyzed Triboration: Straightforward, Atom-Economical Synthesis of 1,1,1-Triborylalkanes from Terminal Alkynes and HBpin, Angew. Chem., Int. Ed., 2019, 58, 18923–18927 CrossRef CAS PubMed.
- (a) R. T. Ruck, M. A. Huffman, M. M. Kim, M. Shevlin, W. V. Kandur and I. W. Davies, Palladium-Catalyzed Tandem Heck Reaction/C-H Functionalization—Preparation of Spiro-Indane-Oxindoles, Angew. Chem., Int. Ed., 2008, 47, 4711–4714 CrossRef CAS PubMed; (b) S. S. Ichake, A. Konala, V. Kavala, C.-W. Kuo and C.-F. Yao, Palladium-Catalyzed Tandem C−H Functionalization/Cyclization Strategy for the Synthesis of 5-Hydroxybenzofuran Derivatives, Org. Lett., 2017, 19, 54–57 CrossRef CAS PubMed; (c) M. K. Armstrong, M. B. Goodstein and G. Lalic, Diastereodivergent Reductive Cross Coupling of Alkynes throughTandem Catalysis: Z- and E-Selective Hydroarylation of Terminal Alkynes, J. Am. Chem. Soc., 2018, 140, 10233–10241 CrossRef CAS PubMed; (d) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Concurrent Tandem Catalysis, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed; (e) Y. You and S. Ge, Cobalt-Catalyzed One-Pot Asymmetric Difunctionalization of Alkynes to Access Chiral gem-(Borylsilyl)alkanes, Angew. Chem., Int. Ed., 2021, 60, 20684–20688 CrossRef CAS PubMed.
- M. Glatz, B. Stoeger, D. Himmelbauer, L. F. Veiros and K. Kirchner, Chemoselective Hydrogenation of Aldehydes under Mild, Base-Free Conditions: Manganese Outperforms Rhenium, ACS Catal., 2018, 8, 4009–4016 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, characterization data, NMR spectroscopic data. See DOI: https://doi.org/10.1039/d3ra08747f |
| This journal is © The Royal Society of Chemistry 2024 |