
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ imaging of mRNA transcripts based on split-aptamer and split protein in living cells†
Yan Peng‡
 *ab,
Mengqi Li‡a,
Fuyu Gong*ab,
Xiaofei Liuac,
Jirou Xionga and
Keran Wanga
*ab,
Mengqi Li‡a,
Fuyu Gong*ab,
Xiaofei Liuac,
Jirou Xionga and
Keran Wanga
aThe Affiliated First Hospital of Fuyang Normal University, Fuyang Normal University, Fuyang, Anhui 236037, P.R. China. E-mail: pengyan@fynu.edu.cn; gong2022@fynu.edu.cn
bFuyang Women and Children's Hospital, Fuyang, Anhui 236037, P.R. China
cFuyang People's Hospital, Fuyang, Anhui 236000, P.R. China
First published on 26th March 2024
Abstract
Messenger RNA (mRNA) is an essential component of cell development and growth. However, the detection of endogenous mRNA in living cells is currently limited. To address this issue, we have developed a novel strategy that comprises split-aptamer and split fluorescent protein dual-color, “turn-on” probes that specifically target mRNA with complementary sequences. Our split-aptamer and split-protein-initiated fluorescence complementation (sAPiFC) approach for live-cell imaging has demonstrated selectivity, stability, and capability for targeting various mRNAs.
Introduction
The visualization of RNA distribution and dynamics provides important information about RNA localization and cellular trafficking, which affect gene expression and regulation. Understanding the temporal dynamics and spatial localization of mRNA is therefore essential to understanding gene expression regulation. For a long time, it was believed that bacterial cells had randomly distributed mRNAs due to their lack of organization. However, recent studies have shown that they do exhibit spatial mRNA organization.1,2 Such localized mRNAs can also be found in both prokaryotic and eukaryotic cells, where they regulate gene expression involved in developmental physiology and cellular activity. Consequently, there has been a growing need to visualize endogenous mRNA and other types of RNA in living cells for a better understanding of the mechanisms underlying various RNA functions and dynamics, as well as for addressing biologically relevant questions to develop diagnostic and therapeutic applications.Efforts have been made to develop probes and strategies for detecting RNA molecules within live cells. Various approaches implement organic fluorophores, fluorescence protein (FP), or split fluorescence protein as reporters.3–5 Single-stranded oligonucleotide RNA fluorescence in situ hybridization (FISH),6 molecular beacon (MB)7,8 or fluorescence resonance energy transfer (FRET)9 probes, which hybridize to complementary target RNA strands, are utilized for organic probe targeting. Additionally, FPs' fusion to RNA binding proteins, such as bacterial phage coat proteins MS2 or PP7,10,11 λN,12 dCas9, or dCas13 (ref. 13 and 14) within the CRISPR system, is frequently utilized. Similarly, nascent chains are labeled by fluorescent antibody-based probes such as SunTags, FLAG and influenza hemagglutinin (HA) epitopes; either single-chain variable fragments (scFv) or fragmented antibodies (Fab).16 These proteins fused to the N-terminus bind to their RNA-interacting sequences of interest. Unlike more traditional fluorescent fusion tags such as GFP, which take minutes to fold and fluoresce,15 the use of FPs, instead of organic fluorophores, has the advantage of delivering molecules into living cells with minimum disruption or probe degradation.17 However, the high background from unbound probes restricts the use of FISH or FP probes in imaging. Recently, hybridization chain reactions (HCR),18,19 click chemistry-based amplification (clampFISH),20 and signal amplification by exchange reaction (SABER)21,22 have been combined with in situ amplification approaches for imaging RNA molecules in living cells. The sensitivity and efficiency of FISH have been greatly enhanced using these methods. However, these approaches require the delivery and multistep washing of fluorophore-modified synthetic oligonucleotides, making them mostly suitable for fixed-cell imaging only.
A more recent technique involves the use of fluorogen-activating RNA aptamers that have an affinity for pro-fluorescent chromophores and can enhance their quantum yield.23–25 These RNA aptamers are typically situated at the 3′ end of the target RNA. Only when the fluorophore complexes with the RNA aptamer can fluorescence be detected, thus reducing background signals caused by unbound fluorophores in the cytosol during live-cell imaging.26 Herein, we developed new recognition RNA transcripts based on split-aptamer and split-protein-initiated fluorescence complementation (sAPiFC) dual-color for use in living cells. The turn-on fluorophore binding RNA aptamers were generated by splitting them into two complementary segments consisting of MS2 and Boxb aptamers, each attached to a unique recognition segment. When the two divided mCherry are brought into proximity, the MCP protein fusion N-mCherry and λN protein fusion C-mCherry allow the mCherry reporter to refold into its native conformation and emit a fluorescence signal (Scheme 1).27,28 This genetically encoded sAPiFC method does not necessitate prior modifications of RNA transcripts and can selectively target any RNA sequence of interest, opening up new avenues for accurate in situ RNA imaging.
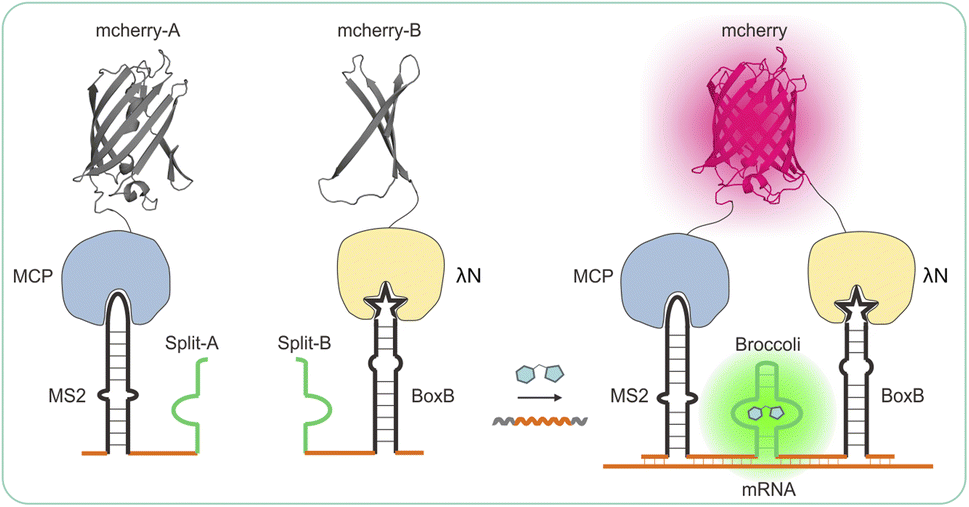 | ||
| Scheme 1 Design and expression of a dual-color recognition system: split-aptamer with split-fluorescent protein for live-cell RNA transcripts imaging. | ||
Experimental
Reagents
DNAs utilized in this study were synthesized and HPLC purified by Sangon Biotechnology Co., Ltd. (Shanghai, China). The DNA sequences are listed in Table S1.† The stock solution of each DNA (100 μM) was prepared with TE buffer containing 10 mM Tris–HCl, 1 mM EDTA, and 12.5 mM MgCl2 (pH 7.4). The 40% acrylamide mix solution, ammonium persulfate (APS), 1,2-bis(dimethylamino)- ethane (TEMED), and DNA ladder were acquired from Sangon Biotechnology Co., Ltd. (Shanghai, China). All the chemicals were of analytical grade and utilized as received without further purification. All oligonucleotides were purchased from Sangon Biotechnology Co., Ltd. (Shanghai, China). All sequencing experiments were performed at Sangon Biotechnology Co., Ltd. (Shanghai, China). DFHBI-1 T (MedChemExpress) and BI were synthesized by our laboratory, SDS-PAGE gel preparation kit, DAPI, and agarose were obtained from Beyotime (Shanghai, China). The HeLa cells (human cervical carcinoma cell line), and CHO cells (Chinese Hamster Ovary) were purchased from Procell, Inc.Equipment
The Leica TCS SP8 laser confocal microscope (Leica Microsystems Inc., Exton, PA), 20 mm glass-bottom dish (Cellvis, Shanghai), Thermo Scientific™ Varioskan™ LUX (Thermo Scientific), and the UV-Vis absorption spectra were recorded on a UV-Vis spectrometer UV-3600 (Shimadzu, Japan).RNA transcription
All RNA sequences for in vitro assays were transcribed in vitro using DNA templates that contained a T7 promoter. The DNA template sequences can be found in Table S1.† In vitro transcription was carried out following the instructions provided by the transcription kit. The transcription reaction contained T7 RNA polymerase, in vitro transcription buffer (20 mM Tris–HCl pH 7.5, 15 mM MgCl2, 5 mM DTT, and 2 mM spermidine), and 2 mM of each ATP, CTP, GTP, and UTP was used in the transcription procedures (New England Biolabs). The reaction was incubated at 37 °C for at least 4 hours, followed by stopping with RNA loading solution (95% formamide and 5 mM EDTA, with trace amounts of Xylene Cyanol FF and Bromophenol Blue). RNA purification was carried out using denaturing polyacrylamide gel electrophoresis (0.75 mm 6% TBE-PAGE, 8 M urea), and the bands corresponding to the expected product size were extracted and eluted by tumbling overnight in 300 mM Sodium Acetate pH 5.4. The elutes were precipitated in ethanol, resuspended in buffer (1 mM EDTA, 10 mM Tris–HCl pH 8.0), and the purified RNA was diluted to 50 μM and stored at −20 °C for further use.In vitro assays
For in vitro assembly reactions on ice, a buffer (1 mM MgCl2, 100 mM KCl, 40 mM HEPES pH 7.5) of 50 μL was prepared. Purified RNA products were also prepared on ice in the same buffer (1 mM MgCl2, 100 mM KCl, 40 mM HEPES pH 7.5) containing BI at 10 μM. The fluorescence spectrum of the RNA products was compared to the fluorophore alone using a Multimode plate reader (ex = 472 nm). The samples were then preincubated at 37 °C before being analyzed for fluorescence (ex = 472 nm, em = 490–700 nm or ex = 472 nm, em = 507 nm) using a Multimode plate reader at room temperature. Additional information regarding FISH probe sequences can be found in Table S2.†The purification of in vitro transcribed RNA was carried out using Trizol reagent, following the instructions provided by the manufacturer. 100 ng RNA was placed into a well of precast 10% TBE-Urea Gel and run at 300 V in 1 × TBE buffer. Following electrophoresis, the gel was rinsed three times for five minutes with water before being stained with 50 μM BI solution containing 1 mM MgCl2, 100 mM KCl, and 40 mM HEPES pH 7.5. Visualization of the RNA was performed using a ChemiDoc MP (Bio-Rad) with excitation at 470 ± 15 nm and emission at 532 ± 14 nm. Subsequently, the gel was imaged using the SYBR Green channel (497 nm excitation and 520 ± 55 nm emission) on the same instrument.
Plasmid construction
The plasmid that expresses sAPiFC probes is obtained from the pSilence 2,1-U6 and contains a U6 promoter and pcDNA3.1+ plasmid. Before plasmid extraction and subsequent experiments, all constructs are accurately sequenced by Sangon Biotechnology Co., Ltd. (Shanghai, China), as documented in the Recombinant Vector Construction Information presented in Table S2.†Cell culture
Plasmid transfection and expression were carried out in HeLa or CHO cells at 37 °C in a humidified incubator with 5% CO2. Cells were cultured in MEM medium (Gibco), supplemented with 100 unit per mL penicillin, 100 g mL−1 streptomycin sulfate, 2 mM L-glutamine, and 10% (v/v) fetal bovine serum (Invitrogen). Cells used for imaging analysis should be no more than 10 passages old. When cells reached 70–80% confluence, they were harvested by washing with 1 × PBS buffer and incubated with 1 mL of Trypsin–EDTA (0.25%) for 3–5 minutes at 37 °C. The cells were then resuspended in 2 mL pre-warmed media and seeded in 12-well plates at a density of 8 × 105 cells per mL. After 24 hours of incubation, the cells should be 50% confluent.Staining assay
On day 0, the cells were seeded into 12-well plates at a density of 1.0 × 105 cells per mL. On day 1, the cells were transfected with two vectors encoding sAPiFC probes. On day 2, the cells were detached with 0.25% trypsin and re-seeded at 1.5–3.0 × 103 cells per dishes, precoated with fibronectin solution in PBS (50 μg mL−1) in the complete growth medium, and then incubated at 37 °C for 10 min in a humidified 5% CO2 incubator. Cy3-DNA probes were co-transfected into cells, and target gene information is presented in Table S3,† while Table S3† contains the sequence for Cy3-DNA probe information. After attaching the cells to the plate, they were stained with BI (final concentration: 10 μM), either with 10 μM BI with or without 10 μM DAPI (final concentration: 1 μg mL−1, Life Technologies) by simply adding 1 from a stock solution in DMSO (1 mM) and DAPI from a stock solution in H2O (1 mg mL−1), growth medium (60 μL), and incubated at 37 °C for 10 min in a humidified 5% CO2 incubator, and subsequently imaged on a Leica TCS SP8 confocal microscope (Leica Microsystems Inc., Exton, PA).Observation of fluorescence using confocal laser scanning microscope (CLSM)
To stabilize the microscope, activate the temperature control system of the incubation chamber at least 30 minutes before imaging. Before imaging with the final imaging solution, which may consist of PBS buffer or MEM supplemented with 1 mM MgCl2, 100 mM KCl, and 40 mM HEPES pH 7.5, cells should be incubated for 10 minutes. The imaging solution should include 10 μM BI, with or without 10 μM DAPI (Life Technologies). Images should be captured at 37 °C using a laser confocal microscope, specifically a Leica TCS SP8 fitted with a HyD CCD camera with a 40 oil objective. The Green channel (ex: 470 ± 15 nm; em: 510 ± 20 nm) is utilized to activate the reassembled aptamer, with an exposure duration of 500 ms. The Red channel (ex: 579 ± 12 nm; em: 599 ± 20 nm), exposure time (100 ms); Blue channel (ex: 358 ± 15 nm; em: 461 ± 20 nm), exposure time: 50 ms. To avoid overexposure, the laser light intensity should be set to 10–15%. Cells should be seeded and transfected with plasmid in a glass-bottom dish for 24 hours before the UHRF1 or survivin mRNA imaging experiment (refer to Table S4†).Results
Design and optimization of the split-aptamer for RNA detection in vitro
From a variety of fluorogen-activating aptamers (FLAP), we selected split-Broccoli as the model system due to its shorter probe length (49 nt) and is free of undesirable intramolecular structures.29 In order to investigate whether the split-aptamer can efficiently and stably recombine into a functioning folded fluorophore binding unit in vitro, we utilized Split-Broccoli-A to connect MS2 and Split-Broccoli-B to connect BoxB, while the recognition sequence served as the complementary link to the target RNA. In the presence of the target sequence (as shown in Fig. 1A), we observed the efficient and proper folding of the split-aptamer into a functioning fluorophore binding unit. The findings indicate that even when there is an excess of fluorophore, the two probe pairs generate minimal fluorescence signals, demonstrating a low background. The fluorophore BI (benzo[d] imidazole-3,5-difluoro-4-hydroxy benzylidene imidazolinone), an improved version of DFHBI-1T, resulted in approximately a 4.4-fold increase in cellular imaging (Fig. S1†),30,31 and the experiment was analyzed using a Multimode plate reader. Green fluorescence was only detected in the presence of the target RNA. The fluorescence signal-to-background ratio (S/B) of MS2-splitA and BoxB-splitB reached 5, respectively, when an equivalent molar amount of target RNA was added to the system (Fig. 1B and S2†). PAGE (Polyacrylamide gel electrophoresis) was utilized to assess the assembly of the two RNA probes in vitro, which demonstrated the creation of a single band with a slowed migration rate in the presence of target RNA and BI, revealing the formation of a complex structure (Fig. 1B). Conversely, in the absence of the target RNA, the combination of probes MS2-splitA and BoxB-splitB exhibited a rate of movement equivalent to that of a single probe. Consequently, the target RNA was able to effectively bring the two split aptamer fragments into proximity (Fig. 1B).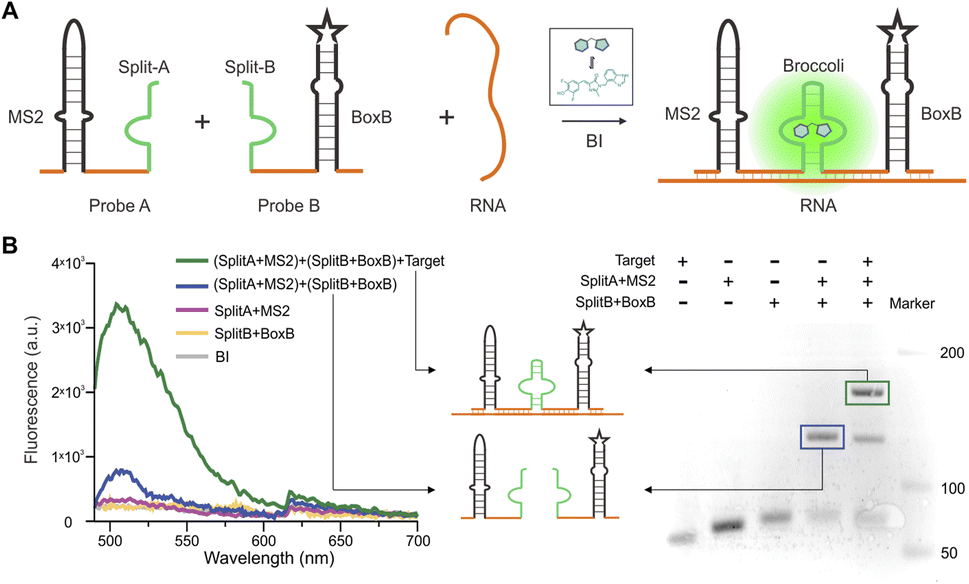 | ||
| Fig. 1 Design and in vitro characterization of a split-aptamer sensor. (A) Schematic representation of the split-aptamer sensor for RNA transcript imaging. (B) (Left) fluorescence intensity measurements of the split-aptamer (1 μM) with 10 μM fluorophore (BI), in the presence or absence of 1 μM target RNA. (Right) PAGE gel (15%) electrophoresis analysis of the split-aptamer sensor, with and without target sequence. A fluorescent structure (green frame) of 1 μM split-aptamer probe is generated in the presence of equal molar target RNA. | ||
We conducted further research on the impact of two base mismatches on the sAPiFC system. Two mutation sites were introduced to the recognition target probes. Our findings reveal that the punctate fluorescence observed in the mutant group was significantly reduced compared with the group that exhibited fully complementary to the target RNA (Fig. 2). These results indicated that the sAPiFC system possesses a high degree of sequence selectivity in targeting substrate nucleotides.
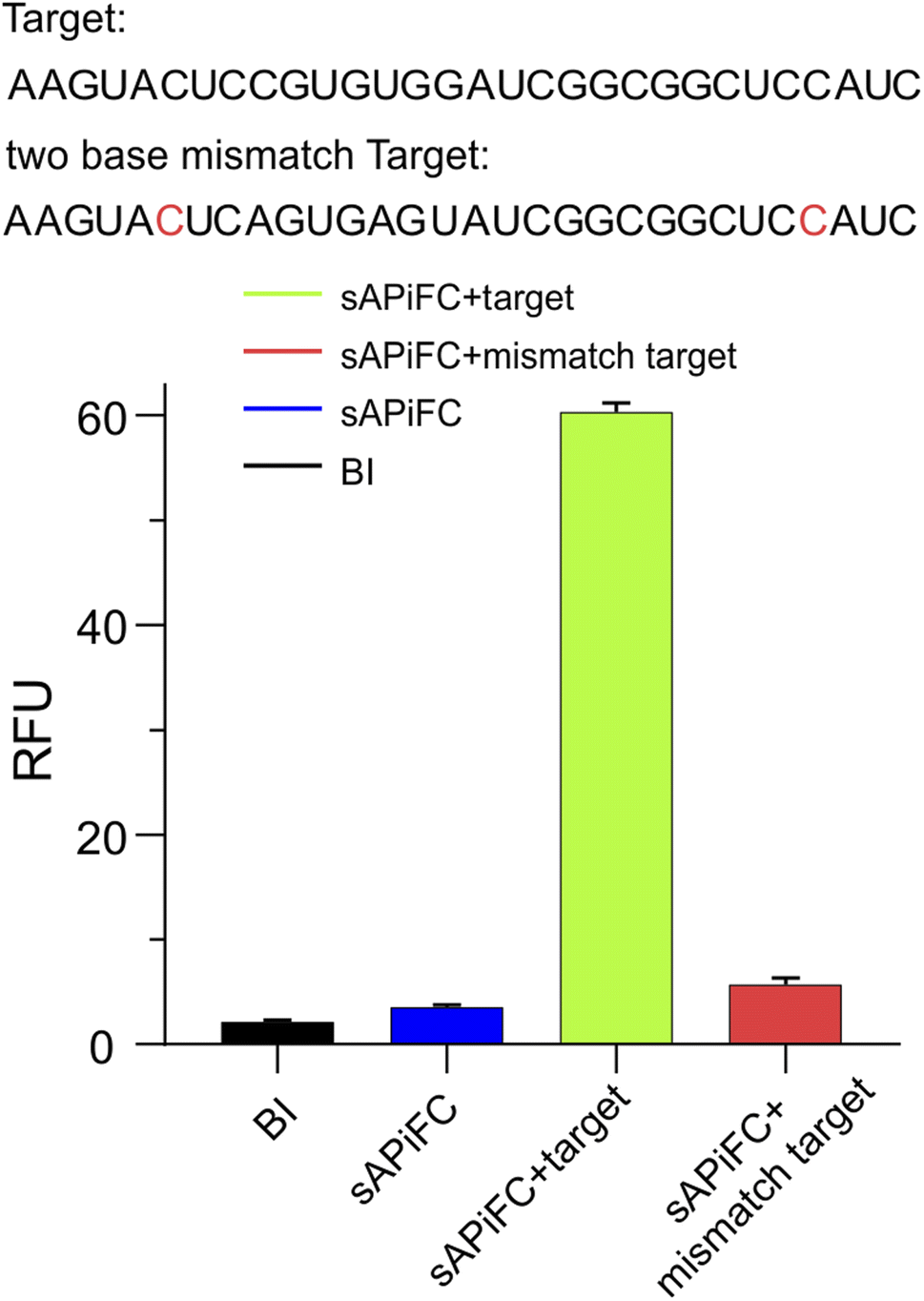 | ||
| Fig. 2 Fluorescence intensities of 1 μM sAPiFC in the presence of 10 μM BI and either the correct or mismatched target RNA (indicated in green and red, respectively). Sites of mismatched are labeled in red. | ||
RNA imaging with split-aptamer sensors in mammalian cells
The present study investigated the ability of Split-probes to detect endogenous mRNA in mammalian cells. A pair of split-aptamer probes were designed with complementary targeting sequences for human β-actin mRNA. Individual recognition split probes were generated in vitro and co-transfected into HeLa cells. Fluorescence signals were observed using a confocal microscope in the presence of BI fluorophore (Fig. 3A). The cytoplasm of split-probe transfectants displayed high fluorescence signals, while untransfected control cells demonstrated negligible background fluorescence. To determine the specificity of the sAPiFC probe target β-actin mRNA, a Cy3-labeled DNA probe complementary to a different sequence but targeting β-actin mRNA was employed (Fig. 3A). The green and red signals were found to co-localize well, thus confirming the identity of the observed signals as β-actin mRNA. Notably, the fluorescence intensity profiles indicated synchronization across the cell (Fig. 3B). These findings suggest that split-aptamer technologies may be effectively used to image mRNA in mammalian cells. | ||
| Fig. 3 RNA imaging in mammalian cells using split-aptamer sensors. (A) Confocal images of split-aptamer sensors (green) co-localized with Cy3-labeled DNA probes (red), targeting β-actin mRNA in HeLa cells. The green channel represents split-aptamer transaction or control, while the red channel displays Cy3-DNA probe, and the blue channel shows DAPI staining. The merged channel displays the overlap between green and red signals in yellow. Scale bars: 10 μm. (B) Normalized fluorescence intensity profiles of regions of interest (white line in left panels). | ||
RNA imaging with split-aptamer and split-protein initiated fluorescence complementation dual-color methods in living cells
Next, we investigated the use of the genetically encoded sAPiFC method for imaging endogenous mRNAs within living cells. We established a technique allowing for high expression of two probes and two fusion proteins (Fig. 4A) and then generated two plasmids encoding two split-aptamers, with the other plasmid encoding two fusion proteins. These plasmids were co-transfected in equal amounts into HeLa cells, which were subsequently treated with BI and monitored using live cell imaging after 48 hours (Fig. 4B). Although untransfected cells showed negligible fluorescence signals, cells transfected with sAPiFC plasmids exhibited clear fluorescence (Fig. 4B). To confirm the increase in fluorescence within cells, a multimode plate reader was utilized (Fig. 4C). | ||
| Fig. 4 Design and optimization of expression vectors for sAPiFC probes. (A) Multiple sAPiFC plasmids were constructed, driven by either the U6 or CMV promoters. The original vector is shown below. (B) Illustration of control vector, NC (negative control did not express the target RNA) vector or sAPiFC vector with confocal images of HeLa cells expressing sAPiFC, respectively. Blue channel (DAPI). Scale bars: 10 μm. (C) Fluorescence emission was measured in 1 × 106 total cells transfected with the two plasmids encoding sAPiFC, respectively. 10 μM BI was added for the measurement, with red representing red signals, green representing green signals, and gray representing BI. | ||
The sAPiFC strategy's general applicability was then investigated. The target sequences were changed to complement additional endogenous mRNAs, including ubiquitin-like with PHD and ring finger domains 1 (UHRF1) and baculoviral inhibitor of apoptosis repeat-containing 5 (survivin), which are crucial cancer biomarkers. Upon transfection with corresponding sAPiFC systems, fluorescence signals were observed in the cytoplasm, demonstrating the versatility of the sAPiFC strategy (Fig. 5). The performance of the sAPiFC strategy in CHO cells was also investigated. Three pairs of sAPiFC probes produced fluorescence signals equivalent to those used in HeLa cells. These results indicated that the sAPiFC strategy may be suitable for imaging various endogenous mRNAs in both human and non-human cells (Fig. 5A). Indeed, these results indicated that the sAPiFC dual-color system can be employed for target mRNA imaging in mammalian cells, achieve more precise localization and semi-quantitative detection analysis.
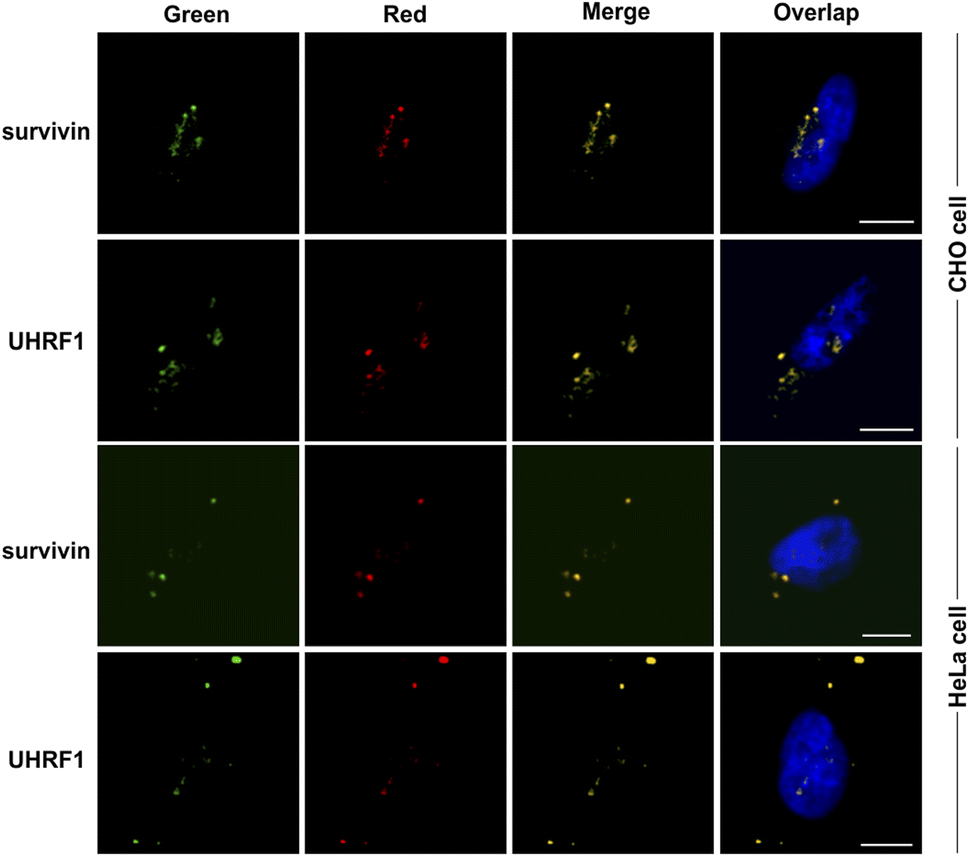 | ||
| Fig. 5 Imaging of endogenous heterologous mRNA in living cells. Visualization of the endogenous mRNAs (UHRF1, and survivin) in HeLa or CHO cells using corresponding sAPiFC probes and Cy3-labeled DNA probe (red). Scale bars: 10 μm. | ||
Conclusions
In summary, we developed and evaluated a dual-color system-based fluorescence “turn-on” probe for mRNA imaging by combining a split aptamer with the target's complementary sequences and split fluorescent protein in live cells. The selectivity and robustness of these fluorescent “turn-on” probes for various mRNA targets have been demonstrated. The sAPiFC strategy can be genetically encoded and directly transcribed for endogenous mRNA targeting, live-cell compatibility, minimal background from the unbound probe, and dual-color fluorescence colocalization. In addition to cellular imaging, this genetically encoded sAPiFC method has the potential to detect intracellular RNA for programmable regulation of cellular activities.Our study's results revealed interesting findings on target mRNA distribution and abundance. We observed a predominant localization of these specific mRNAs around the nucleus, indicating their involvement in critical cellular processes such as transcription and translation. This spatial arrangement emphasizes their significance in regulating gene expression. Additionally, compared to housekeeping mRNAs involved in basic cellular functions, the target mRNAs were relatively less abundant, implying their specialized roles within the cell, possibly participating in more specific and regulated pathways. These findings illuminate the intricate mechanisms underlying gene regulation and emphasize the importance of understanding mRNA localization patterns to decipher cellular processes. Further research is warranted to explore the functional implications of this distinct distribution pattern and its impact on various biological phenomena.
By utilizing genetically encoded plasmids for cellular expression, invasive probe delivery is avoided. However, due to the relatively diminished fluorescence intensity of cytoplasmic fluorescent RNA aptamers, direct visualization of low-copy number mRNA is compensated by accurate co-localization analysis using fluorescent proteins. We have successfully demonstrated the qualitative observation of mRNA in living cells using sAPiFC; however, quantification of mRNA solely through sAPiFC imaging remains a challenge. We have noted that the arrangement of targeting sequence pairs does not necessarily require a direct connection between complementary sequences on the target RNA molecule and can effectively accommodate a certain level of spatial separation. As a result, it should be feasible to expand the potential applications of this technique to visualize functional and regulatory RNA/RNA interactions in vivo. Therefore, we anticipate that this sAPiFC method will be widely used for sensing intracellular RNA of interest. The logical design of sAPiFC probes could also prove effective in regulating metabolic pathways and cellular activity.
Author contributions
Yan Peng: conceptualization, methodology, software, visualization, investigation, funding acquisition, supervision, writing – original draft preparation, writing – reviewing and editing, project administration. Mengqi Li: methodology, software, validation, visualization. Xiaofei Liu: investigation, visualization. Jirou Xiong: visualization, investigation. Keran Wang: visualization, investigation. Fuyu Gong: visualization, investigation, data curation, funding acquisition, writing – original draft preparation, writing – reviewing and editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the Key Project of Scientific Research of Higher Education Institutions in Anhui Province (2023AH050423), the scientific research project of Fuyang Normal University (2022KYQD0016), Fuyang Normal University Horizontal Medical Research Project (2024FYNUEY09, 2024FYNUEY19), the Horizontal Medical Research Project of Fuyang Normal University (2021HXYXZX05ZD), Modern Pharmaceutical Industry Chain Research and Innovation Team of Fuyang Normal University (CYLTD202208), Quality Engineering Project of Fuyang Normal University (2022ZYRCPY02), Fuyang Science and Technology Planning Project (FK202081119), Fuyang Normal University Undergraduate Research Project (XSXM22-069, XSXM22-070).References
- P. M. Llopis, A. F. Jackson, O. Sliusarenko, I. Surovtsev, J. Heinritz, T. Emonet and C. Jacobs-Wagner, Nature, 2010, 466, 77–U90 CrossRef CAS PubMed.
- C. Y. Yan, L. Miao, Y. Zhang, X. L. Zhou, G. Y. Wang, Y. Q. Li, Q. L. Qiao and Z. C. Xu, Sens. Actuators, B, 2023, 386, 133731 CrossRef CAS.
- K. Rau and A. Rentmeister, ACS Cent. Sci., 2017, 3, 701–707 CrossRef CAS PubMed.
- A. dos Santos, R. Heydenreich, C. Derntl, A. R. Mach-Aigner, R. L. Mach, G. Ramer and B. Lendl, Anal. Chem., 2020, 92, 15719–15725 CrossRef PubMed.
- C. Q. Dong and J. C. Ren, Acc. Chem. Res., 2023, 56, 2582–2594 CrossRef CAS PubMed.
- D. Bressan, G. Battistoni and G. J. Hannon, Science, 2023, 381, 499–508 CrossRef PubMed.
- C. Wu, S. Cansiz, L. Zhang, I. T. Teng, L. Qiu, J. Li, Y. Liu, C. Zhou, R. Hu, T. Zhang, C. Cui, L. Cui and W. Tan, J. Am. Chem. Soc., 2015, 137, 4900–4903 CrossRef CAS PubMed.
- P. J. Santangelo, A. W. Lifland, P. Curt, Y. Sasaki, G. J. Bassell, M. E. Lindquist and J. E. Crowe, Nat. Methods, 2009, 6, 347–U346 CrossRef CAS PubMed.
- S. Ranjit, L. Lanzanò, A. E. Libby, E. Gratton and M. Levi, Nat. Rev. Nephrol., 2021, 17, 128–144 CrossRef CAS PubMed.
- S. Das, M. Vera, V. Gandin, R. H. Singer and E. Tutucci, Nat. Rev. Mol. Cell Biol., 2021, 22, 483–504 CrossRef CAS PubMed.
- P. Dave and J. A. Chao, Curr. Opin. Struct. Biol., 2020, 65, 89–95 CrossRef CAS PubMed.
- J. Sponer, G. Bussi, M. Krepl, P. Banás, S. Bottaro, R. A. Cunha, A. Gil-Ley, G. Pinamonti, S. Poblete, P. Jureacka, N. G. Walter and M. Otyepka, Chem. Rev., 2018, 118, 4177–4338 CrossRef CAS PubMed.
- D. A. Nelles, M. Y. Fang, M. R. O'Connell, J. L. Xu, S. J. Markmiller, J. A. Doudna and G. W. Yeo, Cell, 2016, 165, 488–496 CrossRef CAS PubMed.
- O. O. Abudayyeh, J. S. Gootenberg, P. Essletzbichler, S. Han, J. Joung, J. J. Belanto, V. Verdine, D. B. T. Cox, M. J. Kellner, A. Regev, E. S. Lander, D. F. Voytas, A. Y. Ting and F. Zhang, Nature, 2017, 550, 280–284 CrossRef PubMed.
- T. Morisaki and T. J. Stasevich, Cold Spring Harbor Perspect. Biol., 2018, 10, a032078 CrossRef CAS PubMed.
- C. A. Cialek, A. L. Koch, G. Galindo and T. J. Stasevich, Curr. Opin. Genet. Dev., 2020, 61, 75–82 CrossRef CAS PubMed.
- W. Chen, X. Y. Zhao, N. Y. Yang and X. Li, Angew. Chem., Int. Ed., 2023, 62, e202209813 CrossRef CAS PubMed.
- H. M. T. Choi, J. Y. Chang, L. A. Trinh, J. E. Padilla, S. E. Fraser and N. A. Pierce, Nat. Biotechnol., 2010, 28, 1208–U1103 CrossRef CAS PubMed.
- H. M. T. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef CAS PubMed.
- S. H. Rouhanifard, I. A. Mellis, M. Dunagin, S. Bayatpour, C. L. Jiang, I. Dardani, O. Symmons, B. Emert, E. Torre, A. Cote, A. Sullivan, J. A. Stamatoyannopoulos and A. Raj, Nat. Biotechnol., 2019, 37, 84–89 CrossRef CAS PubMed.
- J. Y. Kishi, S. W. Lapan, B. J. Beliveau, E. R. West, A. Zhu, H. M. Sasaki, S. K. Saka, Y. Wang, C. L. Cepko and P. Yin, Nat. Methods, 2019, 16, 533–544 CrossRef CAS PubMed.
- S. K. Saka, Y. Wang, J. Y. Kishi, A. Zhu, Y. Zeng, W. Xie, K. Kirli, C. Yapp, M. Cicconet, B. J. Beliveau, S. W. Lapan, S. Yin, M. Lin, E. S. Boyden, P. S. Kaeser, G. Pihan, G. M. Church and P. Yin, Nat. Biotechnol., 2019, 37, 1080–1090 CrossRef CAS PubMed.
- A. Climent-Catala, I. Casas-Rodrigo, S. Iyer, R. Ledesma-Amaro and T. E. Ouldridge, ACS Synth. Biol., 2023, 12, 3754–3765 CrossRef CAS PubMed.
- J. J. Liang, X. Liu, W. Xiao, P. J. Teng, P. Guan, W. L. Liang, L. S. Hu, G. B. He, H. R. He, G. Li, S. Y. Zou, C. Lu, Q. F. Song, J. F. Zhao, D. L. Cao, B. Zhu, Y. Li and Y. Tang, Chem. Eng. J., 2024, 481, 148651 CrossRef CAS.
- X. W. Liu, N. N. Zhao, H. M. Yuan, D. L. Li, M. Liu and C. Y. Zhang, Biosens. Bioelectron., 2024, 247, 115966 CrossRef CAS PubMed.
- Y. Peng, X. L. Ai, Y. Yuan, J. Dong, X. Cui, F. Du, X. Huang and Z. Tang, Anal. Chim. Acta, 2022, 1209, 339816 CrossRef CAS PubMed.
- S. Feng, S. Sekine, V. Pessino, H. Li, M. D. Leonetti and B. Huang, Nat. Commun., 2017, 8, 370 CrossRef PubMed.
- Y. Peng, L. J. Shu, X. F. Deng, X. Huang, X. M. Mo, F. Du and Z. Tang, Anal. Chem., 2023, 95, 13762–13768 CrossRef CAS PubMed.
- Z. Wang, Y. Luo, X. Xie, X. Hu, H. Song, Y. Zhao, J. Shi, L. Wang, G. Glinsky, N. Chen, R. Lal and C. Fan, Angew. Chem., Int. Ed., 2018, 57, 972–976 CrossRef CAS PubMed.
- X. Li, H. Kim, J. L. Litke, J. Wu and S. R. Jaffrey, Angew. Chem., Int. Ed., 2020, 59, 4511–4518 CrossRef CAS PubMed.
- X. Li, L. Mo, J. L. Litke, S. K. Dey, S. R. Suter and S. R. Jaffrey, J. Am. Chem. Soc., 2020, 142, 14117–14124 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08475b |
| ‡ Equal first author: Yan Peng and Mengqi Li contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2024 |