DOI:
10.1039/D3RA08727A
(Paper)
RSC Adv., 2024,
14, 6476-6493
A synthetic approach towards drug modification: 2-hydroxy-1-naphthaldehyde based imine-zwitterion preparation, single-crystal study, Hirshfeld surface analysis, and computational investigation†
Received
21st December 2023
, Accepted 15th February 2024
First published on 21st February 2024
Abstract
The current work is about the modification of primary amine functionalized drugs, pyrimethamine and 4-amino-N-(2,3-dihydrothiazol-2-yl)benzenesulfonamide, via condensation reaction with 2-hydroxy-1-naphthaldehyde to produce new organic zwitterionic compounds (E)-1-(((4-(N-(2,3-dihydrothiazol-2-yl)sulfamoyl)phenyl)iminio)methyl)naphthalen-2-olate (DSPIN) and (E)-1-(((4-amino-5-(4-chlorophenyl)-6-ethylpyrimidin-2-yl)iminio)methyl)naphthalen-2-olate (ACPIN) in methanol as a solvent. The crystal structures of both compounds were confirmed to be imine-based zwitterionic products via single-crystal X-ray diffraction (SC-XRD) analysis which indicated that the stabilization of both crystalline compounds is achieved via various noncovalent interactions. The supramolecular assembly in terms of noncovalent interactions was explored by the Hirshfeld surface analysis. Void analysis was carried out to predict the crystal mechanical response. Compound geometries calculated in the DFT (Density Functional Theory) study showed reasonably good agreement with the experimentally determined structural parameters. Frontier molecular orbital (FMO) analysis showed that the DSPIN HOMO/LUMO gap is by 0.15 eV smaller than the ACPIN HOMO/LUMO gap due to some destabilization of the DSPIN HOMO and some stabilization of its LUMO. The results of the charge analysis implied formation of intramolecular hydrogen bonds and suggested formation of intermolecular hydrogen bonding and dipole–dipole and dispersion interactions.
1. Introduction
Diseases like influenza, typhoid, typhus, and malaria are conventionally considered as fatal sicknesses due to the unapproachability of operative medications.1 Pyrimidine-based chemical building blocks have been effectively used as medications for malaria.2 The physicians no more prescribe pyrimethamine as a medication for malaria because of the drug resistance phenomenon.3 As pyrimethamine alone has lost its efficacy, its combinations with various chemical building blocks like sulfadoxine-pyrimethamine, sulfamethoxazole-trimethoprim, and sulfalene-pyrimethamine have been used as new antimalarial drugs but they also lost their effectiveness against malaria as time has passed.4 These drugs can further be modified for the sake of new medicinal applications. Previously, we have reported the modification of pyrimethamine via co-crystallization and the structural and electronic features of the modified compounds were studied by computational methods.5
Another important chemical architecture from the point of view of the synthesis and drug development is represented by the sulfonamide derivatives. This class of compounds has presented diverse pharmacological activities including anti-carbonic anhydrase,6 antibacterial,7 hypoglycaemic,8 diuretic,9 antithyroid,10 and antitumor activity in vitro and/or in vivo.11 The modification of these two pharmacologically significant compound classes is important for the syntheses of diversely functionalized chemical building blocks that have medicinal applications as well as interesting optoelectronic properties. For studies of structures, electronic, optical, and reactivity properties of various compounds, the density functional theory (DFT) has been developed as a very promising tool based on computer-based calculations. There are several features of organic compounds that can be investigated via DFT studies, namely, energies of the frontier molecular orbitals, HOMO (the highest occupied molecular orbital) and LUMO (the lowest unoccupied molecular orbital), HOMO–LUMO and optical gaps, noncovalent interactions involving weak attractive forces, and non-linear optical (NLO) properties.12,13 Our collaborative research group has been working on the syntheses and theoretical exploration via DFT analysis of different classes of compounds with organic origin such as β-hydroxycarbonyl compounds and chalcones,14 hydrazones,15,16 piperidone derivatives,17 peptoids,18 phosphonates,19 functionalized esters,20 monocarbonyl curcuminoids,21,22 functionalized pyrimidines,23,24 functionalized indoles,25 unsymmetrical acyl thioureas,26 as well as organic salt systems.27,28
In the current study, the primary amine functionalized drugs, that is, pyrimethamine and 4-amino-N-(2,3-dihydrothiazol-2-yl)benzenesulfonamide, have been modified via the condensation reaction with 2-hydroxy-1-naphthaldehyde which is also an important chemical unit known as a versatile fluorophore for naked eye and fluorometric detections of different anions, cations, and neutral molecules. The final products, namely, (E)-1-(((4-(N-(2,3-dihydrothiazol-2-yl)sulfamoyl)-phenyl)iminio)methyl)naphthalen-2-olate (DSPIN) and (E)-1-(((4-amino-5-(4-chlorophenyl)-6-ethylpyrimidin-2-yl)iminio)methyl)naphthalen-2-olate (ACPIN), have been obtained in crystalline imine-zwitterion forms. Due to their crystalline nature, the products have been studied by the SC-XRD approach, and their structural and electronic features have further been investigated using the Hirshfeld surface analysis and DFT calculations.
2. Experimental part
2.1. Chemicals and instrumentation
Solvents and chemicals of the highest possible quality were used without any further purification. Single-crystal analysis was performed at temperature 296 K using Bruckner made diffractometer (Kappa APEX II CCD), equipped with a graphite monochromator. The XRD data collection was performed using the fine focus of Mo Kα X-rays and APEX2 software.29 The data reduction was accomplished using SAINT-27 software. Structural parameters (bond distances, hydrogen bond characteristics, torsion angles, and bond angles) were obtained using PLATON software.30 SHELXL-97 program was used for the structure solution and direct method was employed for the structure refinement.31 ORTEP-3 software32 was used for thermal ellipsoid diagram creation while for cyclic interaction diagram creation Mercury 4.0 (ref. 33) was used.
2.2. The preparation procedure
Both crystalline compounds were prepared by the condensation reaction. Accordingly, 1.2 mmol each of the 2-hydroxy-1-naphthaldehyde and substituted aromatic primary amines were placed in a 50 mL round bottom flask containing 15 mL of methanol as a solvent. The mixture was stirred under reflux conditions for 4 hours. Once the reaction was completed (monitored by thin layer chromatography), the reaction mixture was cooled to room temperature and was left for 24 hours to obtain crystals of the title organic zwitterion compounds DSPIN and ACPIN (Scheme 1).
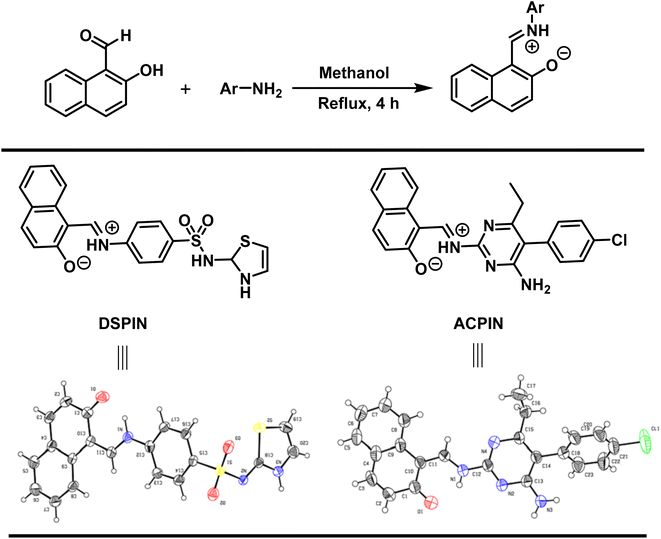 |
| | Scheme 1 Synthetic scheme for the preparation of (E)-1-(((4-(N-(2,3-dihydrothiazol-2-yl)sulfamoyl)phenyl)iminio)methyl)naphthalen-2-olate (DSPIN) and (E)-1-(((4-amino-5-(4-chlorophenyl)-6-ethylpyrimidin-2-yl)iminio)methyl)naphthalen-2-olate (ACPIN). | |
2.3. Computational details
DFT studies were performed with the Gaussian 16 software.34 Using as the starting geometries the structures from the SC-XRD analysis, we optimized the DSPIN and ACPIN molecules without any symmetry constraints and then performed frequency calculations to verify that the optimized structures are true energy minima. All calculations were performed with the hybrid density functional B3LYP35 with the triple-zeta split-valence polarized basis set 6-311+G(d,p)36,37 (with two sets of polarization functions, on hydrogens and on heavier atoms, and one set of diffuse functions). This approach is furthermore referred to as B3LYP/6-311+G(d,p). We did the computational studies and all analyses listed below with the B3LYP/6-311+G(d,p) approach and with the implicit effects from ethanol (dielectric constant ε = 24.852) taken into account, employing the self-reliable IEF-PCM approach38 with the UFF default model as implemented in the Gaussian 16 software, with the electrostatic scaling factor α = 1.0. Below we compare the calculated structural parameters, Natural Population Analysis (NPA) charges, Natural Bond Orbitals (NBO) orbital interactions,39 and frontier molecular orbitals (FMOs) for both compounds. We used the values of the energies of HOMO and LUMO to compute the global reactivity parameters (GRP)40–42 (see eqn (1)–(6)). eqn (1) and (2) were used to calculate the values of the ionization potential (IP) and electron affinity (EA):
For global hardness η and electronegativity X values we used eqn (3) and (4):
| |
 | (3) |
| |
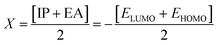 | (4) |
Global electrophilicity ω value was calculated by eqn (5):
| |
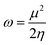 | (5) |
where
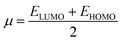
is the chemical potential of the system.
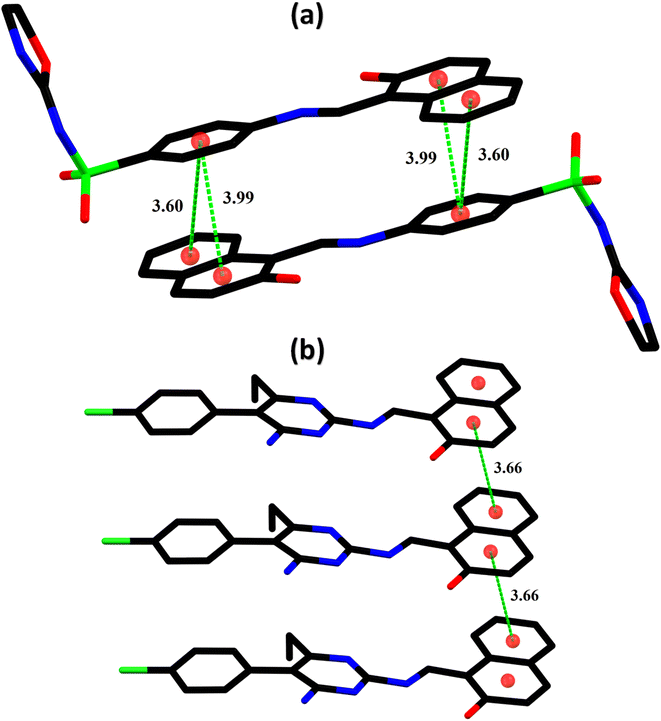
Finally, the global softness σ value was computed with the eqn (6):
| |
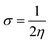 | (6) |
Open GL version of Molden 5.8.2 visualization program was used for the visualization of the structures of the title compounds.43 Avogadro, version 1.1.1, was used to visualize the molecular orbitals and molecular electrostatic potential (MEP) maps.44,45
3. Results and discussions
3.1. Single-crystal XRD study of DSPIN and ACPIN
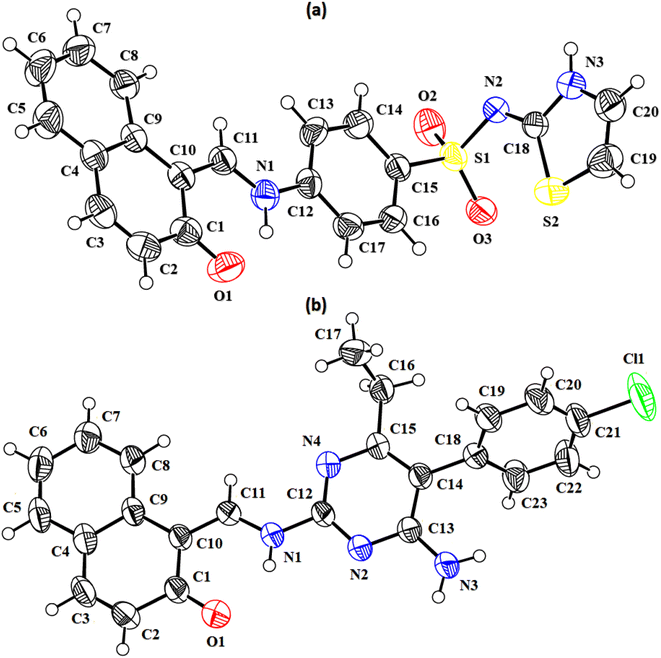
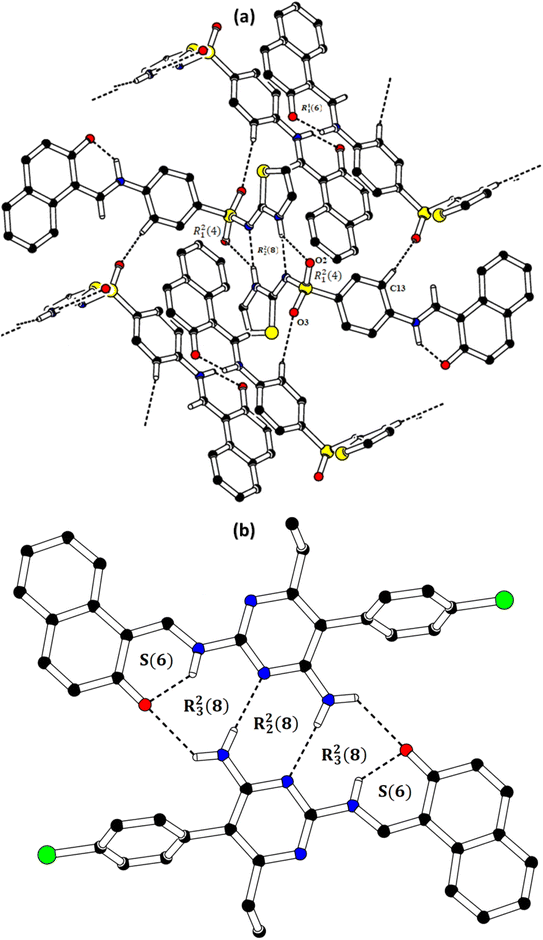
In the zwitterion crystal structure of 1-((Z)-((4-((E)-N-(thiazol-2(3H)-ylidene)-sulfamoyl)phenyl)iminio)methyl)naphthalen-2-olate (Fig. 1a and Table 1), the 1-methylnaphthalen-2-olate group A (C1–C11/O1), 4-aminobenzenethiol group B (C12–C17/N1/S1), and thiazol-2(3H)-imine group C (C18–C20/N2/N3/S2) are roughly planar with root mean square (r.m.s.) deviations of 0.0227, 0.0042, and 0.0078 Å, respectively. The central group B is oriented at the dihedral angle of 3.63(7)° and 77.2(7)° with respect to the groups A and C, respectively. The dihedral angles showed that groups A and B are almost parallel to each other. The geometry around sulphur atom (S1) is a distorted tetrahedron as the value of the structural parameter (τ4) is 0.92. The molecular configuration is stabilized by intramolecular N–H⋯O bonding to form S(6) H-bonded loop. The molecules are interlinked in the form of a dimer through N–H⋯N bonding to form R22(8) loops46 where the N-atom attached to the sulfonyl group acts as H-bond acceptor and NH of the five-membered ring acts as H-bond donor (Fig. 2a and Table 3). The same NH group interacts with one of the O-atoms of the sulfonyl group and completes two bifurcated R12(4) loops. The other O-atom of the sulfonyl group acts as H-bond acceptor for CH of the phenyl ring. As the result of N–H⋯N, N–H⋯O, and C–H⋯O bonding interactions, one-dimensional network of molecules is formed with the base vector [0 0 1]. The crystal packing is further stabilized by off-set π⋯π stacking interactions between aromatic rings of the molecules that are related by inversion symmetry (Fig. 3a). The inter-centroid separation ranges from 3.60 to 3.99 Å and ring off-set ranges from 0.580 to 2.098 Å. Cambridge structural database search provides two hints that have close similarity with the crystal structure of DSPIN with reference codes WUFFUA47 and UTANAG.48 The bond lengths and bond angles of DSPIN (Table 2) are consistent with the corresponding structural parameters in the related structures. The crystal structure with the reference code WUFFUA is crystallized in the monoclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and is a polymorph of DSPIN whereas UTANAG is the hydrated form of DSPIN. Just like WUFFUA and UTANAG, the molecule of DSPIN is non-planar and has intramolecular N–H⋯O bonding. The crystal packing of DSPIN is different from the crystal packing of its polymorph. In WUFFUA, C–H⋯π interactions are present which are absent in DSPIN. Similarly, C–H⋯O bonding is absent in WUFFUA but present in DSPIN. The crystal packing of DSPIN has stronger π⋯π stacking interactions as compared to WUFFUA. The presence of water molecules makes the crystal packing of UTANAG entirely different from the crystal packing of DSPIN and WUFFUA. In UTANAG, both H-atoms of the water molecule act as H-bond donors for the O-atom of the sulfonyl group.
and is a polymorph of DSPIN whereas UTANAG is the hydrated form of DSPIN. Just like WUFFUA and UTANAG, the molecule of DSPIN is non-planar and has intramolecular N–H⋯O bonding. The crystal packing of DSPIN is different from the crystal packing of its polymorph. In WUFFUA, C–H⋯π interactions are present which are absent in DSPIN. Similarly, C–H⋯O bonding is absent in WUFFUA but present in DSPIN. The crystal packing of DSPIN has stronger π⋯π stacking interactions as compared to WUFFUA. The presence of water molecules makes the crystal packing of UTANAG entirely different from the crystal packing of DSPIN and WUFFUA. In UTANAG, both H-atoms of the water molecule act as H-bond donors for the O-atom of the sulfonyl group.
 |
| | Fig. 1 ORTEP diagrams of the compound DSPIN (a) and ACPIN (b) drawn at a probability level of 50%. H-atoms shown by tiny circles of arbitrary radii. | |
Table 1 SC-XRD experimental details of the compounds DSPIN and ACPIN
| Crystal data |
DSPIN |
ACPIN |
| CCDC |
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 263537 263537 |
2263538 |
| Chemical formula |
C20H15N3O3S2 |
C27H21ClN4O |
| Mr |
409.47 |
402.87 |
| Crystal system, space group |
Monoclinic, P21/c |
Triclinic, P |
| Temperature (K) |
296 |
296 |
| a, b, c (Å) |
7.8389 (8), 13.8151 (16), 17.198 (2) |
4.5436 (6), 12.5255 (16), 17.673 (2) |
| α, β, γ° |
94.032 (4) |
88.308 (8), 89.195 (8), 82.611 (9) |
| V (Å3) |
1857.9 (4) |
996.9 (2) |
| Z |
4 |
2 |
| Density (calculated) g cm−3 |
1.464 |
1.324 |
| F(000) |
848 |
420 |
| Radiation type |
Mo Kα |
Mo Kα |
| Wavelength (λ) |
0.71073 Å |
0.71073 Å |
| μ (mm−1) |
0.31 |
0.21 |
| Crystal size (mm) |
0.32 × 0.28 × 0.23 |
0.40 × 0.24 × 0.20 |
|
| Data collection |
| Diffractometer |
Bruker APEXII CCD diffractometer |
Bruker APEXII CCD diffractometer |
| Absorption correction |
Multi-scan (SADABS; Bruker, 2007) |
Multi-scan (SADABS; Bruker, 2007) |
| No. of measured, independent, and observed [I > 2σ(I)] reflections |
15333, 4040, 2547 |
4351, 4351, 2397 |
| Rint |
0.0404 |
0.0454 |
| Theta range for data collection (°) |
2.374 to 27.000 |
|
| Index ranges |
−9 ≤ h ≤ 9, −10 ≤ k ≤ 10, −14 ≤ l ≤ 14 |
−5 ≤ h ≤ 5, −16 ≤ k ≤ 16, −1 ≤ l ≤ 22 |
| (Sin θ/λ)max (Å−1) |
0.639 |
0.649 |
|
| Data refinement |
| R[F2 > 2σ(F2)], wR(F2), S |
0.044, 0.109, 1.02 |
0.076, 0.236, 1.14 |
| No. of reflections |
4040 |
4351 |
| No. of parameters |
259 |
264 |
| No. of restraints |
|
|
| H-atom treatment |
H atoms treated by a mixture of independent and constrained refinement |
H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) |
0.26, −0.27 |
0.32, −0.28 |
 |
| | Fig. 2 Packing diagrams of DSPIN (a) and ACPIN (b). Only selected H-atoms shown for clarity. | |
Table 2 Important bond lengths (Å) and bond angles (°) for DSPIN and ACPIN, X-ray//DFT
| Selected bond lengths (Å) in DSPIN |
Selected bond lengths (Å) in ACPIN |
| S1–O2 |
1.4315 (17)//1.470 |
Cl1–C21 |
1.740 (4)//1.764 |
| S1–O3 |
1.4344 (17)//1.479 |
O1–C1 |
1.270 (5)//1.259 |
| S1–N2 |
1.6056 (18)//1.642 |
N1–C11 |
1.320 (5)//1.339 |
| O1–C1 |
1.269 (3)//1.263 |
N1–C12 |
1.405 (5)//1.402 |
| N1–C11 |
1.325 (3)//1.337 |
N2–C12 |
1.323 (5)//1.331 |
| N3–C20 |
1.370 (3)//1.387 |
N2–C13 |
1.343 (5)//1.342 |
| S1–C15 |
1.762 (2)//1.792 |
N3–C13 |
1.339 (5)//1.356 |
| S2–C19 |
1.719 (3)//1.760 |
N4–C12 |
1.315 (5)//1.324 |
| S2–C18 |
1.724 (2)//1.777 |
N4–C15 |
1.353 (5)//1.352 |
| N1–C12 |
1.402 (3)//1.402 |
— |
— |
| N3–C18 |
1.338 (3)//1.358 |
— |
— |
| N2–C18 |
1.325 (3)//1.312 |
— |
— |
| Selected bond angles (°) in DSPIN |
Selected bond angles (°) in ACPIN |
| O2–S1–O3 |
118.78 (11)//117.89 |
N4–C12–N1 |
118.6 (3)//118.0 |
| O2–S1–N2 |
104.17 (10)//107.24 |
N2–C12–N1 |
112.8 (3)//114.1 |
| O3–S1–N2 |
111.73 (10)//111.72 |
N3–C13–N2 |
115.6 (3)//116.5 |
| N2–C18–S2 |
130.88 (18)//131.67 |
C11–N1–C12 |
126.4 (3)//125.1 |
| N3–C18–S2 |
109.21 (18)//108.18 |
N4–C12–N2 |
128.6 (3)//127.8 |
| O2–S1–C15 |
108.12 (11)//107.52 |
— |
— |
| O3–S1–C15 |
107.28 (10)//107.81 |
— |
— |
| N2–S1–C15 |
106.06 (10)//103.68 |
— |
— |
| N2–C18–N3 |
119.9 (2)//120.15 |
— |
— |
Table 3 Hydrogen-bond geometry (Å, °) in DSPIN and ACPIN, X-ray//DFT
| Symmetry codes: −x + 1, −y + 1, −z + 1. −x + 1, y + 1/2, −z + 1/2. −x + 3, −y + 1, −z. |
| DSPIN |
D–H···A |
D–H |
H⋯A |
D⋯A |
∠(D–H⋯A)° |
| |
N1–H1⋯O1 |
0.98 (2)//1.03 |
1.72 (2)//1.74 |
2.530 (3)//2.594 |
138 (2)//137 |
| |
N3–H3A⋯S1a |
0.89 (2)//1.01 |
2.89 (3) |
3.746 (2) |
163 (2) |
| |
N3–H3A⋯O2a |
0.89 (2)//1.01 |
2.50 (2) |
3.185 (3) |
134 (2) |
| |
N3–H3A⋯N2a |
0.89 (2)//1.01 |
2.07 (2) |
2.922 (3) |
161 (2) |
| |
C11–H11⋯O3b |
0.93//1.08 |
2.64 |
3.521 (3) |
157 |
| |
C13–H13⋯O3b |
0.93//1.08 |
2.58 |
3.463 (3) |
159 |
| ACPIN |
D–H···A |
D–H |
H⋯A |
D⋯A |
∠(D–H⋯A)° |
| |
N1–H1⋯O1 |
0.86//1.03 |
1.86//1.81 |
2.557 (4)//2.613 |
137//132 |
| |
N3–H3A⋯N2c |
0.86//1.01 |
2.19 |
3.037 (5) |
171 |
| |
N3–H3B⋯O1c |
0.86//1.01 |
2.38 |
3.003 (5) |
129 |
 |
| | Fig. 3 Graphical representation of the off-set π⋯π stacking interactions in DSPIN (a) and ACPIN (b). H-atoms not shown for clarity. Distances measured in Å. | |
In the zwitterion crystal structure named as (E)-1-(((4-amino-5-(4-chlorophenyl)-6-ethylpyrimidin-2-yl)iminio)methyl)naphthalen-2-olate, ACPIN (Fig. 1b and Table 1), the 1-methylnaphthalen-2-olate group A (C1–C11/O1), pyrimidine-2,4-diamine ring B (C12–C15/N1–N4), and chlorophenyl ring C (C18–C23/Cl1) are planar with r.m.s. deviations of 0.0264, 0.0303, and 0.0071 Å, respectively. The central ring is oriented at the dihedral angles of 8.85(2)° and 62.4(1)° with respect to the groups A and C, respectively. The atoms of the ethyl group (C16/C17) are at the distances of −0.0253(4) and 1.2138(8) Å, respectively, from the plane of the ring B. The molecular configuration is stabilized by intramolecular N–H⋯O bonding to form S(6) loop. The molecules of the compound ACPIN are also interlinked in the form of dimers through N–H⋯N bonding to complete R22(8) loop, where donor NH is from the amino group and acceptor N-atom is from the pyrimidine ring (Fig. 2b and Table 3). The other H-atom of the amino group acts as H-bond donor for the O-atom and completes two bifurcated R32(8) loops. No C–H⋯O bonding is found in the crystal packing. The ethyl group and one N-atom of the pyrimidine do not participate in the H-bonding. The crystal packing is further stabilized by off-set π⋯π stacking interactions between aromatic rings of the molecules that are related by inversion symmetry (Fig. 3b). The inter-centroid separation is 3.66 Å and ring off-set distance is 1.212 Å. The Cambridge structural database search provides one hint only with reference code SIYBUA. The crystal structure with the reference code SIYBUA was submitted to CCDC as CSD Communication (private communication) in 2019.49 It was crystallized in the monoclinic system with the space group P21/n.
3.2. Hirshfeld surface analysis
The noncovalent interactions represent the one of the most interesting topics for the researchers working in the field of supramolecular chemistry and drug design as the properties of the single crystals are defined by these interactions. Hirshfeld surface analysis was used for the inspection of the noncovalent interactions employing Crystal Explorer version 21.5.50 Hirshfeld surface is acquired by diving the space in the crystal into regions where promolecular electron density dominates over the procrystal electron density. Hirshfeld surface plotted over dnorm consists of three colours, red, white, and blue, to separate long-distance contacts from the other types of contacts.51,52 Red, white, and blue colours indicate the contacts with distance less than, equal to, and greater than sum of van der Waal radii. For DSPIN, the red spots on the surface near NH of the ring, O-atoms of the sulfonyl group, and N-atom directly bonded with the sulfonyl group indicate that these atoms form short contact with the neighbouring molecules (Fig. 4a). The NH group that links naphthalen-2-olate and phenyl ring is not involved in any intermolecular H-bonding. For ACPIN, red spots on the surface near O-atom of the naphthalen-2-olate, N-atoms of the pyrimidine ring, and N-atoms of the amino radical indicate that these atoms are engaged in H-bonding (Fig. 4b).
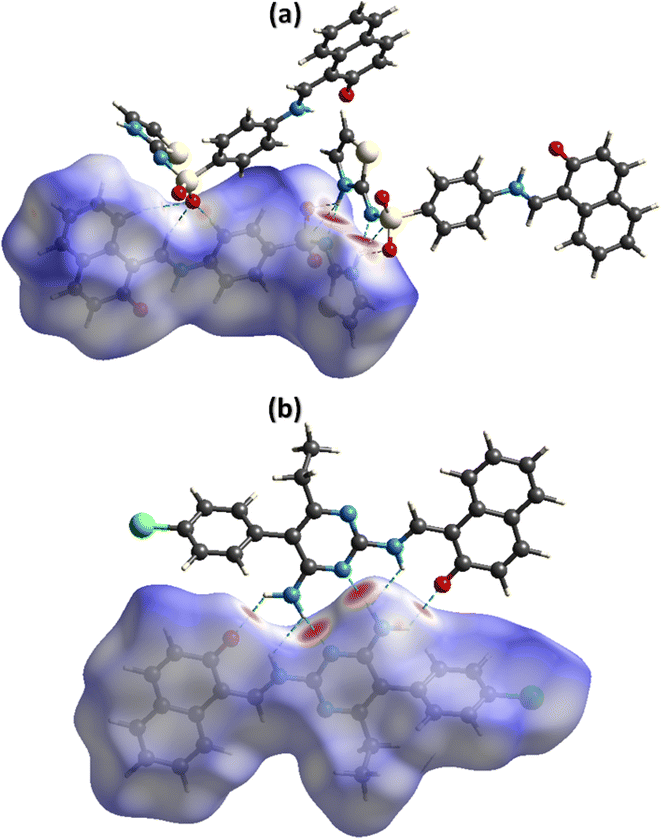 |
| | Fig. 4 Hirshfeld surfaces plotted over dnorm for DSPIN in the range −0.5270 to 1.5020 a. u. (a) and ACPIN in the range −0.4528 to 1.8067 a. u. (b). | |
The role of the interatomic contacts in stabilizing the supramolecular assembly can be explored by 2D fingerprint plots because they provide the quantitative description which is usually remained hidden in the classic way of describing supramolecular assembly.53,54 The plot for overall interactions of DSPIN (Fig. 5a) is different from the corresponding 2D plot in ACPIN (Fig. 6a) showing that crystal packing of DSPIN is different from the crystal packing of ACPIN. Each individual contact is determined by including the reciprocal contacts. For DSPIN, the important interatomic contacts are H⋯H, H⋯C, H⋯O, C⋯C, and H⋯N with percentage contributions of 31.9%, 22.1%, 20%, 7.1%, and 6.6%, respectively. For ACPIN, the major contributors in crystal packing are H⋯H, H⋯C, H⋯Cl, H⋯N, and H⋯O with respective percentage contributions of 45.1%, 19.5%, 11.4%, 6.7%, and 5.2%, respectively. All the contacts have different probabilities to form crystal packing interactions. The probability of a contact to form crystal packing interactions is computed by finding enrichment ratio.55,56 Enrichment ratio is greater than 1 for the pair of chemical species that are favourable to form contacts and less than 0 for not favourable, respectively. For both compounds, C⋯C is the most favourable contact with the enrichment ratio 1.69 for DSPIN and 2.02 for ACPIN. H⋯H contact is not favourable in DSPIN (Table S1†) but is slightly favourable in ACPIN with the enrichment ratio slightly larger than one (Table S2†). H⋯N and H⋯S contacts are favourable for DSPIN whereas O⋯C, H⋯Cl, and H⋯N contacts are favourable for ACPIN.
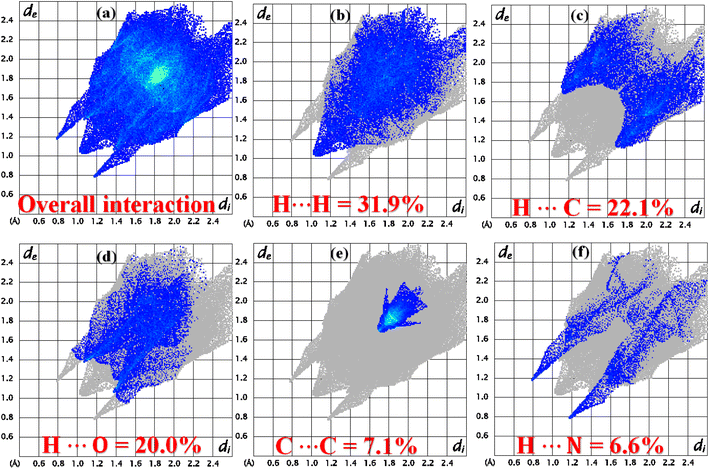 |
| | Fig. 5 2D fingerprint plots for (a) overall interactions, (b–f) important individual interatomic contacts of DSPIN. | |
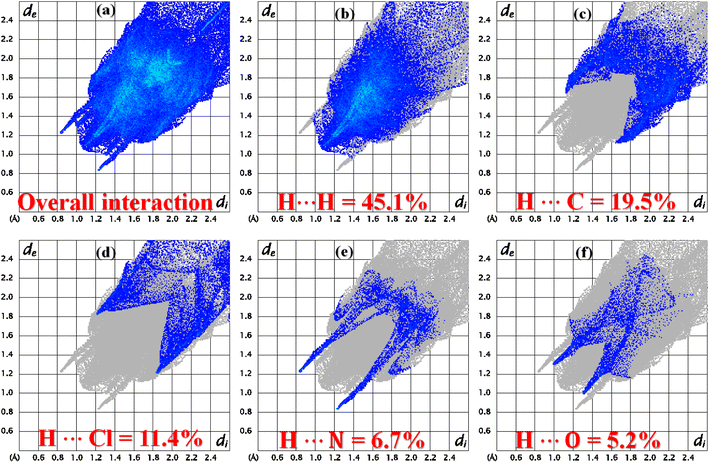 |
| | Fig. 6 2D fingerprint plots for (a) overall interactions, (b–f) important individual interatomic contacts of ACPIN. | |
The reaction of the crystal to applied stress can be investigated by calculating voids in the crystal. Crystals containing large cavities have poor response to applied stress, i.e., such crystals can be broken by applying insignificant stress. For checking mechanical response of DSPIN and ACPIN, voids were calculated by adding the electron density of the atoms that are assumed to have spherical symmetry.57,58 Fig. 7 shows the graphical view of voids in DSPIN and ACPIN. The calculated volumes of voids in DSPIN and ACPIN are 131.95 and 254.65 Å3, respectively. The space used by voids in supramolecular assembly of DSPIN and ACPIN is 13.7% and 13.3%, respectively. The packing index for DSPIN and ACPIN is 86.3% and 86.7%, respectively. In both compounds, the voids occupy a small amount of space, so the crystals have no large cavities and are expected to show good mechanical response.
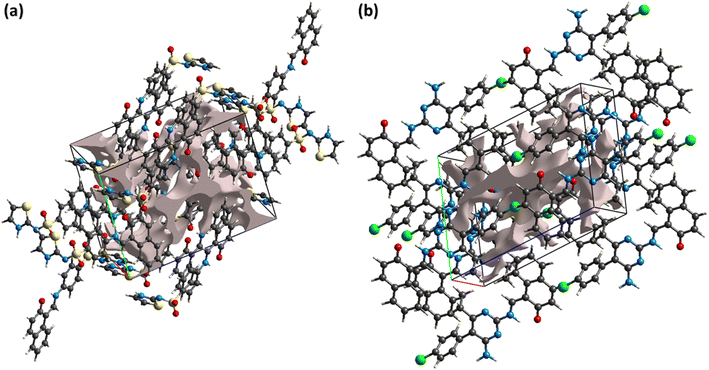 |
| | Fig. 7 Graphical representation of voids in (a) DSPIN, (b) ACPIN. | |
To carry out further investigation of the supramolecular assembly or crystal packing of DSPIN and ACPIN, interaction energies among the molecular pairs were calculated using the B3LYP/6-31G(d,p) electron density model as employed in TONTO built-in in Crystal Explorer version 21.5. A cluster of 3.8 Å around the molecule present in the asymmetric unit was included in interaction energy calculations. Total interaction energy is the sum of Coulomb electrostatic, dispersion, polarization, and repulsion energies.59,60 For organic crystals, Coulomb electrostatic and dispersion energies have dominant role in crystal packing as compared to other kinds of energies. The results of the interaction energy calculations are given in Tables S3 and S4† for DSPIN and ACPIN, respectively. The Coulomb energy is the largest for the molecular pair with intermolecular distances of 11.78 Å and 7.26 Å in DSPIN and ACPIN, respectively. The dispersion energy is the greatest for the molecular pair with intermolecular distances of 6.32 Å and 4.54 Å in DSPIN and ACPIN, respectively. In DSPIN, the Coulomb energy is repulsive for one pair with the intermolecular distance of 13.10 Å whereas in ACPIN the Coulomb energy is repulsive for two pairs with intermolecular distances of 4.54 and 17.67 Å. The pair with the largest total attractive energy contribution in the supramolecular assembly of DSPIN and ACPIN has the intermolecular distances of 11.78 and 7.26 Å, respectively. Fig. 8 shows the energy frameworks for Coulomb electrostatic, dispersion, and total energies for DSPIN and ACPIN. The molecular centers are joined by a cylinder whose width is directly proportional to the strength of the corresponding energy. Although the width of the cylinder for three molecular pairs is greater for the Coulomb energy framework of DSPIN but overall, the contribution of the dispersion energy in stabilization of the crystal packing is larger than the contribution of the Coulomb electrostatic energy. For both compounds, the dispersion energy is the dominant contribution in the crystal packing as compared to other kinds of energies.
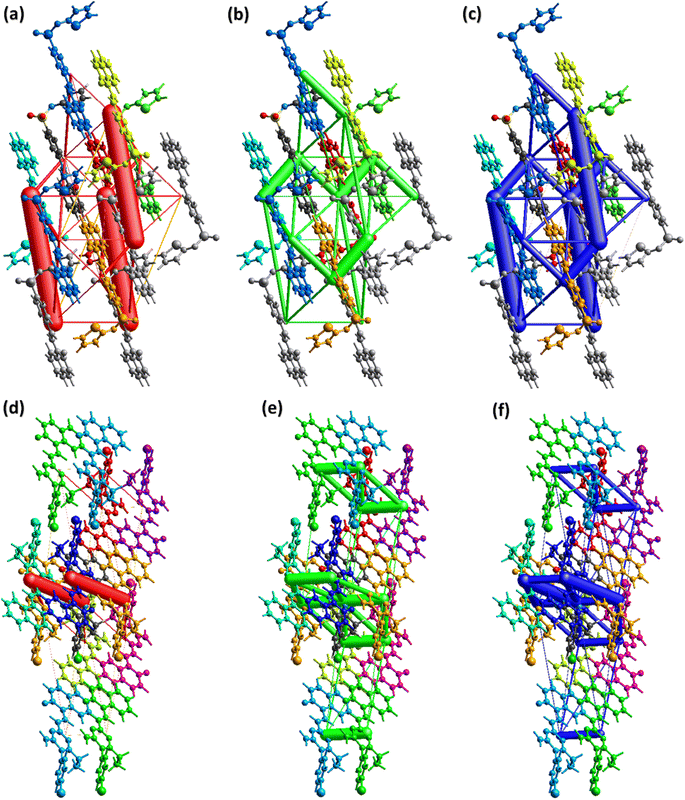 |
| | Fig. 8 Energy frameworks for coulomb, dispersion, and total energy in (a–c) DSPIN, (d–f) ACPIN. | |
3.3. DFT study results
3.3.1. Structural features. In Fig. 9 the structures of DSPIN and ACPIN optimized using the B3LYP/6-311+G(d,p) approach with the implicit solvent effects from C2H5OH are shown, and in Table 2 important bond lengths and bond angles for both compounds, both experimental and calculated, are provided. Comparison of the structural parameters in Table 2 shows reasonably good agreement between the X-ray diffraction and DFT results. Furthermore, in Table S5† more selected structural parameters are given, and in Table 3 several calculated structural parameters related with hydrogen bonding are provided along with the experimentally determined structural parameters. Consideration of the results for DSPIN in Table S5† shows relatively short H11–H13 interatomic distance, 2.07 Å, and even shorter interatomic distance H11–N1, 2.055 Å, along with noticeably longer interatomic distance H13–N1, 2.744 Å; another interatomic distance, N2–H3, is also relatively long, 2.534 Å, however, all three interatomic distances N–H suggest possible formation of intramolecular hydrogen bonding (see below discussion for NBO charges). Furthermore, comparison of the experimental and calculated geometrical parameters for the N1–H1⋯O1 hydrogen bond (Table 3) shows quite good agreement between the experimental and DFT results. Next, consideration of the results for ACPIN in Table S5† shows three interatomic distances N–H being within 2.394–2.468 Å, thus also implying possible formation of intramolecular hydrogen bonding (see below discussion for NBO charges). Moreover, comparison of the experimental and calculated structural parameters for the N1–H1⋯O1 hydrogen bond (see Table 3) again shows quite good agreement between the experimental and DFT results.
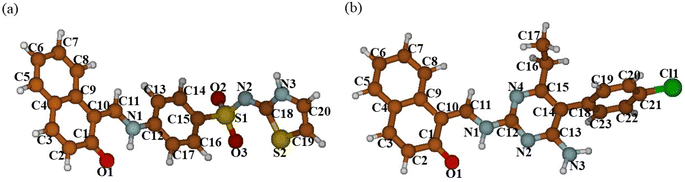 |
| | Fig. 9 Structures of DSPIN (a) and ACPIN (b) optimized with the B3LYP/6-311+G(d,p) approach employing the implicit solvent effects from ethanol. Colour coding: brown for C, light grey for H, blue for N, red for O, dark yellow for S, and green for Cl. Numbering scheme corresponds to Fig. 1. | |
Consideration of the selected dihedral angles calculated for both compounds (Table S5†) shows the molecule of DSPIN being almost flat, the only dihedral angle noticeably different from 0 or 180 degrees is the ∠(H13–C13–C11–H1), ca. 13 degrees. The situation is different for the molecule of ACPIN, where the chlorophenyl group is rotated vs. the rest of the molecule, as supported by the calculated value of the ∠(C15–C14–C18–C19), ca. 76 degrees. It should be noticed that the differences observed between the experimentally determined and DFT optimized structures could be explained by the absence of the crystal structure restrictions in the calculations.
3.3.2. Frontier molecular orbitals (FMOs) analysis. In Fig. 10 the HOMOs and LUMOs of DSPIN and ACPIN computed using the B3LYP/6-311+G(d,p) approach with the implicit solvent effects from ethanol are shown, and Table 4 provides the energies of the selected MOs (HOMO−2–LUMO+2) of DSPIN and ACPIN (eV), the corresponding HOMO/LUMO gaps (eV), calculated at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol, along with the TDDFT gaps (eV). Analysis of the MOs plots and the results from Table 4 shows the following. (i) The DSPIN HOMO is contributed by essentially the whole molecule, with the dominating contributions from the 1-methylnaphthalen-2-olate and 4-aminobenzenethiol groups and much smaller contributions from the thiazol-2(3H)-imine group. The DSPIN LUMO is contributed mostly by contributions from the 1-methyl-naphthalen-2-olate and 4-aminobenzenethiol groups. Both HOMO and LUMO of ACPIN are contributed by the 1-methylnaphthalen-2-olate group and pyrimidine-2,4-diamine ring. Thus, in the electron transfer processes between HOMO and LUMO, in the case of DSPIN essentially the whole molecule is involved, whereas in the case of ACPIN mostly 1-methylnaphthalen-2-olate and 4-aminobenzenethiol groups of the molecule are involved. (ii) The DSPIN HOMO/LUMO gap is by 0.15 eV smaller than the ACPIN HOMO/LUMO gap due to some destabilization of the DSPIN HOMO and some stabilization of its LUMO. (iii) The HOMO−1/LUMO+1 and HOMO−2/LUMO+2 gaps for the compound DSPIN are also smaller compared to the compound ACPIN, by 0.38 and 0.07 eV, respectively. This implies that in the case of DSPIN the HOMO−1 and HOMO-2 are more likely to participate in redox reactions compared to ACPIN. The LUMO+1/LUMO+2 of both compounds are noticeably higher than their LUMOs, by 1.24/1.47 and 1.32/1.34 eV, respectively, and are much less likely to participate in redox reactions. (iv) The TDDFT gaps are, as expected, smaller than the HOMO/LUMO gaps, the TD-ωB97XD/6-311+G(d,p) gaps being larger than the TD-B3LYP/6-311+G(d,p) gaps, by 0.34 and 0.32 eV for DSPIN and ACPIN, respectively.
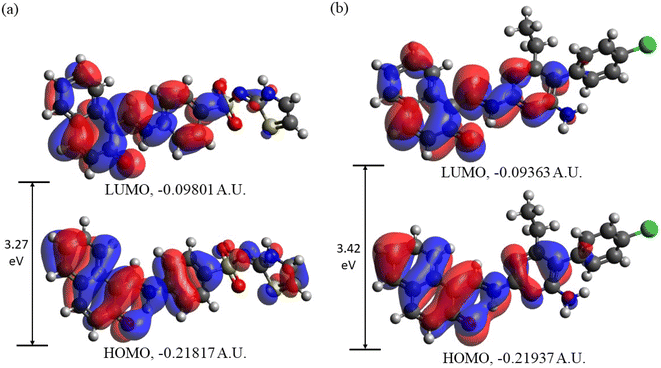 |
| | Fig. 10 Frontier molecular orbitals of DSPIN (a) and ACPIN (b) computed with the B3LYP/6-311+G(d,p) approach employing the implicit solvent effects from ethanol. | |
Table 4 Energies of the selected MOs (HOMO−2–LUMO+2) of DSPIN and ACPIN (eV), the corresponding HOMO/LUMO gaps (eV), calculated at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol, along with the TDDFT gaps (eV)
| MOs |
Energy, eV |
ΔE (HOMO/LUMO), eV |
ΔE (TDDFT-1)a, eV |
ΔE (TDDFT-2)b, eV |
| TD-B3LYP/6-311+G(d,p) approach. TD-ωB97XD/6-311+G(d,p) approach. |
| DSPIN |
| LUMO |
−2.67 |
3.27 |
2.87 |
3.21 |
| HOMO |
−5.94 |
|
|
| LUMO+1 |
−1.43 |
5.01 |
|
|
| HOMO−1 |
−6.44 |
|
|
| LUMO+2 |
−1.20 |
5.50 |
|
|
| HOMO−2 |
−6.70 |
|
|
|
| ACPIN |
| LUMO |
−2.55 |
3.42 |
3.02 |
3.34 |
| HOMO |
−5.97 |
|
|
| LUMO+1 |
−1.23 |
5.39 |
|
|
| HOMO−1 |
−6.62 |
|
|
| LUMO+2 |
−1.21 |
5.57 |
|
|
| HOMO−2 |
−6.78 |
|
|
3.3.3. Natural population analysis (NPA). In Table 5 the NPA charges on the selected atoms of both compounds are provided. The values of these charges imply formation of intramolecular hydrogen bonds, N1–H1⋯O1 and N3–H3⋯N2 in DSPIN and N1–H1⋯O1, N1–H1⋯N2, N3–H3⋯N2, and C11–H11⋯N4 in ACPIN, thus supporting the results of the X-ray diffraction study and Hirshfeld analysis (see above). Furthermore, these charges suggest formation of intermolecular hydrogen bonding and dipole–dipole and dispersion interactions.
Table 5 NPA charges, e, on the selected atoms of the compounds DSPIN and ACPIN, computed at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol
| Atom |
Charge, e |
| DSPIN |
| O1 |
−0.714 |
| N1 |
−0.526 |
| H1(N1) |
0.450 |
| S1 |
2.206 |
| O2 |
−0.954 |
| O3 |
−0.961 |
| S2 |
0.409 |
| N2 |
−0.858 |
| N3 |
−0.548 |
| H3(N3) |
0.441 |
|
| ACPIN |
| Cl1 |
−0.012 |
| O1 |
−0.711 |
| N1 |
−0.547 |
| H1(N1) |
0.456 |
| N2 |
−0.572 |
| N4 |
−0.586 |
| N3 |
−0.761 |
| H3(N3) |
0.411 |
| H11(C11) |
0.226 |
3.3.4. Natural bonding orbital (NBO) analysis. The NBO analysis results provide us with knowledge of orbital interactions of different types, both intra- and intermolecular.39 The NBO analysis is performed by consideration of all possible interactions among the filled, or donor, Lewis-type NBOs and empty, or acceptor, non-Lewis NBOs. Their energetic contributions in interactions are evaluated using the 2nd-order perturbation theory. These interactions result in the decrease of the localized NBOs occupancy in the idealized Lewis structure and corresponding increase of the occupancy of the empty non-Lewis orbitals. As a consequence, they are referred to as ‘delocalization’ corrections to the 0th-order natural Lewis's structure. The stronger donor–acceptor interactions are characterized by higher stabilization energies. The 2nd-order stabilization energy E(2) is computed according to eqn (7):| |
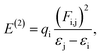 | (7) |
where εi and εj are off-diagonal and Fi. j is the diagonal elements of the NBO Fock matrix, qi is the donor orbital possession, and E(2) is the energy of stabilization.39Second-order perturbation theory analysis results for both compounds, calculated using the B3LYP/6-311+G(d,p) approach with the implicit solvent effects from ethanol, are given in Table S6.† Analysis of these results shows the following. (i) There is a very large number of various donor–acceptor interactions in both compounds with stabilization energies in very broad range, from ca. 10 kcal mol−1 up to ca. 4100 kcal mol−1. (ii) In both compounds, numerous “remote” donor–acceptor interactions between different moieties of the molecule with significant stabilization energies can be noticed, e.g.: in DSPIN, σ(S2–C19)→π*(C13–C14), σ(N1–C11)→σ*(C16–H16), σ(N1–C12)→σ*(C16–H16), σ(N3–C18)→σ*(C16–H16), σ(N3–C20)→π*(C13–C14), σ(N3–C20)→σ*(C16–H16), σ(C1–C2)→π*(C13–C14), π(C7–C8)→σ*(C16–C17), σ(C19-20)→π*(C13–C14), σ(C20–H20)→π*(O1–C1), σ(C20–H20)→σ*(C8–H8), σ(C20–H20)→σ*(C16–C17), with the stabilization energies 488.92, 721.29, 3398.88, 1845.82, 969.38, 2881.41, 652.58, 311.99, 1909.55, 514.97, 483.59, and 1183.19 kcal mol−1, respectively; in ACPIN, σ(N1–C11)→σ*(C16–H16A), σ(N1–C11)→π*(C20–C21), σ(N1–C11)→σ*(C22–C23), σ(N1–C12)→σ*(C8–H8), σ(N1–C12)→σ*(C16–H16A), σ(N1–C12)→σ*(C16–H16B), σ(N1–C12)→σ*(C20–C21), σ(N1–C12)→σ*(C22–C23), σ(C18–C23)→σ*(C8–H8), σ(C18–C23)→σ*(C16–H16B), σ(C23–H23)→σ*(C4–C5), σ(C23–H23)→σ*(C10–C11), with the stabilization energies 213.17, 4137.13, 318.92, 404.57, 486.04, 3645.75, 1853.10, 854.05, 581.82, 604.48, 1349.18, and 654.23 kcal mol−1, respectively. (iii) Also, in both compounds the donor–acceptor interactions within the same moiety of the molecule with noticeable stabilization energies can be seen, e.g.: in DSPIN, σ(N3–C20)→σ*(N3–C18), σ(N3–C20)→σ*(C19–C20), π(C7–C8)→LP*(C10), π(C7–C8)→π*(O1–C1), π(C7–C8)→σ*(C8–H8), σ(C12–C17)→π*(C13–C14), with the stabilization energies 151.39, 207.91, 130.61, 131.51, 170.45, and 333.15 kcal mol−1, respectively; in ACPIN, π(C2–C3)→LP*(C1), π(C10–C11)→LP*(C1), σ(C18–C23)→σ*(C22–C23), σ(C22–H22)→π*(C18–C19), σ(C22–C23)→σ*(C22–C23), LP(O1)→LP*(C1), with the stabilization energies 169.24, 233.38, 389.83, 225.24, 832.74, and 1242.28 kcal mol−1, respectively.
Thus, as can be seen, molecules of both compounds have numerous stabilizing donor–acceptor interactions with noticeable stabilization energies, which would imply high stability of the compounds.
3.3.5. Global reactivity parameters (GRPs) analysis. The global reactivity parameters, ionization potential (IP), electron affinity (EA), global softness (σ), global electrophilicity (ω), global hardness (η), global electronegativity (X), and chemical potential (μ), were computed using the FMOs energies (Table 2) according to eqn (1)–(6) (see Computational details),40–42 and the computed values in eV are presented in Table 6.
Table 6 The calculated GRPs for the compounds DSPIN and ACPIN (eV), computed at the B3LYP/6-311+G(d,p) level with the implicit effects from ethanol
| IP |
EA |
Gap |
X |
η |
μ |
σ |
ω |
| DSPIN |
| 5.94 |
2.67 |
3.27 |
4.305 |
1.635 |
−4.305 |
0.306 |
5.668 |
|
| ACPIN |
| 5.97 |
2.55 |
3.42 |
4.260 |
1.710 |
−4.260 |
0.292 |
5.306 |
Analysis of the calculated GRPs shows the following. (i) Both compounds have significant values of IPs, 5.94 and 5.97 eV, and noticeable values of EAs, 2.67 and 2.55 eV. This implies that both compounds should be quite stable in oxidation processes, that is, would be poor electron donors, but quite good electron acceptors. (ii) The global hardness η values for both compounds, 1.635 and 1.710 eV, should be considered as quite noticeable, whereas their global softness σ values are quite small, 0.306 and 0.292 eV. This implies that both compounds should have quite low reactivity and be quite stable thermodynamically, DSPIN being somewhat more reactive than ACPIN. This is also supported by quite noticeable HOMO/LUMO gap values for both compounds, DSPIN having small HOMO/LUMO gap than ACPIN. The noticeable stability of both compounds is further supported by significant values of their chemical potentials, −4.305 and −4.260 eV. (iii) The global electronegativity X values for both compounds are high, 4.305 and 4.260 eV, as well as their global electrophilicity ω values, 5.668 and 5.306 eV. This implies that both compounds should be good electron acceptors and poor electron donors in redox reactions, which is in line with their IP and EA values.
Thus, from the GRP analysis, it follows that both compounds should be relatively nonreactive and thermodynamically stable. Moreover, they should act as should be good electron acceptors and poor electron donors in redox reactions.
3.3.6. Molecular electrostatic potential (MEP) mapping. Analysis of the MEP plots presented in Fig. 11 shows the following. (i) In both compounds, the accumulation of negative electrostatic potential (as indicated by red color) can be seen in the naphthalenone moiety. (ii) Also, in both compounds noticeable accumulation of positive electrostatic potential (as indicated by blue color) can be seen around the atoms C11–N1, and smaller accumulation of positive electrostatic potential can be seen at the 4-aminobenzenethiol moiety in DSPIN and at the pyrimidine-2,4-diamine moiety and phenyl ring in ACPIN. (iii) In DSPIN noticeable accumulation of negative electrostatic potential can be observed at the thiazol-2(3H)-imine, whereas in ACPIN only weak accumulation of negative electrostatic potential can be seen on the chlorine atom.
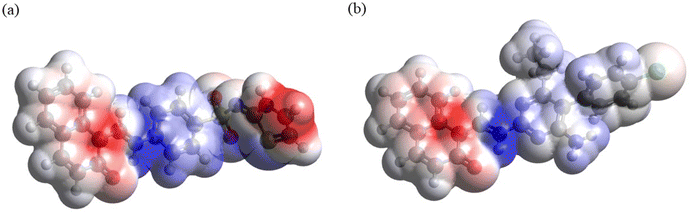 |
| | Fig. 11 MEP plots of DSPIN (a) and ACPIN (b) computed with the B3LYP/6-311+G(d,p) approach employing the implicit solvent effects from ethanol. | |
Thus, both compounds have pronounced areas of accumulation of both positive and negative electrostatic potential, which implies noticeable intermolecular interactions in crystals of these compounds, in line with the Hirshfeld analysis results.
4. Conclusions
Two new organic zwitterionic compounds DSPIN and ACPIN have been prepared via the condensation reaction. The structures of both compounds having crystalline nature were investigated and confirmed via SC-XRD studies which showed that the molecules were mainly connected in the form of dimers through N–H⋯N bonding in both compounds. Further stabilization of the supramolecular assembly was due to noncovalent interactions (N-H⋯S, N–H⋯O, C–H⋯O) in DSPIN and N–H⋯O interaction in ACPIN. Hirshfeld surface analysis was carried out to further investigate the noncovalent interactions showing that H⋯H contacts were the most significant contributor in the crystal packing of both compounds with percentage contributions of 31.9% in DSPIN and 45.1% in ACPIN. Void analysis inferred that both compounds would have good mechanical response. Interaction energy calculations showed that the dispersion energy was the prominent contributor for the stabilization of the supramolecular assembly in both compounds. DFT calculated geometries showed reasonably good agreement with the experimentally determined structural parameters. MOs analysis showed that in the electron transfer processes between HOMO and LUMO, in the case of DSPIN essentially the whole molecule would be involved, whereas in the case of ACPIN mostly 1-methylnaphthalen-2-olate and 4-aminobenzenethiol groups of the molecule would be involved. The DSPIN HOMO/LUMO gap was found to be by 0.15 eV smaller than the ACPIN HOMO/LUMO gap due to some destabilization of the DSPIN HOMO and some stabilization of its LUMO. In the case of DSPIN the HOMO−1 and HOMO−2 are more likely to participate in redox reactions compared to ACPIN. The LUMO+1 and LUMO+2 of both compounds are much less likely to participate in redox reactions. The results of the NPA analysis imply formation of intramolecular hydrogen bonds, thus supporting the results of the X-ray diffraction study and Hirshfeld analysis and suggest formation of intermolecular hydrogen bonding and dipole–dipole and dispersion interactions. The NBO analysis results showed that molecules of both compounds have numerous stabilizing donor–acceptor interactions with noticeable stabilization energies, which would imply high stability of the compounds. From the GRP analysis, it followed that both compounds should be relatively nonreactive and thermodynamically stable. Moreover, they should act as good electron acceptors and poor electron donors in redox reactions. The MEP analysis showed that both compounds have pronounced areas of accumulation of both positive and negative electrostatic potential, which implies noticeable intermolecular interactions in crystals of these compounds, in line with the Hirshfeld analysis results.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors express their gratitude to the Deanship of Scientific Research at King Khalid University for funding this work through the Large Research Group Project under grant number RGP.2-504-44. Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02).
References
- Q. Viviane and P. G. Jean, The Era of Biomedicine: Science, Medicine, and Public Health in Britain and France after the Second World War, Med. Hist., 2008, 52(4), 441–452 CrossRef PubMed.
-
(a) E. Pradhan, S. Bhandari, R. E. Gilbert and M. Stanford, Antibiotics versus no treatment for toxoplasma retinochoroiditis, Cochrane Database Syst. Rev., 2016, CD002218, DOI:10.1002/14651858.CD002218.pub2;
(b) V. Sethuraman and P. T. Muthiah, Hydrogen-bonded supramolecular ribbons in the antifolate drug pyrimeth-amine, Acta Cryst. E, 2002, 58, o817–o818, DOI:10.1107/S1600536802011133.
- R. Cally, P. Richard, N. Shalini, S. Brian, N. François and A. Tim, Intercontinental spread of pyrimethamine-resistant malaria, Science, 2004, 305, 1124, DOI:10.1126/science.1098876.
- L. G. Michelle, B. M. Laura and C. Qin, Evolution of Resistance to Sulfadoxine-Pyrimethamine in Plasmodium falciparum, Antimicrob. Agents Chemother., 2004, 48, 2116–2123, DOI:10.1128/AAC.48.6.2116-2123.2004.
-
(a) M. Ashfaq, G. Bogdanov, V. Glebov, A. Ali, M. N. Tahir and S. Abdullah, Single-crystal investigation, Hirshfeld surface analysis and DFT exploration of the pyrimethamine-based novel organic salt: 2,4-di- amino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 3-carboxybenzoate hydrate (1:1:1), J. Mol. Struct., 2021, 1224, 129309 CrossRef CAS;
(b) M. Ashfaq, A. Ali, A. Kuznetsov, M. N. Tahir and M. Khalid, DFT and single-crystal investigation of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium-4-methylbenzoate hydrate (1:1:1) (DEMH), J. Mol. Struct., 2021, 1228, 129445 CrossRef CAS;
(c) M. Ashfaq, M. N. Tahir, A. Kuznetsov, S. H. Mirza, M. Khalid and A. Ali, DFT and single-crystal X-ray diffraction analysis of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium:4-hydroxybenzoate:methanol:hydrate (1:1:1:1) (DEHMH), J. Mol. Struct., 2020, 1199, 127041 CrossRef CAS;
(d) M. N. Tahir, S. H. Mirza, M. Khalid, A. Ali, M. U. Khan and A. A. C. Braga, Synthesis, single-crystal X-ray diffraction analysis and DFT based computational studies of 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrim idin-1-ium-3,4,5-trihydroxybenzoate-methanol (DETM), J. Mol. Struct., 2019, 1180, 119–126 CrossRef CAS.
- C. T. Supuran and A. Scozzafava, Carbonic anhydrase inhibitors, Curr. Med. Chem., 2001, 1, 61–97 CAS.
- J. Drews, Drug discovery: a historical perspective, Science, 2000, 287, 1960–1964 CrossRef CAS PubMed.
- A. E. Boyd, Sulfonylurea receptors, ion channels, and fruit flies, Diabetes, 1988, 37, 847–850 CrossRef CAS PubMed.
- T. H. Maren, Relations between structure and biological activity of sulfonamides, Annu. Rev. Pharmacol. Toxicol., 1976, 16, 309–327 CrossRef CAS PubMed.
- C. W. Thornber, Isosterism and molecular modification in drug design, Chem. Soc. Rev., 1979, 8, 563–580 RSC.
- A. Casini, A. Scozzafava, A. Mastrolorenzo and C. T. Supuran, Sulfonamides and sulfonylated derivatives as anticancer agents, Curr. Cancer Drug Targets, 2002, 2, 55–75 CrossRef CAS PubMed.
-
(a) G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Naming Interactions from the Electrophilic Site, Cryst. Growth Des., 2014, 14, 2697 CrossRef CAS;
(b) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Non-covalent interactions in the synthesis of coordination compounds: Recent advances, Coord. Chem. Rev., 2017, 345, 54 CrossRef CAS;
(c) H.-J. Schneider, Binding mechanisms in supramolecular complexes, Angew Chem. Int. Ed. Engl., 2009, 48, 3924–3977, DOI:10.1002/anie.200802947;
(d) S. G. Patel, R. M. Vala, P. J. Patel, D. B. Upadhyay, V. Ramkumar, R. L. Gardas and H. M. Patel, Synthesis, crystal structure and in silico studies of novel 2,4-dimethoxy-tetrahydropyrimido[4,5-b]quinolin-6(7H)-ones, RSC Adv., 2022, 12, 18806–18820 RSC;
(e) M. G. Sharma, R. M. Vala, D. P. Rajani, V. Ramkumar, R. L. Gardas, S. Banerjee and H. M. Patel, Crystal structure, antibacterial and antifungal evaluation of 5-bromothiophene based 3,4-dihydropyrimidin-2-(1H)-(thi)ones, Phosphorus, Sulfur, Silicon Relat. Elem., 2023, 198, 145–153, DOI:10.1080/10426507.2022.2121397;
(f) D. B. Upadhyay, R. M. Vala, S. G. Patel, P. J. Patel, C. Chi and H. M. Patel, Water mediated TBAB catalyzed synthesis of spiro-indoline-pyrano[3,2-c]quinolines as α-amylase inhibitor and in silico studies, J. Mol. Struct., 2023, 1273, 134305, DOI:10.1016/j.molstruc.2022.134305.
-
(a) M. Khalid, A. Ali, S. Asim, M. N. Tahir, M. U. Khan, L. C. C. Vieira, A. F. de la Torre and M. Usman, Persistent prevalence of supramolecular architectures of novel ultrasonically synthesized hydrazones due to hydrogen bonding [X–H⋯O; X=N]: Experimental and density functional theory analyses, J. Phys. Chem. Solids, 2021, 148, 109679 CrossRef CAS;
(b) M. Khalid, A. Ali, S. Haq, M. N. Tahir, J. Iqbal, A. A. C. Braga, M. Ashfaq and S. U. H. Akhtar, O -4-Acetylamino-benzenesulfonylated pyrimidine derivatives: synthesis, SC-XRD, DFT analysis and electronic behaviour investigation, J. Mol. Struct., 2021, 1224, 129308 CrossRef CAS;
(c) A. Ali, M. Khalid, S. Abid, M. N. Tahir, J. Iqbal, M. Ashfaq, F. Kanwal, C. Lu and M. F. U. Rehman, Green Synthesis, SC-XRD, Non-Covalent Interactive Potential and Electronic Communication via DFT Exploration of Pyridine-Based Hydrazone, Crystals, 2020, 10, 778, DOI:10.3390/cryst10090778.
- A. Ali, M. Khalid, Z. U. Din, H. M. Asif, M. Imran, M. N. Tahir, M. Ashfaq and E. Rodrigues-Filho, Exploration of structural, electronic and third order nonlinear optical properties of crystalline chalcone systems: Monoarylidene and unsymmetrical diarylidene cycloalkanones, J. Mol. Struct., 2021, 1241, 130685 CrossRef CAS.
- M. Khalid, A. Ali, S. Abid, M. N. Tahir, M. U. Khan, M. Ashfaq, M. Imran and A. Ahmad, Facile Ultrasound-Based Synthesis, SC-XRD, DFT Exploration of the Substituted Acyl-Hydrazones: An Experimental and Theoretical Slant towards Supramolecular Chemistry, ChemistrySelect, 2020, 5, 14844–14856 CrossRef CAS.
- M. Khalid, A. Ali, S. Asim, M. N. Tahir, M. U. Khan, L. C. C. Vieira, F. Alexander and M. Usman, Persistent prevalence of supramolecular architectures of novel ultrasonically synthesized hydrazones due to hydrogen bonding [X–H⋯ O; X= N]: Experimental and density functional theory analyses, J. Phys. Chem. Solids, 2021, 148, 109679 CrossRef CAS.
- M. Khalid, A. Ali, M. F. U. Rehman, M. Mustaqeem, S. Ali, M. U. Khan, S. Asim, N. Ahmad and M. Saleem, Exploration of noncovalent interactions, chemical reactivity, and nonlinear optical properties of piperidone derivatives: a concise theoretical approach, ACS Omega, 2020, 5, 13236–13249 CrossRef CAS PubMed.
- O. Concepcion, A. Ali, M. Khalid, A. F. de la Torre, M. U. Khan, A. R. Raza, G. M. Kamal, M. F. U. Rehman, M. M. Alam and M. Imran, Facile Synthesis of Diversely Functionalized Peptoids, Spectroscopic Characterization, and DFT-Based Nonlinear Optical Exploration, ACS Omega, 2021, 6, 26016–26025 CrossRef CAS PubMed.
- M. Khalid, A. Ali, A. F. de la Torre, K. P. Marrugo, O. Concepcion, G. M. Kamal, S. Muhammad and A. G. Al-Sehemi, Facile Synthesis, Spectral (IR, Mass, UV−Vis, NMR), Linear and Nonlinear Investigation of the Novel Phosphonate Compounds: A Combined Experimental and Simulation Study, ChemistrySelect, 2020, 5, 2994–3006 CrossRef CAS.
- B. Khan, M. Khalid, M. R. Shah, M. N. Tahir, M. U. Khan, A. Ali and S. Muhammad, Efficient synthesis by mono-carboxy methylation of 4,4′-biphenol, X-ray diffraction, spectroscopic characterization and computational study of the crystal packing of ethyl-2-((4′-hydroxy-[1,1′-biphenyl]-4-yl)oxy)acetate, ChemistrySelect, 2019, 4, 9274–9284 CrossRef CAS.
- M. Khalid, A. Ali, M. Adeel, Z. U. Din, M. N. Tahir, E. Rodrigues-Filho, J. Iqbal and M. U. Khan, Facile preparation, characterization, SC-XRD and DFT/DTDFT study of diversely functionalized unsymmetrical bis-ayl-α, β-unsaturated ketone derivatives, J. Mol. Struct., 2020, 1206, 127755 CrossRef.
- A. Ali, Z. U. Din, M. Khalid, M. N. Tahir, E. Rodrigues-Filho, B. Ali, S. Asim and S. Muhammad, Crystal and Quantum Chemical Exploration of the Potent Monocarbonyl Curcuminoids to Unveil Their Structural and Intriguing Electronic Properties, ChemistrySelect, 2020, 5, 3735–3745 CrossRef CAS.
- I. Khan, M. Khalid, M. Adeel, M. U. Khan, M. S. Khan, N. Ahmad, A. Ali and M. Akram, Palladium-catalyzed synthesis of pyrimidine substituted diaryl ethers through Suzuki Miyaura coupling reactions: Experimental and DFT studies, Optik, 2020, 219, 165285 CrossRef CAS.
- A. Ali, M. Khalid, M. F. U. Rehman, S. Haq, A. Ali, M. N. Tahir, M. Ashfaq, F. Rasool and A. A. C. Braga, Efficient synthesis, SC-XRD, and theoretical studies of O-Benzenesulfonylated pyrimidines: Role of noncovalent interaction influence in their supramolecular network, ACS Omega, 2020, 5, 15115–15128 CrossRef CAS PubMed.
- S. Tariq, A. R. Raza, M. Khalid, S. L. Rubab, M. U. Khan, A. Ali, M. N. Tahir and A. A. C. Braga, Synthesis and structural analysis of novel indole derivatives by XRD, spectroscopic and DFT studies, J. Mol. Struct., 2020, 1203, 127438 CrossRef CAS.
- M. Ashfaq, M. N. Tahir, S. Muhammad, K. S. Munawar, A. Ali, G. Bogdanov and S. S. Alarfaji, Single-Crystal Investigation, Hirshfeld Surface Analysis, and DFT Study of Third-Order NLO Properties of Unsymmetrical Acyl Thiourea Derivatives, ACS Omega, 2021, 6, 31211–31225 CrossRef CAS PubMed.
- M. N. Tahir, S. H. Mirza, M. Khalid, A. Ali, M. U. Khan and A. A. C. Braga, Synthesis, single crystal analysis and DFT based computational studies of 2, 4-diamino-5-(4-chlorophenyl)-6-ethylpyrim idin-1-ium 3, 4, 5-trihydroxybenzoate-methanol (DETM), J. Mol. Struct., 2019, 1180, 119–126 CrossRef CAS.
- M. N. Tahir, M. Ashfaq, F. Alexander, J. Caballero, E. W. Hernández-Rodríguez and A. Ali, Rationalizing the stability and interactions of 2, 4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium 2-hydroxy-3, 5-dinitrobenzoate salt, J. Mol. Struct., 2019, 1193, 185–194 CrossRef CAS.
- Bruker, APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
-
(a) G. M. Sheldrick. SHELXS97 and SHELXL97. University of Göttingen, Germany, Acta Crystallogr., Sect. E: Struct. Rep. Online, 1997, pp. 1600–5368 Search PubMed;
(b) G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, WinGX and ORTEP for Windows: an update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: from visualization to analysis, design and prediction, J. Appl. Crystallogr., 2020, 53 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. B. Axel, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- A. D. McLean and G. S. Chandler, Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z=11-18, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, J. S. Raymond and J. A. People, Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Conceptual density functional theory, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- A. Chakraborty, S. Pan and P. K. Chattaraj, Biological activity and toxicity: a conceptual DFT approach, Struct. Bond., 2013, 150, 143–180 CrossRef CAS PubMed.
- S. Jorio, M. Salah, H. Abou El Makarim and M. Tabyaoui, Reactivity indices related to DFT theory, the electron localization function (ELF) and non-covalent interactions (NCI) calculations in the formation of the non-halogenated pyruvic esters in solution, Mediterr. J. Chem., 2019, 8, 476–485 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, Molden: a pre- and post-processing program for molecular and electronic structures, J. Comput.-Aided Mol. Design., 2000, 14, 123–134 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 41, 1–17, DOI:10.1186/1758-2946-4-17.
- Avogadro: An Open-Source Molecular Builder and Visualization Tool. Version 1.1.1, http://avogadro.cc/ Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N. L. Chang, Patterns in hydrogen bonding: functionality and graph set analysis in crystals, Angew Chem. Int. Ed. Engl., 1995, 34, 1555–1573, DOI:10.1002/anie.199515551.
- M. Shahid, M. N. Tahir, M. Salim, M. A. Munawar and H. A. Shad, 1-{[(E)-(4-{[(2Z)-2, 3-Dihydro-1,3-thiazol-2-ylidene]sulfamoyl}phenyl)iminiumyl]methyl}naphthalen-2-olate, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2015, 71, o421–o422 CrossRef CAS PubMed.
- S. Mondal, S. M. Mandal, T. K. Mondal and C. Sinha, Spectroscopic characterization, antimicrobial activity, DFT computation and docking studies of sulfonamide Schiff bases, J. Mol. Struct., 2017, 1127, 557–567, DOI:10.1016/j.molstruc.2016.08.011.
- H. Elghamry, CSD Communication, 2019 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, Crystal Explorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011, DOI:10.1107/S1600576721002910.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32, 10.1039/B818330A.
- A. N. Malik, A. Kuznetsov, A. Ali, M. Ashfaq, M. N. Tahir and A. Siddique, Imine-based Zwitterion: Synthesis, single-crystal characterization, and computational investigation, J. Mol. Struct., 2022, 1253, 132237, DOI:10.1016/j.molstruc.2021.132237.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces, Chem. Comm., 2007, 3814–3816, 10.1039/B704980C.
- M. Ashfaq, K. S. Munawar, G. Bogdanov, A. Ali, M. N. Tahir, G. Ahmed, A. Ramalingam, M. M. Alam, M. Imran, S. Sambandam and B. Munir, Single crystal inspection, Hirshfeld surface investigation and DFT study of a novel derivative of 4-fluoroaniline: 4-((4-fluorophenyl)amino)-4-oxobutanoic acid (BFAOB), J. Iran. Chem. Soc., 2022, 19, 1953–1961, DOI:10.1007/s13738-021-02432-4.
- C. Jelsch, K. Ejsmont and L. Huder, The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis, IUCrJ, 2014, 1, 119–128, DOI:10.1107/S2052252514003327.
-
(a) M. Ashfaq, M. Khalid, M. N. Tahir, A. Ali, M. N. Arshad and A. M. Asiri, Synthesis of Crystalline Fluoro-Functionalized Imines, Single Crystal Investigation, Hirshfeld Surface Analysis, and Theoretical Exploration, ACS Omega, 2022, 7, 9867–9878, DOI:10.1021/acsomega.2c00288;
(b) A. Ali, M. Khalid, M. Ashfaq, A. N. Malik, M. N. Tahir, M. A. Assiri, M. Imran, S. F. de Alcântara Morais and A. A. C. Braga, Preparation, QTAIM and Single-Crystal Exploration of the Pyrimethamine-Based Co-Crystal Salts with Substituted Benzoic Acids, ChemistrySelect, 2022, 7, e202200349, DOI:10.1002/slct.202200349.
- M. J. Turner, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Visualisation and characterisation of voids in crystalline materials, CrystEngComm, 2011, 13, 1804–1813, 10.1039/C0CE00683A.
-
(a) M. Ashfaq, A. Ali, M. N. Tahir, M. Khalid, M. A. Assiri, M. Imran, K. S. Munawar and U. Habiba, Synthetic approach to achieve halo imine units: Solid-state assembly, DFT based electronic and non-linear optical behavior, Chem. Phys. Lett., 2022, 803, 139843, DOI:10.1016/j.cplett.2022.139843;
(b) A. Ali, M. Ashfaq, Z. U. Din, M. Ibrahim, M. Khalid, M. A. Assiri, A. Riaz, M. N. Tahir, E. Rodrigues-Filho, M. Imran, A. Kuznetsov and Synthesis, Structural, and Intriguing Electronic Properties of Symmetrical Bis-Aryl-α,β-Unsaturated Ketone Derivatives, ACS Omega, 2022, 7, 39294–39309, DOI:10.1021/acsomega.2c05441.
-
(a) M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, Accurate and efficient model energies for exploring intermolecular interactions in molecular crystals, J. Phys. Chem. Lett., 2014, 5, 4249–4255, DOI:10.1021/jz502271c;
(b) C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, Crystal Explorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems, IUCrJ, 2017, 4, 575–587, DOI:10.1107/S205225251700848X.
-
(a) A. Ali, Z. U. Din, M. Ibrahim, M. Ashfaq, S. Muhammad, D. Gull, M. N. Tahir, E. Rodrigues-Filho, A. G. Al-Sehemi and M. Suleman, Acid catalyzed one-pot approach towards the synthesis of curcuminoid systems: unsymmetrical diarylidene cycloalkanones, exploration of their single crystals, optical and nonlinear optical properties, RSC Adv., 2023, 13, 4476–4494, 10.1039/D2RA07681K;
(b) M. N. Tahir, A. Ali, M. Khalid, M. Ashfaq, M. Naveed, S. Murtaza, I. Shafiq, M. A. Asghar, R. Orfali and S. Perveen, Efficient Synthesis of Imine-Carboxylic Acid Functionalized Compounds: Single Crystal, Hirshfeld Surface and Quantum Chemical Exploration, Molecules, 2023, 28, 2967–2984, DOI:10.3390/molecules28072967.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Muhammad Ashfaq*a,
Muhammad Nawaz Tahira,
Mohammad Mahtab Alamc,
Mohamed S. Mostafa
*b,
Muhammad Ashfaq*a,
Muhammad Nawaz Tahira,
Mohammad Mahtab Alamc,
Mohamed S. Mostafa



 is the chemical potential of the system.
is the chemical potential of the system.












