DOI:
10.1039/D3RA07836A
(Paper)
RSC Adv., 2024,
14, 1062-1071
Electronic substituent effect on the conformation of a phenylalanine-incorporated cyclic peptide†
Received
16th November 2023
, Accepted 19th December 2023
First published on 2nd January 2024
Abstract
The Phe-incorporated cyclic peptide [cyclo(-Phe1-oxazoline2-D-Val3-thiazole4-Ile5-oxazoline6-D-Val7-thiazole8-)] is in a conformational equilibrium between square and folded forms in solution. In the folded form, a CH⋯π interaction between the Phe1 aromatic ring and the Oxz2 methyl group is observed. We endeavored to control the local conformation and thus modulate the CH⋯π interaction and flexibility of the Phe1 side chain by controlling the electronic substituent effects at the 4-position of the aromatic ring of the Phe1 residue. The effect of the 4-substituent on the global conformation was indicated by the linear relationship between the conformational free energies (ΔGo) determined through NMR-based quantification and the Hammett constants (σ). Electron-donating substituents, which had relatively strong CH⋯π interactions, promoted peptide folding by restraining the loss in entropy. Local control by the 4-substituent effects suggested that the Phe side chain exerts an entropic influence on the folding of these cyclic peptides.
Introduction
The conformational energy is an important factor when interpreting a variety of phenomena. The conformational free energy (ΔGo) combines enthalpy (ΔHo) and entropy (ΔSo) into a single value, but the entropic term generally accounts for only a small portion of the ΔGo value; the enthalpic term is dominant. We previously performed a structural analysis of a cyclic peptide containing oxazoline (Oxz) and thiazole (Thz) as a heterocycle, cyclo(-Xaa1-Oxz2-D-Val3-Thz4-Ile5-Oxz6-D-Val7-Thz8-).1 When the backbone of this cyclic peptide folds at the two Oxz rings, it is stabilized by four intramolecular hydrogen bonds, so the enthalpy term greatly contributes to the spontaneous folding.2–8 On the other hand, the entropic term is also important for molecular recognition, and receptor affinity can reportedly be improved by minimizing the loss in entropy needed to fold an active conformer.9–13
In the present study, to focus on the contribution of the entropic term to peptide folding, we attempted local control of the Phe1 side chain orientation (χ space) mediated by the CH⋯π interaction using a Phe1-incorporated cyclic peptide (4), cyclo(-Phe1-Oxz2-D-Val3-Thz4-Ile5-Oxz6-D-Val7-Thz8-), as a scaffold (Fig. 1). In solution, peptide 4 is in an equilibrium between its “square” form, in which the peptide backbone is open, and its “folded” form (Fig. 2), where a CH⋯π interaction between the aromatic ring of the Phe1 residue and methyl group of the Oxz2 ring is observed.1 We introduced the following substituents at the 4-position of the Phe(X)1 aromatic ring of peptide 4: [X: OCH3 (1), CH3 (2), C(CH3)3 (3), F (5), I (6), Cl (7), Br (8), CF3 (9) and NO2 (10)] (Fig. 1). With the exception of peptides 4 and 5, these peptides were all newly synthesized. Electronic substituent effects are often used in the studies of non-covalent interactions,14–19 and modulation of the CH⋯π interaction is expected in the present study as well. In other words, indirect modulation of the Phe1 side chain dihedral angle by the 4-position substituents may become possible. Here we describe the structural characterization of peptides 1–10 based on X-ray diffraction, circular dichroism (CD) spectroscopy and variable temperature (VT) 1H NMR measurements. We then used estimated thermodynamic parameters (ΔHo, ΔSo and ΔGo) to address the local control of CH⋯π interactions and the associated flexibility of the Phe side chain by the 4-position substituents and their effects on global conformational equilibrium.
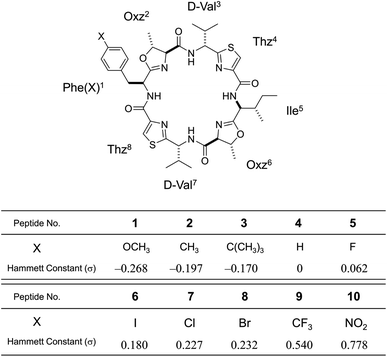 |
| | Fig. 1 Chemical structure of Phe(X)-incorporated cyclic peptide, 4-substituents (X) in each peptide (1–10) and their Hammett constants (σ).22 Peptides 4 and 5 were synthesized previously.2,5 Peptides 1–3 and 6–10 are newly synthesized. | |
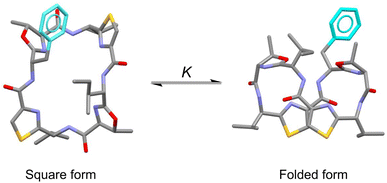 |
| | Fig. 2 Conformational equilibrium between the square and folded forms of Phe(X)-incorporated cyclic peptide. Carbon, nitrogen, oxygen and sulfur atoms and the aromatic ring of Phe residue are represented in gray, blue, red, yellow and light blue, respectively. | |
Results and discussion
Crystal structures
The X-ray structures of 2, 3, 6, 7, 8 and 9 are shown in Fig. 3. Peptides 2, 6, 7 and 8 were crystallized in acetonitrile (CH3CN) solution. Within the crystal, the peptide backbone of 2 was folded at the two Oxz rings, and the two Thz rings faced each other. The asymmetric units of 6, 7 and 8 each contained two independent molecules, but because their structures were similar, only molecule A is shown in Fig. 3. The crystal structures of 6, 7 and 8 also exhibited structural features very similar to 2, and all four folded forms were stabilized by four hydrogen bonds: N[Phe(X)1]–H⋯Oγ(Oxz6), N(D-Val3)–H⋯O(Thz8), N(Ile5)–H⋯Oγ(Oxz2) and N(D-Val7)–H⋯O(Thz4) (Table S4†). By contrast, the peptide backbones of 3 and 9 were in the square form, which unfolded such that the Oxz and Thz rings were separated from one another. Crystals of 3 and 9 were grown in CH3CN and acetone (Ac2O) solution, respectively. A water molecule was held at the center of the peptide ring of 3 by four hydrogen bonds, and similarly, an Ac2O molecule was held at the center of the peptide ring of 9 by two hydrogen bonds (Table S4†).
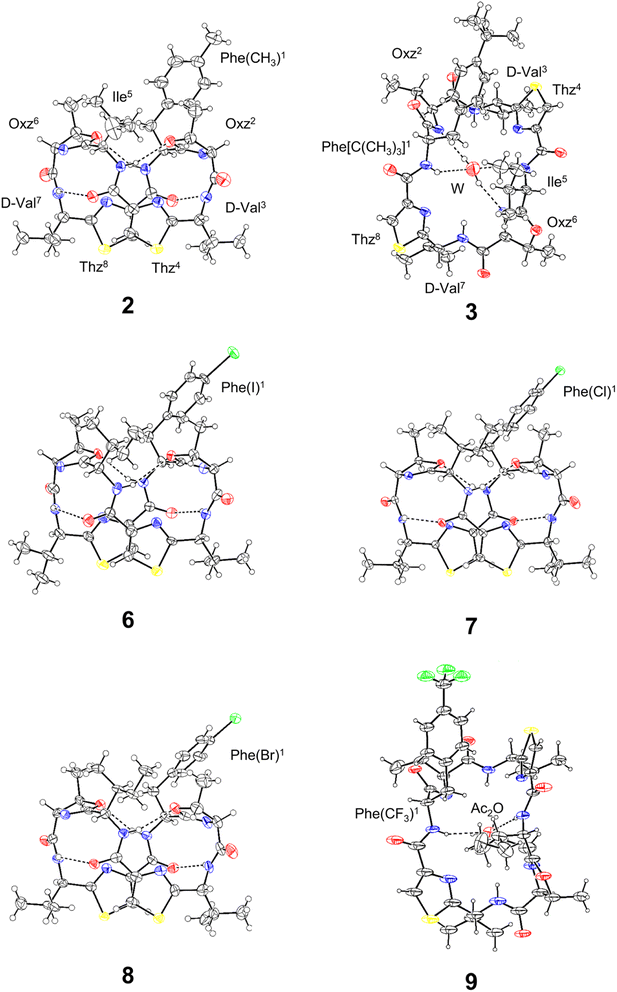 |
| | Fig. 3 Crystal structures of 2, 3, 6, 7, 8 and 9. Carbon, nitrogen, oxygen, sulfur and halogen atoms are represented in black, blue, red, yellow and green, respectively. The dashed lines represent hydrogen bonds. Peptides 2, 3, 6, 7 and 8 were crystallized as CH3CN solvated forms, but the CH3CN molecules are not shown because they are not involved in any hydrogen bonds. The asymmetric units of 6–8 each contained two independent molecules (molecule A and B), but because their conformations were similar, only molecule A is shown. | |
The superimposition of all the folded forms, including the previously determined folded X-ray structures of 4 (ref. 2) and 5,5 is shown in Fig. 4. Each molecular fitting was carried out for the peptide backbone of 4 to that of 2, 5, 6, 7 or 8 [root mean square deviation (RMSD) = 0.074 Å, 0.100 Å, 0.099 Å, 0.084 Å and 0.072 Å, respectively]. Overall, the crystallographic folded forms were remarkably similar, except for the side chain moieties of the Phe(X)1 residue. The observed side chain dihedral angles χ1 (N–Cα–Cβ–Cγ) of Phe(X)1 are listed in Table 1. Although all χ1 angles adopted a trans conformer, the aromatic planes of 2 and 4 had positive values and were oriented toward the methyl group of Oxz2, whereas the aromatic rings of 5–8 had negative values and were oriented away from the methyl group of Oxz2. Furthermore, the distances between the methyl group of Oxz2 and the Phe(X)1 aromatic ring were estimated by surveying the CH⋯π contacts for the six-membered π-system20,21 (Table 1). The CH⋯π contacts of 2 and 4 were determined to be 3.233 Å and 2.893 Å, respectively, but in 5–8 the methyl groups of Oxz2 were located outside the π-orbital of the Phe(X)1 aromatic ring, and CH⋯π contacts were not observed. These results suggest that the more electron-rich aryl groups of 2 and 4 can interact with the methyl group of Oxz2, whereas peptides 5–8, which have more electron-poor aryl groups due to their electron-withdrawing substituents, cannot. On the other hand, because the X-ray structures of both 3, which bears a relatively strong electron-donating substituent [C(CH3)3], and 9, which bears a relatively strong electron-withdrawing substituent (CF3), assumed the square form, no clear correlation between their conformation and the 4-substituent of Phe(X)1 residue was exhibited in the crystals. In these square forms, the aromatic rings of the Phe(X)1 residue were oriented toward the alkyl groups of the Ile5 side chain, but no CH⋯π contact by its alkyl groups was observed. The χ1 angles of the Phe(X)1 side chains in 3 and 9 also adopted the trans conformer, and few differences in the χ1 angle were observed among the crystal structures due to the 4-substituents.
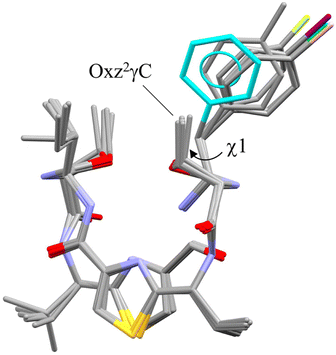 |
| | Fig. 4 Superimposition of the folded crystal structures of 2, 4, 5, 6 (molecule A), 7 (molecule A) and 8 (molecule A). Molecular fitting was carried out for the peptide backbone, which showed that the backbones of 2, 5, 6, 7 and 8 differed from 4 with the RMSD = 0.074, 0.100, 0.099, 0.084 and 0.072 Å, respectively. The crystal structures of 4 and 5 are taken from previous reports.2,5 The aromatic ring of the Phe residue (4) and the methyl group carbon (2), fluorine (5), iodine (6), chlorine (7) and bromine (8) atoms are represented in blue, gray, yellow, purple, green and pink, respectively. | |
Table 1 Summary of the crystal structures of 2–9
| Peptide |
χ1a (°) |
CH⋯π contactb (Å) |
| χ1 is the dihedral angle in the side chain of the Phe(X)1 residue, N–Cα–Cβ–Cγ. The CH⋯π contacts were estimated using the method described by Umezawa et al.20,21 This crystal structure is taken from a previous report.2 This crystal structure is taken from a previous report.5 The distances between γH of Oxz2 and the aromatic ring of the Phe(X)1 residue. |
| Folded form |
| 2 |
172.31 |
3.233e |
| 4c |
160.23 |
2.893e |
| 5d |
−169.75 |
ND |
| 6 – molecule A |
−177.82 |
ND |
| 6 – molecule B |
−179.15 |
ND |
| 7 – molecule A |
−173.90 |
ND |
| 7 – molecule B |
−171.95 |
ND |
| 8 – molecule A |
−172.84 |
ND |
| 8 – molecule B |
−174.26 |
ND |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Square form |
| 3 |
166.20 |
ND |
| 9 |
175.41 |
ND |
CD spectra
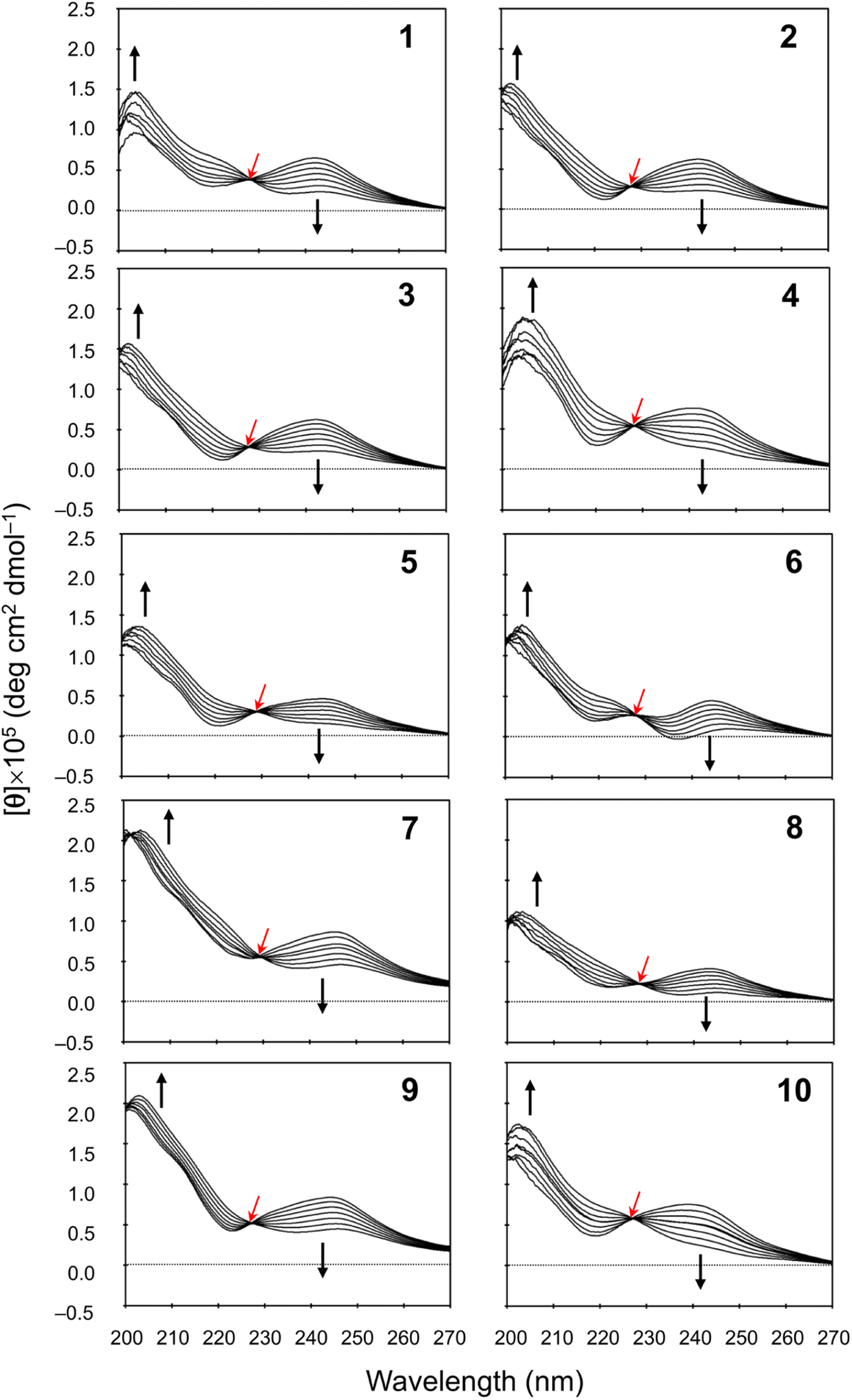
We measured the temperature dependence of the CD spectra for peptides 1–10 in CH3CN solution at 273–333 K. Their spectral changes as temperature was increased in 10 K steps are shown in Fig. 5. In earlier spectral data, a square form was assigned when [θ]245 was negative and a folded form was assigned when [θ]245 was positive.3–8 In the present study, the CD spectra for all peptides showed characteristics of the folded form at 273 K, and the increasing temperature led to reductions in [θ]245. Furthermore, a single isosbestic point was observed at around 230 nm in all spectra. These observations suggest that increasing temperature leads to a conformational change from the folded to the square form due to weakening of four intramolecular hydrogen bonds, N[Phe(X)1]–H⋯Oγ(Oxz6), N(D-Val3)–H⋯O(Thz8), N(Ile5)–H⋯Oγ(Oxz2) and N(D-Val7)–H⋯O(Thz4), which contribute to stabilizing the folded form. However, none of the peptides exhibited a negative [θ]245, even at 333 K. No significant difference was observed in the temperature dependence of the CD spectra in CH3CN solution for any of the peptides, including peptides 3 and 9, which had square X-ray structures. While a crystal structure represents a specific energy state, the structure observed in solution is an average structure based on the abundance of each possible conformation. Therefore, disparities between the solution and crystal structures may arise, as peptides 3 and 9.
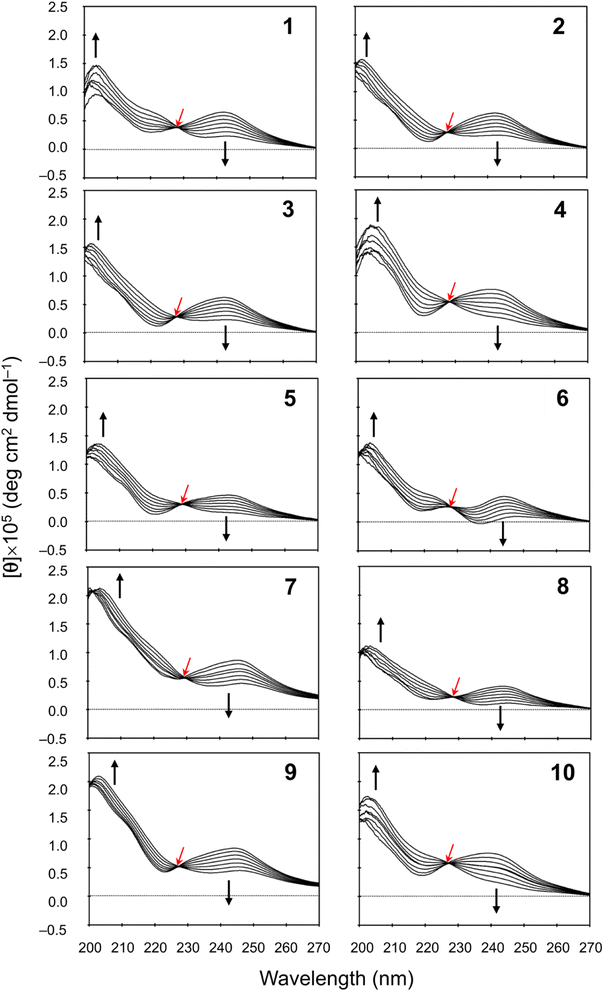 |
| | Fig. 5 Temperature dependence of the CD spectra for peptides 1–10. The CD spectra for 5 were taken from a previous report.5 The spectra were measured every 10 K in CH3CN solution at 273–333 K. The isosbestic point and the direction of spectral change with increasing temperature are represented by red and black arrows, respectively. | |
NMR-based quantification of the conformational equilibrium
The conformational equilibrium constants (K) of the peptides were determined with a previously described method6 using the chemical shifts of Thz protons at various temperatures in CH3CN-d3. The enthalpy (ΔHo) and entropy (ΔSo) were obtained from a linear van't Hoff plot (lnK versus 1/T), after which the Gibbs free energy (ΔGo) was evaluated as ΔGo = −RTlnK. These thermodynamic parameters (ΔHo, ΔSo and ΔGo298 K) are listed in Table 2. The folding of all peptides was enthalpically favorable and entropically unfavorable. It is reasonable that the ΔHo values were negative because four hydrogen bonds stabilizing the conformation are formed during folding. Peptide 3 exhibited the highest ΔHo value (−13.09 kJ mol−1), while 9 exhibited the second highest ΔHo value (−14.27 kJ mol−1). This may be related to the fact that only these two crystal structures were square forms. In comparison, among the folded crystal structures, the ΔHo values of 2 (−14.89 kJ mol−1) and 4 (−14.99 kJ mol−1), which have the potential for CH⋯π interactions between the Phe(X)1 aromatic ring and the Oxz2 methyl group were slightly higher than those of 5 (−15.65 kJ mol−1), 6 (−15.62 kJ mol−1), 7 (−15.73 kJ mol−1) and 8 (−15.66 kJ mol−1). These findings suggest that the CH⋯π interaction does not contribute to the stability of the folded form.
Table 2 Thermodynamic parameters of peptides 1–10
| Peptide |
ΔHo (kJ mol−1) |
ΔSo (J K−1 mol−1) |
ΔGo298 K (kJ mol−1) |
| The thermodynamic parameters of 4 are taken from a previous report.6 |
| 1 |
−14.48 |
−41.37 |
−2.15 |
| 2 |
−14.89 |
−42.90 |
−2.10 |
| 3 |
−13.09 |
−37.00 |
−2.07 |
| 4a |
−14.99 |
−45.06 |
−1.57 |
| 5 |
−15.65 |
−46.51 |
−1.79 |
| 6 |
−15.62 |
−45.74 |
−1.99 |
| 7 |
−15.73 |
−46.70 |
−1.81 |
| 8 |
−15.66 |
−46.46 |
−1.81 |
| 9 |
−14.27 |
−42.40 |
−1.64 |
| 10 |
−15.69 |
−48.05 |
−1.37 |
Plots of the temperature versus ΔGo, ΔHo or TΔSo for all peptides are shown in Fig. 6. The ΔGo values of all peptides at every measured temperature exhibited negative or zero values, except for 10 at 333 K, which displayed a slightly positive ΔGo value (0.31 kJ mol−1). In previous CD measurements, a slight negative band appeared at 245 nm for peptides with a ΔGo value of approximately 1.0 kJ mol−1, and a clear negative band emerged for peptides with a ΔGo value of approximately 2.0 kJ mol−1 or higher.6–8 Therefore, a robust correlation between the ΔGo value and the CD spectra was observed. The folding process of 1–3 had |ΔHo| > |TΔSo| at all measured temperatures, which indicates they were able to fold spontaneously (ΔGo < 0). During the folding of 4–9, the |TΔSo| value neared the |ΔHo| value as the temperature increased, and the ΔGo value became zero at 333 K. In the folding process of 10, the |ΔHo| and |TΔSo| were equal at 323 K, and no spontaneous folding (ΔGo > 0) was observed at 333 K. Thus, peptides with electron-withdrawing substituents appear to be more strongly influenced by the entropy term during the folding process than peptides with electron-donating substituents.
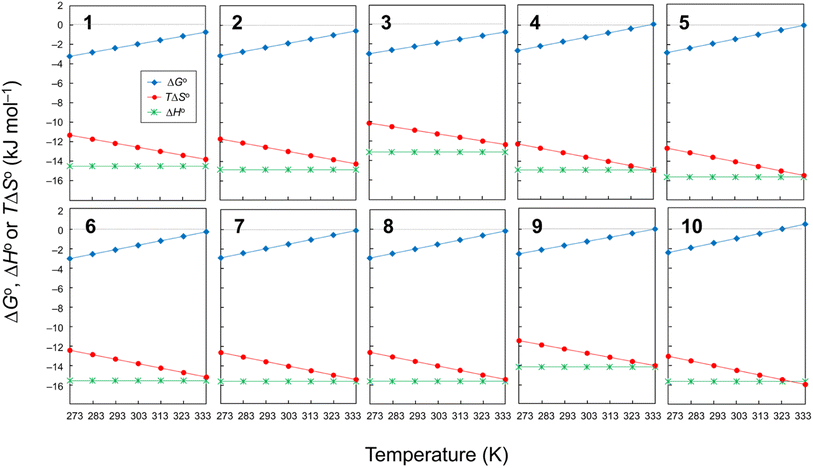 |
| | Fig. 6 Plots of the temperature dependences of ΔGo, ΔHo and TΔSo for peptides 1–10. The data for 4 were taken from a previous report.6 | |
We also examined the 4-substituent effect on conformation using the linear relationship between the ΔGo298 K and the Hammett constant σ (ref. 22) (Fig. 7). With the exception of 4–6, there was a very good linear correlation (R2 > 0.98) between ΔGo298 K and σ, though even when 5 and 6 were included, the correlation coefficient was >0.90. The most significant departure from this correlation arose with the unsubstituted peptide 4. This peptide was more difficult to fold by 0.39 kJ mol−1 than the Hammett substituent effects would predict. The observed linear correlation suggests that the peptide folding was dependent on the electronic properties of the Phe(X)1 aromatic ring. However, as described above, the CH⋯π interactions between the Phe(X)1 aromatic ring and the Oxz2 methyl group did not contribute enthalpically to the peptide folding, and in fact no correlation between the ΔHo value and σ constant was indicated. We therefore focused next on the entropic term and examined the temperature dependence of the vicinal coupling constants (3J) for the Phe(X)1 side chain.
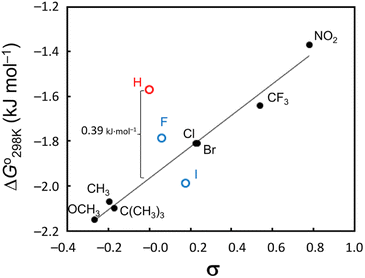 |
| | Fig. 7 Hammett plot of the ΔGo298 K values vs. σ constants. A very strong linear correlation (R2 > 0.98) was observed when 4–6 were excluded, and even when 5 and 6 (blue markers) were included, the correlation coefficient was >0.90. Peptide 4 (red marker) was more difficult to fold by 0.39 kJ mol−1 than the Hammett substituent effects would predict. | |
Restriction of the Phe(X)1 χ1 dihedral angle
As shown in Fig. 8, the side chains of the amino acids within the peptides usually adopt one of three dihedral angles χ1 (N–Cα–Cβ–Cγ) [−60°, gauche(−); −180°, trans; or 60°, gauche(+)], which are separated by relatively low energy rotational barriers.23,24 The two vicinal coupling constants (3Jαβ1 and 3Jαβ2) of the Phe(X)1 residue deliver dihedral angles θ1 (Hα–Cα–Cβ–Hβ1) and θ2 (Hα–Cα–Cβ–Hβ2), respectively (Fig. 8). In all peptides at 298 K, these two angles were assumed to be gauche(−) and trans based on observations of one large (>11 Hz) and one small (>6 Hz) 3Jαβ coupling constant for the Phe(X)1 residue. Additionally, the respective assignments of θ1 and θ2 were determined by comparing the rotating-frame Overhauser effect (ROE) cross-peaks between the NH and Hβ and between the Hα and Hβ in the Phe(X)1 residues. The ROE intensities of the two Hβ resonances with NH (dNβ1 and dNβ2) were nearly equal. On the other hand, when comparing the ROE intensities of the two Hβ resonances with Hα (dαβ1 and dαβ2), dαβ1 was stronger than dαβ2. As an example, the ROE cross-peaks between NH and Hβ and between Hα and Hβ in the Phe(OCH3)1 residue in peptide 1 are shown in Fig. 9. These observed features of the ROE cross-peaks enabled us to assign θ1 and θ2 as gauche(−) and trans, respectively, and to ultimately conclude that the χ1 angles for the Phe(X)1 side chains in all peptides are trans at 298 K.
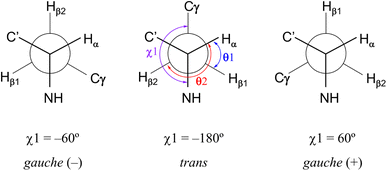 |
| | Fig. 8 Three low-energy conformations around χ1 (N–Cα–Cβ–Cγ) in an amino acid residue and the dihedral angles θ1 (Hα–Cα–Cβ–Hβ1) and θ2 (Hα–Cα–Cβ–Hβ2) related to the vicinal coupling constants (3Jαβ1 and 3Jαβ2) for the Phe(X)1 residue. χ1 (trans), θ1 [gauche(+)] and θ2 (trans) observed with all peptides in CD3CN solution at 298 K are shown in the central conformation. | |
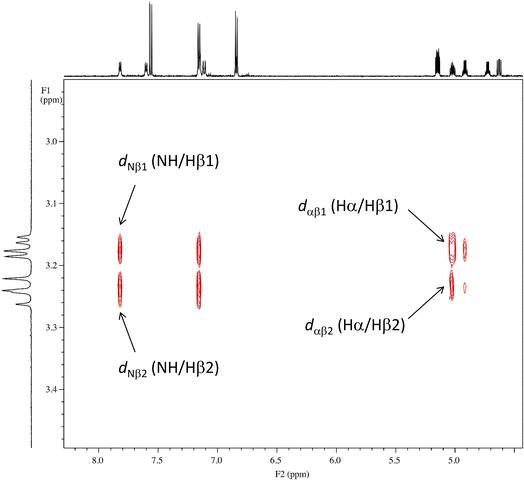 |
| | Fig. 9 ROE cross-peaks between NH and Hβ (dNβ1 and dNβ2) and between Hα and Hβ (dαβ1 and dαβ2) of the Phe(OCH3) residue in peptide 1. The intensities of dNβ1 and dNβ2 are equal, while the intensity of dαβ1 is stronger than that of dαβ2. | |
The temperature dependence of the vicinal coupling constants (3Jαβ1 and 3Jαβ2) and geminal coupling constants (2Jβ1β2) for Phe(X)1 residues are shown in Fig. 10. Naturally, the geminal coupling constants did not exhibit temperature dependence, whereas the vicinal coupling constants were classified into those exhibiting large or small temperature dependences. The vicinal coupling constants for 1, 2, 4 and 5 did not change much with increasing temperature; in particular, those of 1 did not change at all from 273 K to 293 K. For peptides 3, 6, 7, 8, 9 and 10, the 3Jαβ1 value increased and the 3Jαβ2 value decreased with increasing temperature. For peptides 3, 6, 7, 8 and 10, the two constants were equal (7.8 Hz) at 333 K, whereas for peptide 9, the constants were equal at 323 K, and 3Jαβ1 was larger than 3Jαβ2 at 333 K. Equality between 3Jαβ1 and 3Jαβ2 means that the θ1 and θ2 angles are equal. These observations suggest that the χ1 angles of Phe(X)1 in peptides 3, 6, 7, 8, 9 and 10 change from trans to gauche(+) with increasing temperature. In other words, the Phe(X)1 side chains in these peptides are more flexibility than those in peptides 1, 2, 4 and 5.
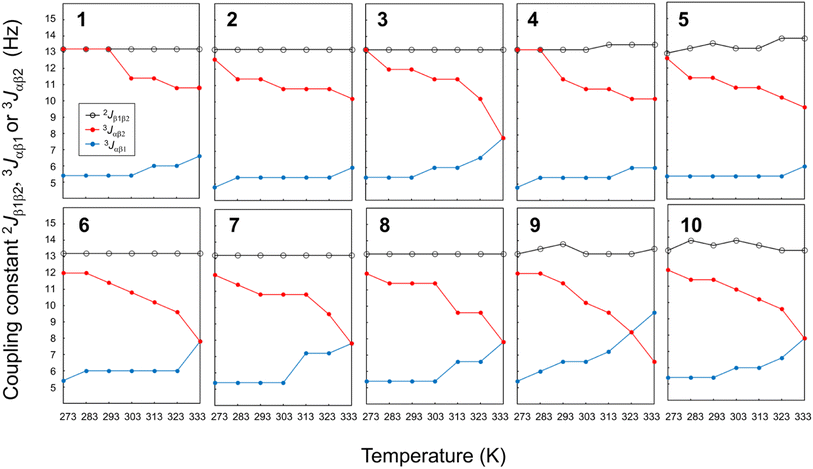 |
| | Fig. 10 Plots of the temperature dependences of the germinal coupling constant (2Jβ1β2) and vicinal constants (3Jαβ1 and 3Jαβ2) for peptides 1–10. The data for 4 were taken from an earlier report.6 | |
The relationship between the σ constants and the flexibility of the Phe(X)1 side chains (except for peptide 3) allows us to conclude that the flexibility of the Phe(X)1 side chain is controlled by the CH⋯π interactions between the Phe(X)1 aromatic ring and the Oxz2 methyl group. Peptides 6–10, bearing substituents with relatively small σ values, have little or no CH⋯π interaction, which increases the flexibility of the Phe(X)1 side chain. By contrast, the Phe(X)1 side chains of 1, 2, 4 and 5, bearing substituents with relatively large σ constants, are fixed by the CH⋯π interaction. As shown in Table 2, the entropic costs of folding peptides 6, 7, 8 and 10, which have flexible Phe(X)1 side chains, was higher (ΔSo = −46 to −49 J K−1 mol−1) than entropic cost of folding peptides 1 and 2, which have less flexible Phe(X)1 side chains (ΔSo = −41.37 and −42.90 J K−1 mol−1, respectively). These findings are consistent with our observations. On the other hand, there were discrepancies between the flexibility of the Phe(X)1 side chain and ΔSo for peptides 3, 4, 5 and 9. Even though the flexibility of the Phe(X)1 side chains of 4 and 5 was low, the entropic costs of folding were ΔSo = −45.06 and −46.51 J K−1 mol−1, respectively. The deviations from the linear relationship between the σ constant and ΔGo value in these two peptides (Fig. 7) may be due to the entropic cost of folding, which was larger than expected. There was also no correlation between the flexibility of the Phe(X)1 side chains and ΔSo values for peptides 3 and 9. Although the Phe(X)1 side chains of 3 and 9 showed flexibility, they had the lowest and second lowest folding entropic cost among all peptides (ΔSo = −37.00 and −42.40 J K−1 mol−1, respectively). In the folding of 3 and 9, the contribution of the enthalpic term was smaller than with the other peptides (ΔHo = −13.09 and −14.27 kJ mol−1, respectively). These results indicate that for peptides 3 and 9, there is a correlation between the σ constant and ΔGo value (Fig. 7), but this correlation may be a coincidence. The thermodynamic folding mechanism for these two peptides may differ from that of the other peptides.
Conclusion
Using a Phe-incorporated cyclic peptide as a scaffold, we controlled the CH⋯π interaction of substituents in the 4-position of the aromatic ring of the Phe residue and, accordingly, the flexibility of Phe side chain. We also investigated in detail the contribution of the entropic term to the peptide's folding. During the folding process in these peptides, four intramolecular hydrogen bonds contribute to the enthalpic term. In addition, CH⋯π interactions between the Phe(X)1 aromatic ring and Oxz2 methyl group appear to develop in the folded form. However, this CH⋯π interaction does not make an enthalpic contribution to peptide folding, but does appear to affect the entropic term. The relatively strong CH⋯π interactions in peptides bearing electron-donating substituents reduce the flexibility of the Phe(X)1 side chains, resulting in a relatively small entropic cost to folding. By contrast, because less CH⋯π interaction is observed in peptides bearing electron-withdrawing substituents, the flexibility of the Phe(X)1 side chains increases the entropic cost, making the peptides more difficult to fold. These findings, although containing some contradictions, were confirmed to some extent by the linear correlation between the σ constant and the ΔGo value.
Experimental
Peptide syntheses
The Thz unit (Boc-D-Val(Thz)-OMe) was synthesized as previously described.25,26 The linear peptide Boc-Phe(X)-allo-Thr-D-Val-Thz-Ile-allo-Thr-D-Val-Thz-OMe was synthesized using 1-hydroxybenzotriazole (Watanabe Chemical lnd. Ltd., Hiroshima, Japan) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (Watanabe Chemical lnd. Ltd., Hiroshima, Japan) in a liquid phase. All 4-substituted phenylalanines were purchased from Watanabe Chemical lnd. Ltd. (Hiroshima, Japan). Macrocyclization was performed using benzotriazolyloxy-tris(pyrrolidino)phosphonium hexafluorophosphate (Watanabe Chemical lnd. Ltd., Hiroshima, Japan) in the presence of 4-dimethylaminopyridine (Nacalai Tesque, Kyoto, Japan). Two Oxz rings were formed by reacting the Phe(X)-allo-Thr and Ile-allo-Thr moieties with bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) (Sigma-Aldrich Co. LLC, St. Louis, USA).27 The synthesis and characterization of 1, 2, 3, 6, 7, 8, 9 and 10 are detailed in the ESI.†
X-ray diffraction
Peptides 2, 3, 6, 7 and 8 were crystallized in CH3CN, and peptide 9 was crystallized in Ac2O. X-ray diffraction data for 2 were collected with a Bruker Smart APEXII (Bruker Corp., Massachusetts, USA). X-ray diffraction data for 3, 6, 7, 8 and 9 were collected with a Rigaku CrysAlisPro (Rigaku Corp., Tokyo, Japan). The structure of 2 was solved using SHELXS-97 (ref. 28) and refined using SHELXL-97.28 The structures of 3, 6, 7, 8 and 9 were solved using SHELXT29 and refined using SHELXL-2018/3.30 The crystal data have been deposited with the Cambridge Crystallographic Data Center under deposition numbers 2289960, 2289964, 2289961, 2289963, 2289962 and 2289965, corresponding to peptides 2, 3, 6, 7, 8 and 9, respectively. The crystal data for the six peptides are given in the ESI.†
Circular dichroism
CD spectra were measured using a JASCO 500A dichrograph (JASCO, Tokyo, Japan) with a 1 cm quartz cell at room temperature. The spectra were scanned in the range of 200–280 nm at a speed of 5 nm min−1 with a 0.1 nm interval for uptake to a computer. Data were averaged over each 1 nm and plotted. The temperature dependence of the CD spectra in CH3CN solution was assessed at 273–333 K. Peptide concentrations were about 0.04 mM.
1H NMR
1H NMR spectra were recorded on an Agilent DD2 600 MHz NMR spectrometer (Agilent Technologies, California, USA). Peptide concentrations were about 5.0 mM in CH3CN-d3. Chemical shifts were measured relative to internal tetramethylsilane at 0.00 ppm. The protons were assigned using two-dimensional correlated spectroscopy (2D-COSY) and rotating-frame Overhauser effect spectroscopy (ROESY; mixing time = 500 ms). VT-1H NMR measurements were made every 10 K from 273 K to 333 K. The assignment lists and 1D, 2D-COSY and ROESY spectra for 1, 2, 3, 6, 7, 8, 9 and 10 are given in the ESI.†
NMR-based quantitative studies
The conformational equilibrium constants (K) (Fig. 2) of the peptides were determined using the chemical shifts of Thz protons measured with VT-1H NMR.6 When the Thz rings were face to face in the folded form, the chemical shifts for the Thz4 and Thz8 protons appear in a higher magnetic field than those in the square form due to the ring-current effects of the Thz8 and Thz4 rings, respectively. The chemical shifts of Thz protons thus reflect the conformational changes. The K values of the peptides were estimated using eqn (1),| | |
K = (δS − δobs)/(δobs − δF)
| (1) |
where δobs is the chemical shift of the Thz protons in the equilibrating peptide, δS (=8.09 ppm) is the chemical shift of the Thz protons in T3ASC as a square reference peptide,31 and δF (=7.35 ppm) is the chemical shift of the Thz protons in dASC as a folded reference peptide.32 The chemical structures of T3ASC and dASC are given in the ESI.†
The enthalpy (ΔHo) and entropy (ΔSo) were determined from a linear van't Hoff plot, after which the Gibbs free energy (ΔGo) at each temperature was calculated using eqn (2).
| | |
ΔGo = ΔHo − TΔSo = −RTlnK
| (2) |
Conflicts of interest
There are no conflicts to declare.
References
- Y. Hamamoto, M. Endo, M. Nakagawa, T. Nakanishi and K. Mizukawa, Chem. Commun., 1983, 6, 323–324 RSC
 .
. - M. Doi, F. Shinozaki, Y. In, T. Ishida, D. Yamamoto, M. Kamigauchi, M. Sugiura, Y. Hamad, K. Kohda and T. Shioiri, Biopolymers, 1999, 49, 459–469 CrossRef CAS PubMed .
- A. Asano, K. Minoura, T. Yamada, A. Numata, T. Ishida, Y. Katsuya, Y. Mezaki, M. Sasaki, T. Taniguchi, M. Nakai, H. Hasegawa, A. Terashima and M. Doi, J. Pept. Res., 2002, 60, 10–22 CrossRef CAS PubMed .
- A. Asano, T. Yamada and M. Doi, Bioorg. Med. Chem., 2011, 19, 3372–3377 CrossRef CAS PubMed .
- A. Asano, T. Yamada and M. Doi, J. Pept. Sci., 2014, 20, 794–802 CrossRef CAS PubMed .
- A. Asano, K. Minoura, Y. Kojima, T. Yoshii, R. Ito, T. Yamada, T. Kato and M. Doi, RSC Adv., 2020, 10, 33317–33326 RSC .
- A. Asano, M. Nakagawa, C. Miyajima, M. Yasui, K. Minoura, T. Yamada and M. Doi, J. Pept. Sci., 2021, 27, e3363 CrossRef CAS PubMed .
- A. Asano, K. Minoura, T. Yamada and M. Doi, RSC Adv., 2023, 13, 2458–2466 RSC .
- H. Gohlke and G. Klebe, Angew. Chem., Int. Ed., 2002, 41, 2644–2674 CrossRef CAS PubMed .
- A. Velazquez-Campoy, M. J. Todd and E. Freire, Biochemistry, 2000, 39, 2201–20207 CrossRef CAS PubMed .
- D. G. Udugamasooriya and M. R. Spaller, Biopolymers, 2008, 8, 653–667 CrossRef .
- J. Wang, Z. Szewczuk, S. Yue, Y. Tsuda, Y. Konishi and E. O. Purisima, J. Mol. Biol., 1995, 253, 473–492 CrossRef CAS PubMed .
- V. Lafont, A. A. Armstrong, H. Ohtaka, Y. Kiso, L. M. Amzel and E. Freire, Chem. Biol. Drug Des., 2007, 69, 413–422 CrossRef CAS PubMed .
- H. Suezawa, T. Hashimoto, K. Tsuchinaga, T. Yoshida, T. Yuzuri, K. Sakakibara, M. Hirota and M. Nishio, J. Chem. Soc., Perkin Trans. 2, 2000, 1243–1249 RSC .
- F. Hof, D. M. Scofield, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2004, 43, 5056–5059 CrossRef CAS PubMed .
- W. F. Bailey, K. M. Lambert, K. B. Wiberg and B. Q. Mercado, J. Org. Chem., 2015, 80, 4108–4115 CrossRef CAS PubMed .
- R. K. Raju, J. W. G. Bloom, Y. An and S. E. Wheeler, ChemPhysChem, 2011, 12, 3116–3130 CrossRef CAS PubMed .
- I. K. Mati and S. L. Cockroft, Chem. Soc. Rev., 2010, 39, 4195–4205 RSC .
- F. J. Carver, C. A. Hunter, D. J. Livingstone, J. F. McCabe and E. M. Seward, Chem.–Eur. J., 2002, 8, 2847–2859 CrossRef CAS .
- Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207–1213 CrossRef CAS .
- Y. Umezawa, S. Tsuboyama, H. Takahashi, J. Uzawa and M. Nishio, Bioorg. Med. Chem., 1999, 7, 2021–2026 CrossRef CAS PubMed .
- D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS .
- V. J. Hruby, G. Li, C. Haskell-Luevano and M. Shenderovich, Pept. Sci., 1997, 48, 189–266 CrossRef .
- V. J. Hruby, Acc. Chem. Res., 2001, 34, 389–397 CrossRef CAS PubMed .
- Y. Hamada, S. Kato and T. Shioiri, Tetrahedron Lett., 1985, 26, 3223–3226 CrossRef CAS .
- Y. Hamada, M. Shibata, T. Sugiura, S. Kato and T. Shioiri, J. Org. Chem., 1987, 52, 1252–1255 CrossRef CAS .
- A. J. Phillips, Y. Uto, P. Wipf, M. J. Reno and D. R. Williams, Org. Lett., 2000, 2, 1165–1168 CrossRef CAS PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
- A. Asano, T. Yamada, T. Taniguchi, N. Sasaki, K. Yoza and M. Dio, J. Pept. Sci., 2018, e3120 CrossRef PubMed .
- A. Asano, M. Doi, K. Kobayashi, M. Arimoto, T. Ishida, Y. Katsuya, Y. Mezaki, H. Hasegawa, M. Nakai, M. Sasaki, T. Taniguchi and A. Terashima, Biopolymers, 2001, 58, 295–304 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yukiko Kawanami,
Mao Fujita,
Yuta Yano,
Rio Ide,
Katsuhiko Minoura
*,
Yukiko Kawanami,
Mao Fujita,
Yuta Yano,
Rio Ide,
Katsuhiko Minoura