
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMebendazolium mesylate anhydride salt: rational design based on supramolecular assembly, synthesis, and solid-state characterization†
Eduardo L. Gutiérrez *a,
Agustín A. Godoyb,
Elena V. Brusaub,
Daniel Vegac,
Griselda E. Nardab,
Sebastián Suárezd and
Florencia Di Salvod
*a,
Agustín A. Godoyb,
Elena V. Brusaub,
Daniel Vegac,
Griselda E. Nardab,
Sebastián Suárezd and
Florencia Di Salvod
aINQUISAL-CONICET, Área de Química Física, Facultad de Química, Bioquímica y Farmacia, Universidad Nacional de San Luis, Chacabuco y Pedernera, CP 5700, San Luis, Argentina. E-mail: egutierrez@unsl.edu.ar
bInstituto de Investigaciones en Tecnología Química (INTEQUI), Área de Química General e Inorgánica “Dr G. F. Puelles”, Facultad de Química, Bioquímica y Farmacia, Universidad Nacional de San Luis, Almte. Brown 1500-1402, D5700APA, Chacabuco y Pedernera, CP 5700, San Luis, Argentina
cGerencia de Investigación y Aplicaciones, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica, Av. Gral. Paz 1499, 1650 San Martín, Buenos Aires, Argentina
dINQUIMAE-CONICET y Departamento de Química Inorgánica, Analítica y Química Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, CABA, Argentina
First published on 2nd January 2024
Abstract
The design mebendazole (MBZ) multicomponent systems is important to obtain new materials that incorporate the API (active pharmaceutical ingredient) with better thermal stability, avoiding the interconversion of desmotropes. Interestingly, the presence of water molecules in the mebendazolium mesylate monohydrate prevents the formation of the R22(8) supramolecular synthon, found in all mebendazolium salts with polyatomic counterions. Here, we designed a new mebendazolium mesylate anhydrous salt based on statistical scrutiny of all mebendazole crystal structures identified in the literature and an exhaustive analysis of the conformational and geometrical requirements for the supramolecular assembly. The synthesis of this new salt and its solid-state characterization through single-crystal X-ray diffraction and complementary techniques are presented. As expected, mebendazole recrystallization in methanol with methanesulfonic acid – a Food and Drug Administration accepted coformer – in the absence of water yields a mesylate anhydrous salt with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. This new salt crystallizes in the P212121 (19) space group. The main intermolecular interactions found in the crystal structure are the hydrogen bonds that form a R22(8) supramolecular motif that assembles the ionic pairs. Additional non-classical H-bond, as well as π⋯π and carbonyl⋯cation interactions, contribute to the final stabilization of the crystal packing. This new salt is stable up to 205 °C when it undergoes the endothermic loss of the ester moiety to yield 2-amino-5-benzoylbenzimidazole. Moreover, preliminary dissolution experiments in aqueous 0.1 mol L−1 HCl suggest an apparent solubility of mebendazolium mesylate anhydride 2.67 times higher than that of the preferred for pharmaceutical formulations MBZ form C.
:1 stoichiometry. This new salt crystallizes in the P212121 (19) space group. The main intermolecular interactions found in the crystal structure are the hydrogen bonds that form a R22(8) supramolecular motif that assembles the ionic pairs. Additional non-classical H-bond, as well as π⋯π and carbonyl⋯cation interactions, contribute to the final stabilization of the crystal packing. This new salt is stable up to 205 °C when it undergoes the endothermic loss of the ester moiety to yield 2-amino-5-benzoylbenzimidazole. Moreover, preliminary dissolution experiments in aqueous 0.1 mol L−1 HCl suggest an apparent solubility of mebendazolium mesylate anhydride 2.67 times higher than that of the preferred for pharmaceutical formulations MBZ form C.
1 Introduction
Mebendazole (MBZ, methyl N-(5-benzoylbenzimidazol-2-yl) carbamate, Scheme 1) is a synthetic broad-spectrum benzimidazole-derivative anthelmintic included in the World Health Organization (WHO) Model List of Essential Drugs.1 It was developed by Janssen Pharmaceutica and introduced in the European market in 1972.2 MBZ has been extensively used in human medicine ever since, being administrated orally as tablet formulation or suspension.3–8 In recent years, this active pharmaceutical ingredient (API) has been repurposed and postulated as a possible anticancer drug9–13 and promising advances have been done in this matter.14–18 MBZ is a class II drug of the Biopharmaceutics Classification System19,20 due to its poor solubility either in water or aqueous 0.1 mol L−1 HCl and high permeability.21,22 | ||
| Scheme 1 Structure of MBZ 1H tautomer, the one present in MBZ form C. | ||
Polymorphism of MBZ has long been known, as well as the differences in physicochemical properties and bioavailability arising from it.23,24 So far, three pure MBZ solid forms have been identified and characterized, named A,25 B,26 and C.27 Moreover, since imine–enamine tautomerism28 (Scheme S1†) is responsible for the polymorphism of MBZ, all these three phases should be classified as desmotropes.26,29 MBZ A is the most stable30 and least soluble of the three forms and has shown the same efficacy than placebos in controlling worm infections.31 MBZ B is the least stable30 and the most soluble one,4 demonstrating toxicity during both anthelmintic and anticancer tests. Finally, MBZ C is the preferred polymorph for pharmaceutical formulations since it has shown the best therapeutic results.21,22,31 The dissolution profile and the solubility of MBZ C ensure acceptable efficacy, and concentration values able to cause toxicity are not reached.21,22
The polymorphic interconversion in solid drug formulations is a big problem in the pharmaceutical industry.32 Particularly, the anthelmintic efficacy of MBZ is highly dependent on polymorphism: it was determined that at least 30% w/w of form A in the formulation is enough to suppress the desired pharmacological activity of pharmaceutical materials.21,22,27 Such is the importance of the polymorph choice, that the International Pharmacopoeia in its 11th Edition restricts the suitable polymorphic form to MBZ C for tablets and chewable tablets, emphasizing that the overall manufacturing process should be designed and controlled to minimize its conversion into the more stable polymorph A.33 Polymorphic interconversion has been demonstrated to occur in tablets prepared with form C, which suffer a transformation into products containing form A under the effects of moisture and heat. Due to the same phenomenon, suspensions of polymorph C may also contain some active principle converted to polymorph A, as well.34 This behaviour represents a big issue and brings a great concern since MBZ is extensively used by public health institutions from tropical and subtropical regions around the world.3,5,7
The design and synthesis of multicomponent materials – i.e., salts, cocrystals, solvates, etc., using crystal engineering techniques allows us to modulate the physicochemical and pharmacokinetic properties of the API in the solid, by its supramolecular environment without modifying the intrinsic chemical structure of the drug.35–37 This strategy is widely used to improve the performance, processability, and mechanical properties of APIs – e.g., drug dissolution, absorption and bioavailability, manufacturing reproducibility, stability in storage and transport regarding chemical and polymorph transformations, etc.38–40 Although salt formation is a straightforward approach to obtain multicomponent materials of pharmaceutical compounds,39 supramolecular assembly of polyatomic anions and cations can be rationalized and designed based on crystal engineering. Mebendazole has a very interesting structure in terms of supramolecular chemistry and crystal engineering design. Several salts and cocrystals of MBZ have been synthesized in attempts to modulate the physicochemical properties and pharmacokinetics of this API.41–48 Other strategies to modulate MBZ properties include the inclusion of the API in cyclodextrin complexes49,50 or the load of the drug inside specifically designed metal–organic frameworks.51
Regarding the importance of designing mebendazole multicomponent systems able to avoid polymorphic interconversion, with improved thermal stability and, solubility and dissolution profile similar to those of the therapeutically preferred polymorph MBZ C, we present the rational design, synthesis, and characterization of a novel mebendazolium mesylate anhydride salt. Methanesulfonic acid (MsOH) (Scheme S2,† also known as mesylic acid or methylsulfonic acid, CAS 75752, UNII 12EH9M7279) was chosen for salification of MBZ and further recrystallization as an anhydride mesylate salt. Mesylate anion is used as a counterion of several active ingredients registered in the Food and Drug Administration (FDA) Orange Book 40th Edition, being suitable for capsules, tablets, and suspension formulations among others.52 Methanesulfonic acid exhibits a pKa of −1.9,53 which is appropriate for total proton transfer towards the basic site of the API (pKa of MBZ conjugated acid, the mebendazolium cation, MBZH+, is 4.4).54 According to the empirical pKa rule, a salt should be expected when the difference in pKa between the conjugate acid of the base and the acid (ΔpKa) is greater than approx. 2–3 units.39,55 In this case, ΔpKa is 6.3, suggesting that proton transfer before crystallization is very likely. Methylsulfonate anion (MsO−) has a delocalized negative charge over three oxygen atoms in a tetrahedral geometry around the S atom; this configuration makes this species an appropriate counterion for the formation of the extended heterosynthon presented in the Scheme S3.†
Further characterizations by solid-state techniques were performed for a better understanding of the properties of this new material, as well as for comparing with those of the mebendazolium mesylate monohydrate salt previously reported. Even though this salt exhibits an important increase in solubility and dissolution rate in relation to MBZ A and C, it undergoes an early dehydration process which affects its thermal stability. Previous investigations on this monohydrated salt did not go on to study the nature of the eventual phase transformations that this material undergoes when dehydrated. Based on an extensive structural and statistical analysis of previously reported structures, we proposed the synthesis of an anhydride salt in which both, cation and anion, should assemble in the solid state by a R22(8) supramolecular synthon (Scheme 2).
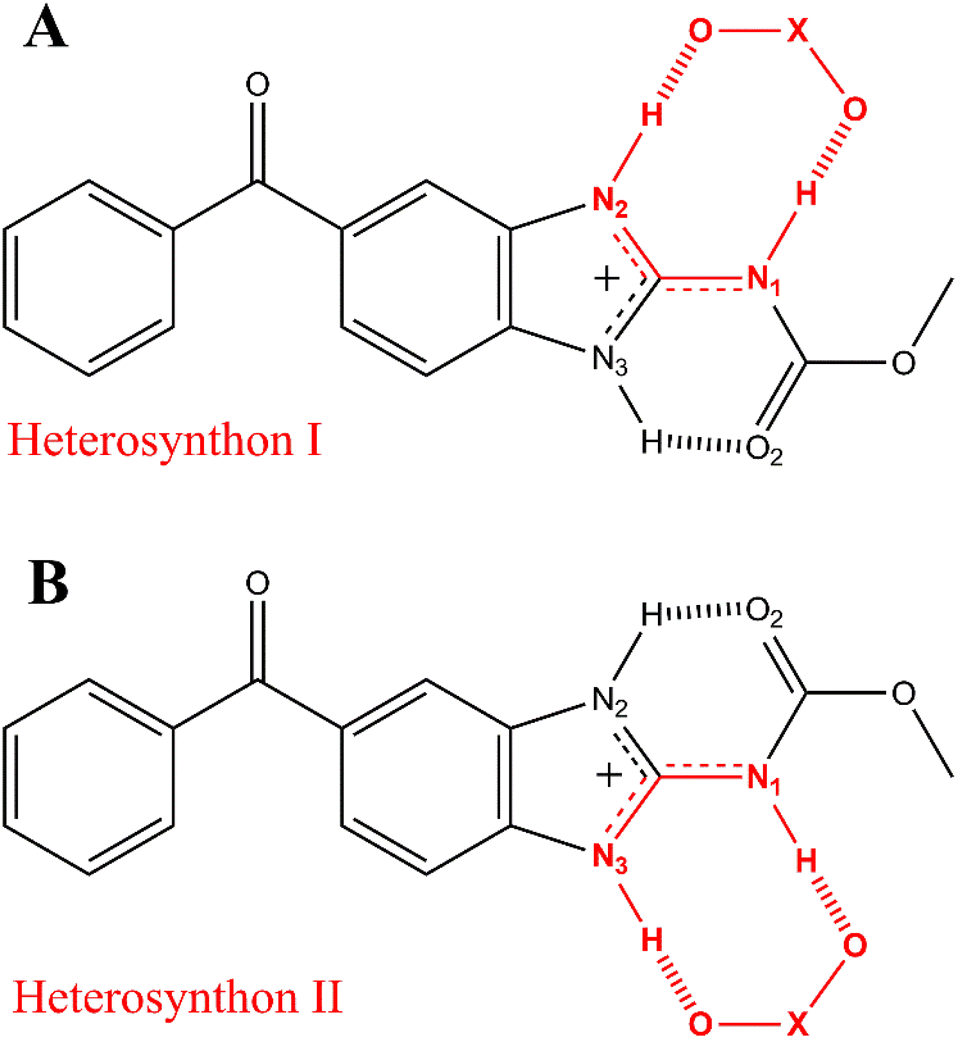 | ||
| Scheme 2 The R22(8) heterosynthons I (A) and II (B) found in all mebendazolium salts with polyatomic counterion (X stands for any atom). | ||
2 Experimental
2.1 Reagents and supramolecular synthesis
Samples of MBZ (295.29 g mol−1) desmotropes A and C were kindly provided by the Laboratorio de Control de Calidad de Medicamentos, UNSL. The purity of these samples was checked using PXRD. Absolute methanol (MeOH, Sintorgan, HPLC grade) was used without any further purification as the solvent for the suspension and dissolution of the drug. Methanesulfonic acid (MsOH, 100.5% w/w, 1.478 g mL−1 (20 °C), 96.11 g mol−1) was obtained from Anedra and used without further purification. n-Hexane was distilled in our laboratory for purification. Ultrapure water (18.3 MΩ cm at 25 °C) was used to prepare aqueous solutions of MsOH for the synthesis of the mesylate monohydrate mebendazolium salt. In the solubility experiments, hydrochloric acid (HCl, 37% w/w, 1.185 g mL−1 (20 °C), 36.46 g mol−1) from Tetrahedron and ultrapure water were used.A sample of MBZ desmotrope C of approx. 30 mg (0.1 mmol) was suspended in 25 mL of methanol at room temperature with constant magnetic stirring (1000 rpm). A volume of 10 μL of pure MsOH (0.13 mmol) was added to complete dissolution of the suspended drug. After 10 min of stirring, the solution was filtered. Finally, the solution contained in a vial was covered with a parafilm foil with small holes, brought to 5 °C and then left to achieve slow evaporation of the solvent. After approx. seven days, the formation of small, colourless prismatic crystals was observed. These crystals were separated by filtration and washed several times with n-hexane. FT-IR spectroscopy and powder X-ray diffraction were performed to check the identity and purity of the solids. Then the crystalline material was stored at room temperature for further characterization.
Mebendazolium mesylate monohydrate salt was obtained in an analogous procedure, using 100 μL of an aqueous solution 1 mol L−1 of MsOH (0.1 mmol) for the dissolution and recrystallization of a suspended sample of MBZ C (30 mg, 0.1 mmol) in methanol. The same steps previously described were followed after the crystallization of the solid material for its separation and characterization.
2.2 Characterization of the bulk material
Powder X-ray diffraction (PXRD) patterns in Section 3.2 were obtained on a Rigaku Ultima IV diffractometer using Cu Kα radiation, in step mode, between 3° and 50° in 2θ with a scanning rate of 2 s per step1 and a step of 0.02°. The powder diffraction patterns were collected and processed with SmartLab Studio-II software, associated with the diffractometer.Fourier transform infrared spectra were recorded on a Nicolet Protégé 460 spectrophotometer equipped with a CsI beamsplitter, in the 4000–400 cm−1 range with 64 scans and a spectral resolution of 2 cm−1, using the KBr pellet technique. Measurements were collected and processed with the “Omnic” software associated with the spectrometer. Variable Temperature Fourier Transform Infrared (VT-FTIR) spectra were recorded in the 25–250 °C, using an accessory developed by Seasing SRL consisting in a variable-temperature IR cell provided with KBr windows operating under high vacuum.
2.3 Structural determination by single-crystal X-ray diffraction
Suitable single crystals were mounted over a mylar loop using paratone oil and data were collected at room temperature on an Oxford Diffraction Xcalibur Gemini Eos diffractometer using graphite-monochromated Cu-Kα radiation (λ = 1.54184 Å) at T = 298.5 K during data collection. CrysAlisPro, from Oxford Diffraction, was used to collect initial frames for the determination of the unit cell, and subsequently, the program was used to plan a data collection. After collection, data reduction was carried out in the CrysAlisPro suite, and multiscan absorption correction was carried out.56 The structures were solved by the intrinsic phasing method with ShelXT57 and refined using a full-matrix least-squares method on an F2 employing ShelXL package, using Olex2 software.58 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were positioned at geometrically calculated positions with thermal parameters derived from the parent atoms.The crystallographic information file (CIF) was deposited in the Cambridge Structural Database59 under the code CCDC 2236578. Geometrical calculations and molecular representations, pictures and tables were performed by and/or generated with MERCURY 4.3.1 60 and PLATON61 software programs.
Hirshfeld surface analysis was performed using the software CrystalExplorer17 62,63 with the CIF as the input file. Hirshfeld surface for MBZH+ was mapped with dnorm function over the range of −0.35 to 0.25 (color code: from blue – distances longer than sum of van der Waals radii – through white to red – distances shorter than sum of van der Waals radii). Hirshfeld surface was also mapped with shape index function, over the ranges of −1.0 to 1.0. 2D-fingerprint plot was obtained for de and di between 0.4 and 2.6.
2.4 Thermal analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were performed with a Shimadzu TGA-51 thermogravimetric analyser and a Shimadzu DSC-60 differential scanning calorimeter respectively, using a platinum pan, flowing N2 at 10 mL min−1, and a heating rate of 10 °C min−1 from room temperature (RT) to 600 °C (DSC) and to 900 °C (TGA). TGA and DSC curves were collected and processed with “TA60” software, associated to the instruments.The thermal evolution of the monohydrated mesylate salt (MBZH·MsO·H2O) was studied by keeping a powder sample of this material in a heating stove at 100 °C for 60 min in air. The heating stove was previously brought and stabilized at 100 °C. The same procedure was followed with a powder sample of the anhydrous salt (MBZH·MsO). The powder X-ray diffraction patterns of both samples, before and after the heating process were compared in order to examine the eventual transformations of the solid materials due to dehydration, phase transitions, amorphization, etc. These powder X-ray diffraction patterns were obtained on a Bruker D8 Advance diffractometer with Bragg–Brentano geometry using Cu Kα radiation between 5° and 40° in 2θ with a scanning rate of 300 s per step and a step of 0.026°.
2.5 Stability and solubility studies
Powder patterns after being 30 days exposed to the atmosphere (Section 3.6) were collected on a PANalytical Empyrean diffractometer equipped with a 4 kW sealed tube Cu Kα X-ray radiation (generator power settings: 60 kV and 100 mA) and a PIXcel3D area detector using parallel beam geometry (1/2–1–8 mm slits, 15 mm incident mask). Samples were packed on a silicon monocrystal sample holder that was then placed on the sample holder attachment. The data were collected over the range 5° to 90° in 2θ with a scanning speed of 23 s per step with 0.026° step.Equilibrium solubility of MBZH·MsO at room temperature was evaluated and compared with those of mebendazole desmotrope C in 0.1 mol L−1 hydrochloric acid aqueous solution, using the saturation shake flask method.64 Saturated solutions of the two solids were prepared by stirring an excess amount of the solids (approx. 75 mg) in 4 mL of the dissolution medium for a period of 48 h (25 °C and 200 rpm) in an orbital shaker from Ferca provided with a Novus N321 temperature controller. After sedimentation, the solutions were filtered through a 0.22 μm/13 mm polytetrafluoroethylene (PTFE) syringe filter (Clarify). The identity of the solid sediments was checked by PXRD. UV-vis spectroscopy was employed to measure the MBZ concentration in the saturated solutions, applying a direct calibration method, using the absorbance maxima at 287 nm as a parameter proportional to the concentration. The saturated solutions were diluted in the dissolution medium to ensure that we work on the method linear range of response. Solubility measurements were made over triplicate. UV-visible absorbance measurements were carried out on an Agilent 8454 UV-vis spectrophotometer (Agilent Technologies), and the results were collected and processed with “8453 UV Visible Chemstation Rev. A.10.01” software associated with the instrument.
3 Results and discussion
3.1 Rational design
It is known that the protonation of MBZ prevents the self-assembly of the API molecules by either of the three homosynthons observed in MBZ A, MBZ B, or MBZ C (Tables S1, S2, and S3† respectively). Moreover, protonation of the API also inhibits the tautomeric equilibrium responsible for polymorphic conversion.25,27,48 In all mebendazolium salts reported in the literature, either with polyatomic carboxylates or other oxyanions, one of the two supramolecular synthons shown in Scheme 2 is always found in their structures.42,44,45,47,48 The formation of one or other synthon depends on the conformation around the bond C3–N1. In one conformation (Scheme 2A), the carbamic carbonyl group is syn to the N3 (heterosynthon I), while in the other conformation (Scheme 2B), this group is syn to the N2 (heterosynthon II). Heterosynthon I is found in all mebendazolium salts with non-carboxylates counterions (nitrate – CCDC 1585622,47 perchlorate – CCDC 1995782,48 and methylsulfate – CCDC 1995783)48 and in two salts with carboxylate counterions (methanoate – CCDC 929536 (ref. 45) and methyloxalate – CCDC 866968).44 Heterosynthon II is found in the structure of the two remaining salts, mebendazolium trifluoracetate – CCDC 929535,45 and maleate – CCDC 866969.44The formation of these R22(8) supramolecular synthons, driven by moderate hydrogen bonding,65,66 presents some conformational and geometrical requirements. For the heterosynthon I (Scheme 2A), not only the atoms N1, C3, and N2 need to be in the same plane, but also, the H atoms on N2 and N1. This conformation (Scheme 3B) is highly favoured by protonation of the API and depends on two factors. On the one hand, the sp2 character of N1 restricts the rotation through the bond C3–N1. On the other hand, the formation of an intramolecular hydrogen bond between N3 and O2 ensures the fixing of the bond N1–H in the expected orientation.
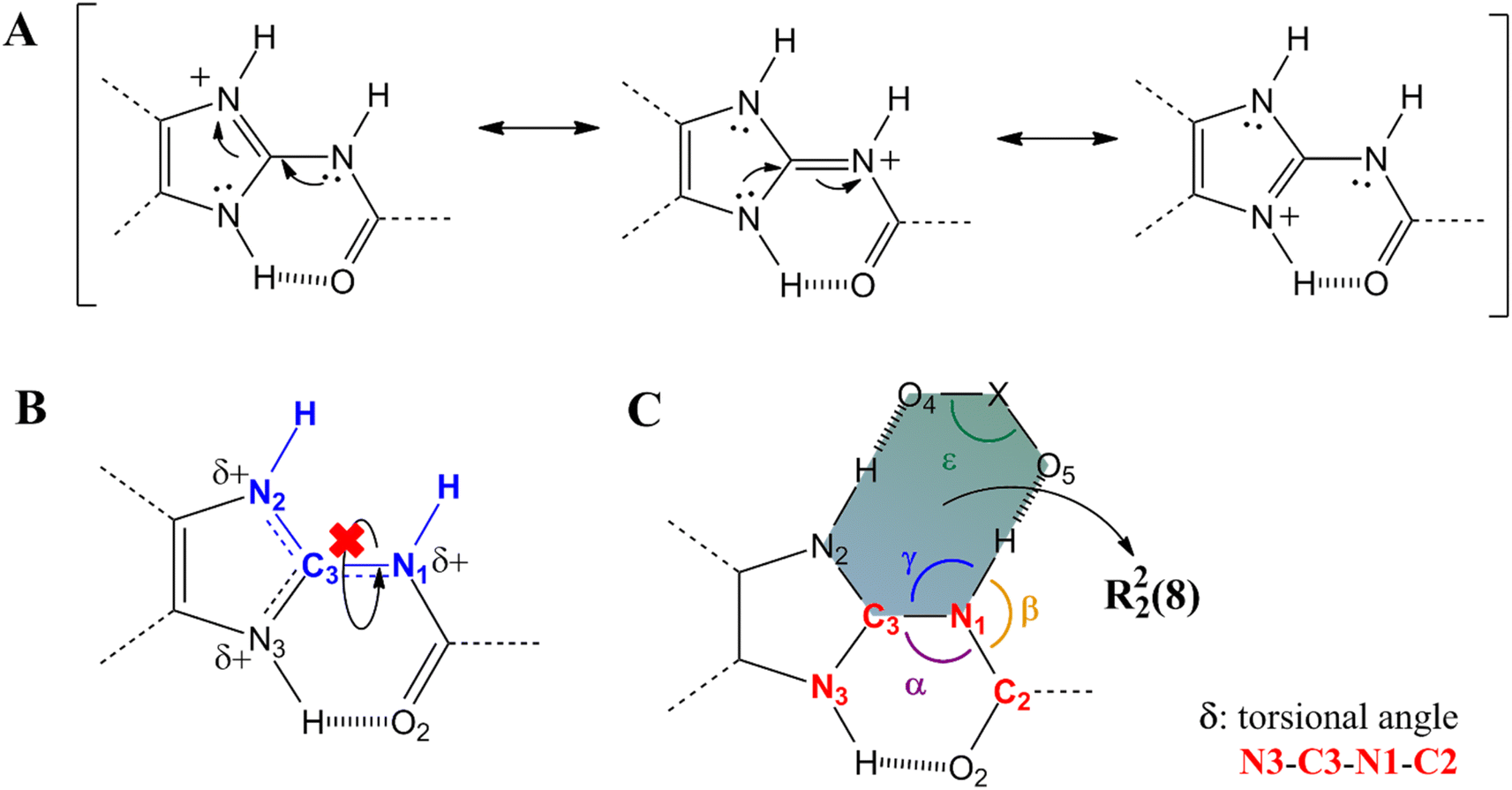 | ||
| Scheme 3 (A) Detail of resonances structures of mebendazolium cation (MBZH+). (B) Conformation around the partial double bond C3–N1 in heterosynthon I. (C) Representative conformational parameters in heterosynthon I. | ||
The sp2 character of N1 can be evidenced by the length of the bond C3–N1. The positive charge on the benzimidazole ring of mebendazolium cation causes the displacement of electron density from N1 towards the ring (Scheme 3A), increasing the double bond character of the bond N1–C3, and decreasing its length. This bond is 1.37(1) Å in MBZ C, while it is shorter in all the reported salts (Table S4†). Further evidence on the hybridization of N1 is provided by the values of the angles α, β, and γ (Scheme 3C), which should be about 120° for an idealistic sp2 atom. Indeed, these angles are close to this value in all the aforementioned compounds. The coplanarity of the atoms H–N2–C3–N1–H is also highly dependent on the torsional angle δ between N3–C3–N1–C2, that should be about 0° for a planar conformation. We found that for all the studied cases, this torsional angle is, in fact, close to 0°. It is also worth noting that this synthon is formed despite the geometry of the counterion (angle ε in Scheme 3A is close to 120° for trigonal planar geometry, and close to 109.5° for tetrahedral geometry). The values of these parameters in the MBZ ionic structures previously reported in the literature, are gathered in Table S4.† The preceding analysis is analogue for the heterosynthon II.
In the crystal structures of the mebendazolium nitrate, perchlorate, and methylsulfate salts, the presence of multiple oxygen atoms in the anions allows the formation of a bifurcated hydrogen bond involving the bond N3–H. As N3 is also implicated as a proton donor in the intramolecular hydrogen bond N3–H⋯O2, the intermolecular hydrogen bond N3–H⋯O6 (shown in red in Scheme S3†) is usually longer, and therefore weaker, than those involved in the R22(8) motif (Table S5†). Even when the R22(8) supramolecular synthon is not found in the structure of the mesylate monohydrate mebendazolium salt (CCDC 934635),46 we did find that the H-bond N3–H⋯O6 indeed is developed, connecting adjacent ionic pairs. With these hydrogen bonds as the main intermolecular interactions of these structures, mebendazolium cations, and their respective anions, assemble in the crystalline state in infinite chains of intercalated ions. Additional non-classical hydrogen bonds C–H⋯O could contribute to the stabilization of these chains, as shown in orange in Scheme S3.†
This empirical evidence suggests that through the use of the appropriate counterion, not only it is possible to predict the formation of the described heterosynthons in the solid-state, but also tuning the development of intermolecular interactions apart from those involved in the R22(8) motif and therefore, designing long-range arrangements of ionic pairs in infinite chains, as we present in Scheme S3.†
As mentioned before, the only exception found in the literature for the formation of the heterosynthons I or II, is the mebendazolium mesylate monohydrate salt.46 Mesylate (MsO−) is the common name for methanesulfonic acid conjugated base, the methanesulfonate anion. In this structure, the presence of water molecules prevents the formation of either of those supramolecular motifs, as shown in Fig. S1.† One water molecule interacts as a hydrogen bond donor (D) with the mesylate anion (acceptor, A) (O4⋯H–O7, A⋯H: 1.82 Å), while another water molecule acts as hydrogen bond acceptor, establishing and intermolecular interaction with the mebendazolium cation through the N2–H bond (O7⋯H–N2: 1.75 Å).
3.2 Crystal habit, identity and purity of the new material
The supramolecular synthesis described in the previous section yielded a solid material that crystallizes as small elongated colourless prismatic crystals (Fig. S2†). Preliminary evidence of a presumably new solid form, prior to the single-crystal X-ray diffraction experiments, was provided by its vibrational spectrum and powder X-ray diffraction pattern. Likewise, the samples of the mebendazolium mesylate monohydrate salt were characterized by its FT-IR spectrum and PXRD pattern.The position of the band corresponding to the stretching mode of the carbamic carbonyl group, in the IR or Raman spectra, is useful to identify the crystalline forms of MBZ.3 This band appears at 1730 cm−1, 1715 cm−1 and 1697 cm−1 for polymorphs A, C and B, in their FTIR spectra respectively,34 due to the different intermolecular interactions in the crystal packing. Similarly, this band appears at 1757 cm−1 in the FTIR spectrum of the mebendazolium mesylate monohydrate. Moreover, the hydrated salt presents in the high-frequency region two well defined bands at 3477 cm−1 and 3371 cm−1 assigned to the antisymmetric and symmetric stretching modes (νO–H) of crystallization water molecules. These values are in agreement with those previously reported.46 The FTIR spectrum of the new material also exhibits the stretching mode of the carbamic group at 1757 cm−1. The shift of this signal to higher frequencies with respect to the MBZ polymorphs provides evidence about the obtaining of a material different from those polymorphs, as this phenomenon was observed in previously reported salts.47,48 The absence of the two bands assigned to water confirms the anhydrous nature of the product. Other important bands arising in the organic moiety are: νN–H (amide), 3211 cm−1; νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (benzoyl), 1655 cm−1; and νC–O–C (methyl ester), 1371 cm−1. The identification of the characteristic IR signals of the mesylate anion, accounts for its incorporation in the crystal structure of the new material. The bands originated in the antisymmetric and symmetric stretching modes (νS–O) of MsO− anion were found at 1167 cm−1 and 1047 cm−1.67 These bands are slightly shifted from those found in the monohydrate salt (1153 cm−1, 1041 cm−1).46 We also observed the bands corresponding to the stretching modes of MsOH (1319 cm−1 and 1090 cm−1),67 most likely due to impurities of remaining coformer in the sample. The bands derived from the stretching modes of the methyl group of the anion appear at 2862 cm−1 and 2816 cm−1, while the stretching modes of the methyl group of the carbamate moiety of MBZ are at 2960 cm−1 and 2922 cm−1. FTIR spectra of both monohydrate and anhydrous salts are shown in Fig. S3.†
O (benzoyl), 1655 cm−1; and νC–O–C (methyl ester), 1371 cm−1. The identification of the characteristic IR signals of the mesylate anion, accounts for its incorporation in the crystal structure of the new material. The bands originated in the antisymmetric and symmetric stretching modes (νS–O) of MsO− anion were found at 1167 cm−1 and 1047 cm−1.67 These bands are slightly shifted from those found in the monohydrate salt (1153 cm−1, 1041 cm−1).46 We also observed the bands corresponding to the stretching modes of MsOH (1319 cm−1 and 1090 cm−1),67 most likely due to impurities of remaining coformer in the sample. The bands derived from the stretching modes of the methyl group of the anion appear at 2862 cm−1 and 2816 cm−1, while the stretching modes of the methyl group of the carbamate moiety of MBZ are at 2960 cm−1 and 2922 cm−1. FTIR spectra of both monohydrate and anhydrous salts are shown in Fig. S3.†
We ruled out the crystallization of any of pure MBZ polymorphs, neither as the main phase nor as impurities, by comparing the calculated XRD patterns of MBZ A, B and C with the experimental XRD diffractogram obtained for the new material. Since none of the characteristic peaks of MBZ pure phases were observed in the experimental pattern of the new material, we discarded the presence of the pure API in any extent (Fig. 1). In addition, the crystallization of the mesylate monohydrate salt along with the new material was also discarded through the comparison of the corresponding XRD patterns and the confirmation of the absences of their characteristic peaks (Fig. S4†). This evidence supported the hypothesis of the obtention of a new solid product with high purity and without the incorporation of water molecules in the crystal structure from the ambient atmosphere, during or after the crystallization process.
 | ||
| Fig. 1 Experimental powder X-ray diffraction pattern MBZH·MsO, compared with those calculated from single-crystal XRD data. | ||
The crystalline purity of the mebendazolium mesylate monohydrate salt samples synthesized for this work, was also evaluated by comparing its PXRD pattern with that simulated form the reported structural data46 and also, with the experimental and calculated ones of the anhydrous salt here reported (Fig. S4†).
3.3 Description of the crystal structure
The resolution of the structure confirmed the crystallization of a new mebendazolium mesylate anhydride salt, MBZH·MsO, with 1:1 stoichiometry. This salt crystallizes in the orthorhombic P212121 (19) space group. Crystal data, data collection and structure refinement parameters are shown in Table 1. An ORTEP type view of the asymmetric unit of MBZH·MsO is shown in Fig. 2A, displaying the 50% probability ellipsoids and the atoms labeling according to the nomenclature used by Martins et al.27 The unit cell contains four asymmetric units, as shown in Fig. 2B. Hydrogen-bonding geometry parameters are shown in Table 2.
| Identification code | Mebendazolium mesylate (MBZH·MsO) |
|---|---|
| Empirical formula | C17H17N3O6S (C16H14O3+ CH3O3S−) |
| Formula weight | 391.39 |
| Temperature/K | 298 |
| Crystal system | Orthorhombic |
| Space group | P212121 (19) |
| a/Å | 7.2177(3) |
| b/Å | 9.2009(4) |
| c/Å | 27.0007(17) |
| α/° | 90 |
| β/° | 90 |
| γ/° | 90 |
| Volume/Å3 | 1793.09(16) |
| Z | 4 |
| ρcalc/g cm−3 | 1.450 |
| μ/mm−1 | 1.975 |
| F(000) | 816.0 |
| Crystal size/mm3 | 0.3 × 0.2 × 0.1 |
| Radiation | CuKα (λ = 1.54184 Å) |
| 2Θ range for data collection/° | 10.156 to 147.262 |
| Index ranges | −8 ≤ h ≤ 8, −10 ≤ k ≤ 11, −33 ≤ l ≤ 33 |
| Reflections collected | 6778 |
| Independent reflections | 3311 [Rint = 0.0440, Rsigma = 0.0547] |
| Data/restraints/parameters | 3311/0/246 |
| Goodness-of-fit on F2 | 1.123 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0628, wR2 = 0.1717 |
| Final R indexes [all data] | R1 = 0.0773, wR2 = 0.1814 |
| Largest diff. peak/hole/e Å−3 | 0.29/–0.41 |
| Flack parameter | 0.12(4) |
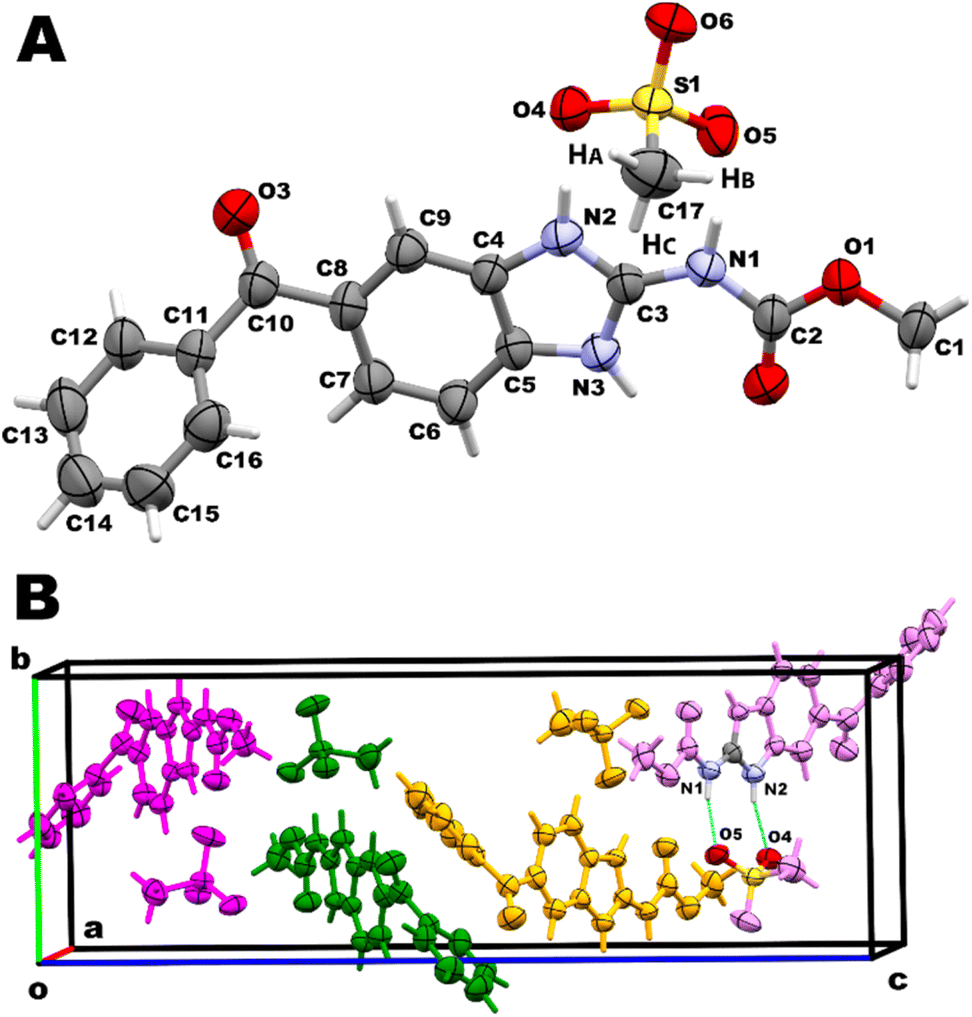 | ||
| Fig. 2 (A) View of MBZH·MsO asymmetric unit, showing the atoms labelling and the 50% probability ellipsoids for non-hydrogen atoms. The hydrogen atoms are shown as sticks of arbitrary radii (colour code. C: grey; H: white; O: red; N: blue; S: yellow). (B) View of the unit cell of MBZH·MsO containing four asymmetric units MBZH+/MsO−. | ||
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | D–H⋯A angle/° |
|---|---|---|---|---|
| N2–H⋯O4 | 0.86 | 1.96 | 2.787(8) | 160 |
| N1–H⋯O5 | 0.86 | 1.87 | 2.714(8) | 167 |
| N3–H⋯O2 (intramolecular) | 0.86 | 2.21 | 2.718(8) | 118 |
| N3–H⋯O6 | 0.86 | 2.00 | 2.714(9) | 139 |
| C6–H⋯O4 | 0.93 | 2.66 | 3.574(10) | 169 |
| C6–H⋯O6 | 0.93 | 2.69 | 3.262(11) | 120 |
| C17–HA⋯O3 | 0.96 | 2.51 | 3.468(13) | 174 |
| D: donor. A: acceptor | ||||
Bond length changes in the imidazole ring and the carbamate moiety confirmed that protonation of the API indeed occurs as expected (Scheme S4†). In the unprotonated form of MBZ, as found in form C, both bonds C3–N2 (1.304(11) Å) and C3–N3 (1.349(10) Å) are significantly different. The bond C3–N2 exhibits a higher double bond character, as shown in the canonical structure in Scheme 3. However, in the mebendazolium cation present in MBZH·MsO, resonance effects are more important since protonation on the molecule leads to the decreasing in the bond length difference [C3–N2 (1.319(9) Å) and C3–N3 (1.336(10) Å)]. Further evidence of the protonation of the API is given by the bond length C3–N1. While this bond is 1.372(11) Å in the neutral molecule, it is shortened in MBZH+ (1.348(10) Å) due to a displacement of electron density from N1 towards the positively charged imidazole ring (Scheme 3A).
The intramolecular resonance-assisted hydrogen bond between O2 and N3 (2.21 Å) found in other MBZ structures, is also present here, in that way that the carbamate moiety is coplanar with the benzimidazole ring. The highest deviation from the least-squares plane passing through these 15 atoms (including C10) is −0.041(8) Å for C8 (root mean square deviation (r.m.s.d.) of fitted atoms is 0.007 Å). The planar conformation around N1 is also observed in the values of the angle C2–N1–C3 (124.3(7)°, close to 120°) and the torsional N3–C3–N1–C2 (2(1)°, close to 0°). Even when that intramolecular H-bond exhibits an angle N2–H⋯O2 of 118°, angles slightly smaller than 120° for this type of intramolecular interactions could be expected as they were experimentally observed in several crystal structures reported,68 including several MBZ structures.27,44–46
Additionally, we observed that the benzene ring from benzoyl group is not coplanar with the benzimidazole/carbamate moiety. There is a dihedral angle of 57.7(4)° between the least-squares planes passing through the benzene (r.m.s.d.: 0.010 Å) and the benzimidazole (r.m.s.d.: 0.007 Å) rings respectively. The carbonyl group C10O3 is not conjugated with either of both aromatic rings, as can be assessed from the torsional angles involving the C9–C8–C10–O3 and C12–C11–C10–O3 atoms, whose values are 25.8(12)° and 39.3(12)° respectively. These values deviate significantly from the expected value for a perfectly conjugated π-system of about 0° (Fig. 3C).
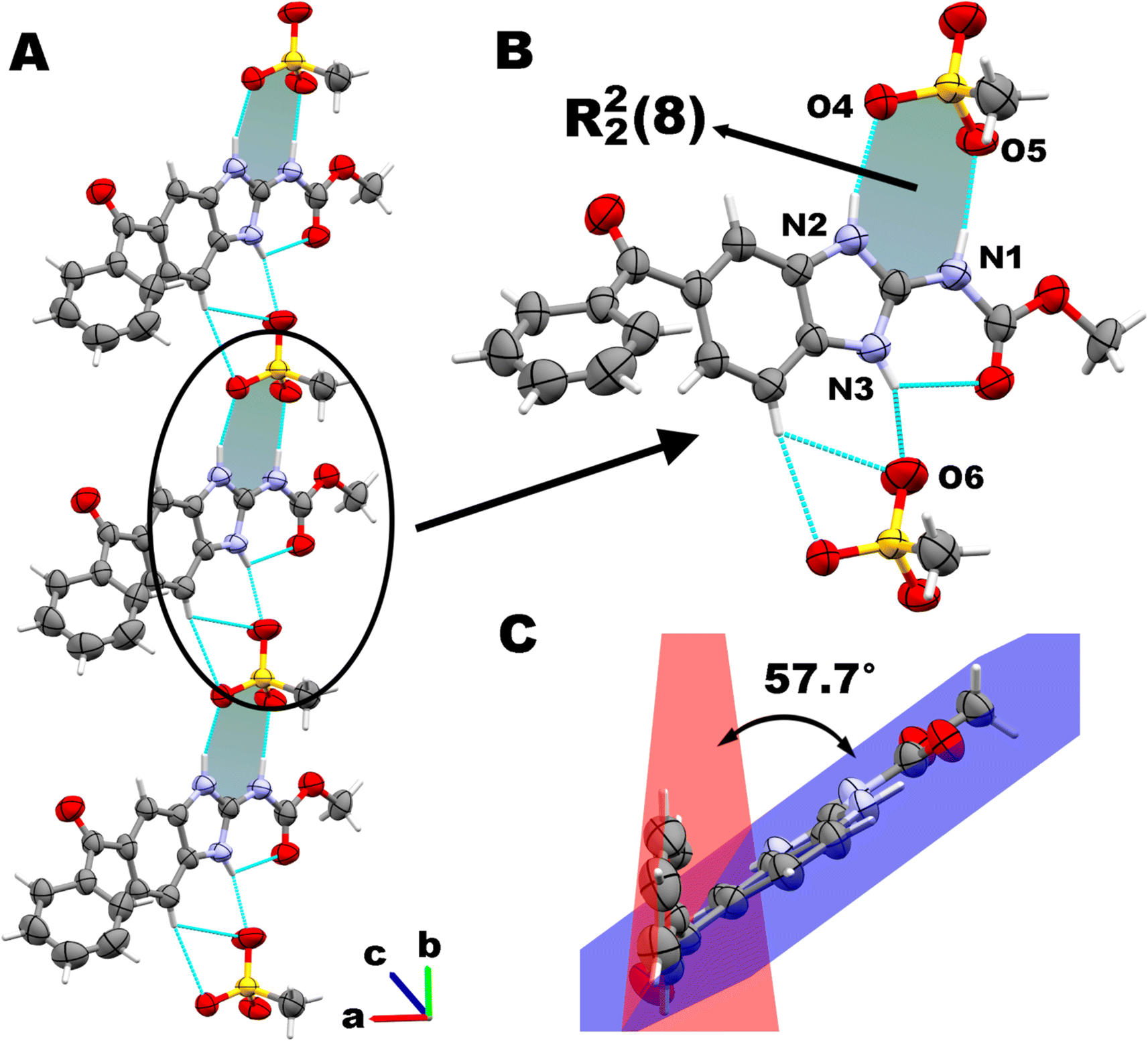 | ||
| Fig. 3 A. Ionic pairs MBZH+/MsO− forming chains assembled by intermolecular H-bonds along the b axis. B. Supramolecular motif R22(8) formed by one MBZH+ cation and one MsO− anion. C. Conformation of the MBZH+ cation in the crystal structure. | ||
According to our assumption, after proton transfer, MBZH+ cations and MsO− anions assemble in the solid state by a R22(8) heterosynthon type I (Scheme 2A) stabilized by moderate hydrogen bonds, according to Jeffrey's classification.65 N2 and N1 act as proton donors, while O4 and O5 are acceptors, establishing two hydrogen bonds schemes: N2–H⋯O4 (1.96 Å) and N1–H⋯O5 (1.87 Å) (Fig. 3B). This supramolecular motif is not planar according to the dihedral angle of 38.0(10)° between the planes determined by the atoms N2, C2, and N1, and O4, S1, and O5, respectively.
Each MBZH+ is involved in a third intermolecular hydrogen bond through N3, which connects the ionic pairs MBZH+/MsO− forming infinite chains along the b axis (N3–H⋯O6, 2.00 Å). This agrees with our additional expectation regarding the arrangement of ionic pairs in chains by a third hydrogen bond. The connection of ionic pairs along this direction is reinforced by two non-classical weaker hydrogen bonds: C6–H⋯O6, 2.69 Å; and C6–H⋯O4, 2.66 Å. These chains along the b axis are shown in Fig. 3A. Consecutive parallel chains in the bc plane are connected by a non-classical H-bond C17–HA⋯O3 (2.51 Å).
Other important interactions lying in the ac plane contribute to the stabilization of the crystal packing. Chains related by a 21 screw axis are connected by a close contact interaction between the C1 atom from one MBZH+ cation and the O5 atom from a MsO− anion belonging from the adjacent chain (3.04(1) Å). Additional interactions in this plane are described in the next section.
3.4 Hirshfeld surface analysis
Hirshfeld surfaces offer a very suitable approach for exploring the intermolecular interactions in the crystalline state, as an alternative for the analysis based on internuclear distances and angles and identifications of close contacts for discrete atoms. Particularly, we have employed the dnorm function to map the Hirshfeld surface69 around the mebendazolium cation. As shown in Fig. 4, it is observed that for the mebendazolium cation as found in the mesylate anhydride salt, the regions where the interatomic distances are shorter than the sum of the van der Waals radii are highly located on the imidazole/carbamate moiety. These regions appear as large red spots on the Hirshfeld surface and correspond to the H-bonds described before. The non-classical H-bonds C6–H⋯O4 and C17–HA⋯O3, and the close contact C1⋯O5 are also identified and plotted as faint smaller red spots (Fig. 4). | ||
| Fig. 4 Mebendazolium cation mapped with the dnorm function on the Hirshfeld surface. | ||
The fingerprint plot also shows that the dominant interactions are the H-bond in which the N atoms act as donors (21%). These interactions correspond to the upper spike (ellipsoid A) in the plot of Fig. S5B.† The lower and smaller spike (ellipsoid B in Fig. S5B†) is associated with the non-classical H-bond C17–HA⋯O3 in which the cation MBZH+ acts as acceptor (9%). Overall, O⋯H contacts account for the 30% of the close contacts on the Hirshfeld surface. The relatively higher density of spots in the centre of the fingerprint plot corresponds to the planar stacking arrangement of MBZH+ cations (ellipsoid C in Fig. S5B†). The relative contributions to the Hirshfeld surface area for the relevant close intermolecular contacts for MBZH·MsO are shown in Fig. S5A.†
In addition to the main intermolecular interactions Hirshfeld surface analysis also revealed two other types of contacts responsible for the stabilization of the crystal packing, i.e., π⋯π and carbonyl⋯π interactions in the ac plane. While these contacts are shown as faint small red spots on the Hirshfeld surface of the MBZH+ cation, they are better visualized through the complementarity of colours orange-to-blue when we mapped the Hirshfeld surface with the shape index function (Fig. S6†). These interactions are responsible for the particular conformation around the benzoyl moiety of the cation described in the previous section. The T-shaped or edge-to-face π⋯π interaction (according to the Hunter and Sanders model)69 is found between the benzene and the benzimidazole rings with an angle of 57.7(4)° while the distance between the centroid of the benzene ring from the benzimidazole moiety and the H atom of C12, is 3.071 Å. A carbonyl⋯π electrostatic interaction (or lone pair⋯π interaction)70 is found between O3 and the positive-charged imidazolium ring (the distance between O3 and the centroid of the imidazole ring is 3.339 Å), while the close contact is disclosed between the O3 and the positive-charged C3 (3.16(1) Å) (Fig. S7†).
3.5 Thermal studies
As expected, the mebendazolium mesylate anhydrous salt presents improved thermal stability compared with its monohydrate salt. Thermal analysis (TGA and DSC curves are shown in Fig. 5) indicate that MBZH·MsO is stable up to 205 °C, revealing that the new salt shows a higher thermal stability than the MBZH+ methylsulfate (160 °C) and similar to the MBZH+ perchlorate (210 °C).48 It is noteworthy to point out that, even though the better thermal stability of the perchlorate salt in comparison to the other reported salts, the anion is not a pharmaceutical-approved counterion, unlike the mesylate.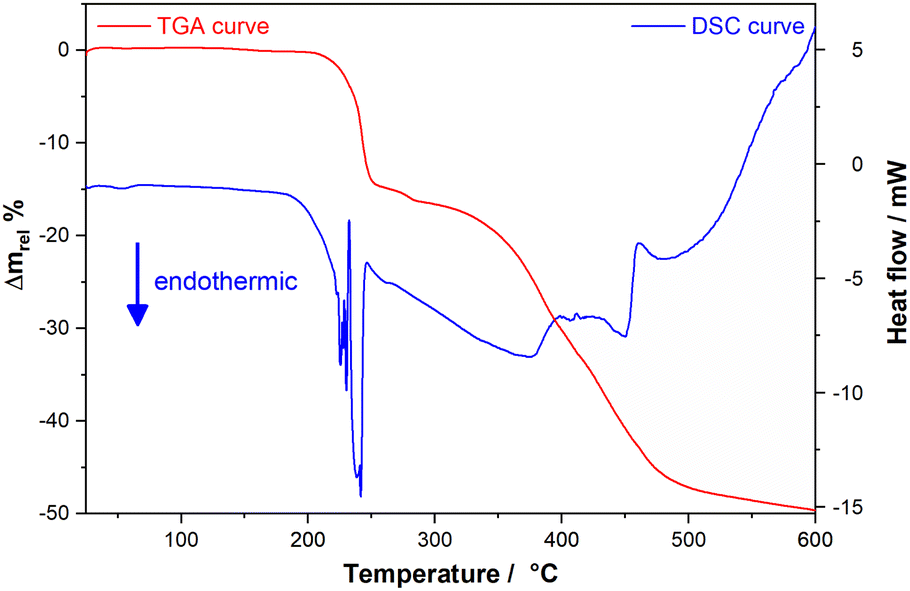 | ||
| Fig. 5 Thermal analysis curves for MBZH·MsO shown in the room temperature – 600 °C range. | ||
DSC curve shows two consecutive endothermic events (temp. onset 205 °C) associated with a relative mass loss of 14.61%, as demonstrated by the TGA experiment. The loss of the methyl ester moiety in the MBZH+ cation implies a theoretical mass loss of 14.83%. Even though for MBZ71 and other mebendazolium salts,48 the first degradation process also implies the loss of the methyl ester moiety, the double DSC signal observed in this case, can be attributed to the different reaction atmosphere since TGA and DSC experiments were carried out under N2 atmosphere instead of air.
To obtain more evidence of this process, we conducted a variable-temperature FT-IR experiment. The progressive reduction in the intensity of the signal at 1757 cm−1 derived from the carbamic carbonyl group up to almost completely disappearing at the highest temperature (approx. 250 °C, Fig. 6A), suggests that, in fact, the methyl ester moiety is being lost during the process to give rise to the 2-amino-5-benzoylbenzimidazolium cation. The disappearance of the bands assigned to the methyl group of the carbamate moiety (2960 cm−1 and 2922 cm−1) and to the C–O–C stretching mode (1371 cm−1, Fig. 6C), and the shift to higher frequencies of the νN–H band (from 3211 cm−1 to 3368 cm−1) also support this proposal. The retention (although blue-shifted to 1680 cm−1) of the benzoyl carbonyl band in the FT-IR spectra recorded at the highest temperature reached (Fig. 6B), suggests that the benzophenone moiety is not eliminated in this stage of the degradation process.
 | ||
| Fig. 6 Temperature-variable FT-IR spectra of MBZH·MsO. | ||
The bands assigned to the MsO− anion (1167 cm−1 and 1047 cm−1) did not show the same behaviour of the carbamate carbonyl stretching band, as the former began to decrease in intensity at higher temperatures and in a slower fashion, as an indication that the elimination of the anion takes place in the following stage (Fig. 6E) posterior to the break of the R22(8) supramolecular motif. We also observed the progressive disappearance as temperature increased of the bands assigned to the remaining MsOH (1319 cm−1 and 1090 cm−1) as impurity in the sample (Fig. 6D and E).
The second event in the TGA curve begins at approx. 260 °C and goes until approx. 575 °C, representing a mass loss of 34.31% (overall weight loss: 49.14%). This event is associated to a very broad endothermic DSC signal followed by an exothermic and less intense one. This mass loss is attributed to the elimination of the MsOH and the breakdown of the benzimidazolium cation to yield probably, benzophenone (theoretical mass loss of these two events: 38.61%). No stability plateau is observed between these two events in the TGA curve, which made difficult to assigned a temperature value for the end of these events, even analysing the TGA first derivate curve. Increasing temperature beyond 575 °C and up to 900 °C in the TGA experiment led the sample to a final relative mass of 35%. In the absence of O2, the final products of degradation are rather carbonized than being eliminated as CO2. The proposed degradation mechanism is shown in Scheme S5.†
In addition, thermal evolution of the sample of MBZH·MsO·H2O kept at 100 °C for 60 min was also investigated. We found that the material experiments a dehydration and an almost completely transformation to the crystalline anhydrous phase, as shown in the experimental powder diffraction patterns in Fig. S8.† The powder diffraction pattern of the sample after the experiment matches perfectly with that of MBZH·MsO kept in the same conditions. The peak of very little intensity observed at 6.10° can be assigned to the small amount of the monohydrated phase remaining in the sample (this signal corresponds to the most intense according to the experiments shown in Fig. 1 and S8† for the monohydrated salt).
3.6 Stability and solubility studies
A freshly synthesized and characterized powder sample of MBZH·MsO was kept in a vial in contact with the atmosphere in a 24–7 temperature-controlled laboratory at 22 °C with humidity maintained approximately constant during 30 days, to evaluate the eventual transformations that the material could undergo under these conditions. The diffraction pattern of the sample recorded after the experiment shows that the anhydrous material is very stable, and the incorporation of water into its crystal structure to yield the monohydrate salt after that period, could be at most, a minor process since we only observed a peak of very lower intensity at 6.08° that, as mentioned before, could be assigned to the most intense diffraction peak of the monohydrated material (Fig. S9†).We also studied the solubility behaviour of mebendazolium mesylate anhydride in 0.1 mol L−1 hydrochloric acid aqueous solution. A modification of the USP saturation shake flask method was followed: we did not employ sodium lauryl sulphate since previous reports stated that not using surfactants is the most discriminating way to distinguish between the solubility differences of MBZ solid forms.21 MBZ UV-vis spectra in 0.1 mol L−1 HCl (aq.) as well as the calibration curve are shown in Fig. S10 and S11.† We determined a solubility of 0.124 (±0.005) mg mL−1 for MBZ C in this medium after 48 h. This value is significantly higher (α = 0.05) than that of 0.04 mg mL−1 previously reported by Swanepoel et al.21 The apparent equilibrium solubility of the mebendazolium mesylate anhydride was 2.67 times higher than that of MBZ form C after 48 h.
Even when the mebendazolium salt synthesized here is very stable in the solid state, in general all mebendazolium salts are metastable phases in aqueous suspension.47,48,72 Our results show that the mebendazolium mesylate anhydride salt completely transformed into MBZ C during the equilibrium solubility experiment (Fig. S11†). This transformation can be justified by the great difference in solubility between the mebendazolium cation and the anions of the salts. The pH of the medium does not prevent the deprotonation of the MBZH+ cation and the precipitation of these molecules to form one or another MBZ desmotrope. Further experiments were needed to study the kinetics of the dissolution process of these salts. On the other hand, MBZ C did not undergo any phase transformation in the conditions of the equilibrium solubility experiment (Fig. S12†).
To get further insights on the solubility behaviour of mebendazolium mesylate anhydride salt, we evaluated its apparent solubility after 24 h in the same medium and in the same conditions. We found that the solubility of this salt is even higher than that of MBZ C after this period of time. While the solubility of MBZ C after 24 h showed no significant difference from the value determined after 48 h, the mebendazolium mesylate anhydride salt was 4.47 times more soluble. These findings let us to evaluate the behaviour of this material in the early stages of the solubility experiment. Fig. S13† shows the solubility of the salt during the experiment, along with that of MBZ C. It is observed that the solubility of the mebendazolium mesylate anhydride salt increased during the first five hours until reaching a maximum after 24 h, and then decreased after 48 h. The diminishing of the solubility of the new material is in agreement with the transformation into MBZ desmotropes observed after 48 h when analysing the remaining solid.
5 Conclusions
Using crystal engineering approaches, we have designed and synthesized a new mebendazole solid form based on supramolecular assembly principles. Recrystallization of MBZ with methanesulfonic acid (an FDA-accepted conformer) by slow solvent evaporation in water-free methanol, yielded the novel salt of 1:1 stoichiometry that crystallizes in the P212121 (19) space group. Total proton transfer was confirmed by single-crystal X-ray diffraction experiments and, we found, as expected, that both ions form a supramolecular motif of the type R22(8) through the formation of two intermolecular N–H⋯O hydrogen bonds. Moreover, we confirmed our prediction on the formation of an extended heterosynthon throughout the crystal structure, creating an infinite chain of intercalated ions driven by a third N–H⋯O hydrogen bond. Another classical and non-classical hydrogen bonds as well as edge-to-face π⋯π and lone pair–cation interactions contribute to the final stabilization of the crystal structure. Like MBZ pure solid forms and other multicomponent solid forms of the API, thermal studies of this new material showed that it undergoes degradation instead of melting. This salt is stable up to 205 °C when the endothermic elimination of the methyl ester moiety of the mebendazolium cation takes place. This new material prevents polymorphic interconversion and shows good stability in the solid state when it is stored at room temperature, since it did not degrade or take moisture from the atmosphere to yield the monohydrated phase. We also found that the monohydrated salt previously reported transforms into our new material upon heating up to 100 °C. Furthermore, this new salt material exhibits a solubility in 0.1 mol L−1 hydrochloric acid aqueous solution slightly higher than that of MBZ preferred form C. Our findings suggest that this new salt is a suitable candidate for pharmaceutical formulations.
Author contributions
E. L. Gutiérrez: conceptualization, formal analysis, investigation, methodology, visualization, writing – original draft. A. A. Godoy: investigation, methodology. E. V. Brusau: investigation, methodology, supervision, writing – review and editing. D. Vega: project administration, resources, investigation, methodology. Writing – review and editing. G. E. Narda: project administration, resources, supervision, writing – review and editing. S. Suárez: formal analysis, investigation, methodology, project administration, resources, supervision, writing – review and editing. F. Di Salvo: formal analysis, investigation, methodology, project administration, resources, supervision, writing – review and editing (based on CRediT authorship contribution).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Universidad Nacional de San Luis (PROICO 02-3218 and PROICO 02-2016), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) (PIP 11220200101956CO), the University of Buenos Aires (UBA) (grants numbers UBACYT 20020170200031BA and 20020190200010BA), and the National Agency for the Promotion of Research, Technological Development, and Innovation (grant number PICT-2019-2019-00123) all in Argentina. The authors also acknowledge Farm. Elbio Saidman from the Laboratorio de Control de Calidad de Medicamentos (UNSL) for kindly supplying the MBZ A and C samples.Notes and references
- World Health Organization, WHO 19th Model List of Essential Medicines (April 2015) (Amended November 2015), http://www.who.int/medicines/publications/essentialmedicines/en/.
- A. A. Al-Badr and M. Tariq, in Analytical Profiles of Drug Substances, 1987, vol. 16, pp. 291–326 Search PubMed.
- A. P. Ayala, H. W. Siesler and S. L. Cuffini, J. Raman Spectrosc., 2008, 39, 1150–1157 CrossRef CAS.
- W. Liebenberg, T. G. Dekker, A. P. Lötter and M. M. de Villiers, Drug Dev. Ind. Pharm., 1998, 24, 485–488 CrossRef CAS PubMed.
- M. S. Palmeirim, S. M. Ame, S. M. Ali, J. Hattendorf and J. Keiser, EClinicalMedicine, 2018, 1, 7–13 CrossRef PubMed.
- A. A. Escobedo, P. Almirall, E. González-Fraile and J. Ballesteros, Acta Trop., 2018, 188, 50–57 CrossRef CAS PubMed.
- B. Levecke, A. Montresor, M. Albonico, S. M. Ame, J. M. Behnke, J. M. Bethony, C. D. Noumedem, D. Engels, B. Guillard, A. C. Kotze, A. J. Krolewiecki, J. S. McCarthy, Z. Mekonnen, M. V. Periago, H. Sopheak, L.-A. Tchuem-Tchuenté, T. T. Duong, N. T. Huong, A. Zeynudin and J. Vercruysse, PLoS Neglected Trop. Dis., 2014, 8, e3204 CrossRef PubMed.
- P. Steinmann, J. Utzinger, Z.-W. Du, J.-Y. Jiang, J.-X. Chen, J. Hattendorf, H. Zhou and X.-N. Zhou, PLoS One, 2011, 6, e25003 CrossRef CAS PubMed.
- J.-Y. Chai, B.-K. Jung and S.-J. Hong, Korean J. Parasitol., 2021, 59, 189–225 CrossRef CAS PubMed.
- T. Guerini, B. Maddalo, B. Frassine, T. Alghisi, P. Borghetti, M. Bresciani and M. Buglione, Cancers, 2019, 11, 1284 CrossRef PubMed.
- M. De Witt, A. Gamble, D. Hanson, D. Markowitz, C. Powell, S. Al Dimassi, M. Atlas, J. Boockvar, R. Ruggieri and M. Symons, Mol. Med., 2017, 23, 50–56 CAS.
- P. Pantziarka, G. Bouche, L. Meheus, V. Sukhatme and V. P. Sukhatme, Ecancermedicalscience, 2014, 8, 443 CrossRef PubMed.
- P. Nygren, M. Fryknäs, B. Ågerup and R. Larsson, J. Cancer Res. Clin. Oncol., 2013, 139, 2133–2140 CrossRef CAS PubMed.
- S. Mansoori, M. Fryknäs, C. Alvfors, A. Loskog, R. Larsson and P. Nygren, Sci. Rep., 2021, 11, 8981 CrossRef CAS PubMed.
- H. S. Choi, Y. S. Ko, H. Jin, K. M. Kang, I. B. Ha, H. Jeong, H.-N. Song, H. J. Kim and B. K. Jeong, Molecules, 2021, 26, 5118 CrossRef CAS PubMed.
- T. Williamson, M. C. de Abreu, D. G. Trembath, C. Brayton, B. Kang, T. B. Mendes, P. P. de Assumpção, J. M. Cerutti and G. J. Riggins, Oncotarget, 2021, 12, 1326–1338 CrossRef PubMed.
- T. Williamson, T. B. Mendes, N. Joe, J. M. Cerutti and G. J. Riggins, Endocr.-Relat. Cancer, 2020, 27, 123–136 CAS.
- A. Maali, E. Ferdosi-Shahandashti, F. Sadeghi and E. Aali, Int. J. Hematol. Oncol. Stem Cell Res., 2020, 14, 257–264 Search PubMed.
- L. X. Yu, G. L. Amidon, J. E. Polli, H. Zhao, M. U. Mehta, D. P. Conner, V. P. Shah, L. J. Lesko, M.-L. Chen, V. H. L. Lee and A. S. Hussain, Pharm. Res., 2002, 19, 921–925 CrossRef CAS PubMed.
- H. V. Chavda, C. N. Patel and I. S. Anand, Sys. Rev. Pharm., 2010, 1, 62–69 CrossRef CAS.
- E. Swanepoel, W. Liebenberg, B. Devarakonda and M. M. De Villiers, Pharmazie, 2003, 58, 117–121 CAS.
- E. Swanepoel, W. Liebenberg and M. M. De Villiers, Eur. J. Pharm. Biopharm., 2003, 55, 345–349 CrossRef CAS PubMed.
- M. Himmelreich, B. J. Rawson and T. R. Watson, Aust. J. Pharm. Sci., 1977, 6(4), 123–125 CAS.
- A. C. Evans, J. E. Fincham, M. A. Dhansay and W. Liebenberg, S. Afr. Med. J., 1999, 89, 1118 CAS.
- F. F. Ferreira, S. Antonio Gutierrez, P. C. Pires Rosa and C. de O. Paiva-Santos, Int. J. Drug Dev. Res., 2011, 3, 26–33 Search PubMed.
- F. Bravetti, S. Bordignon, E. Alig, D. Eisenbeil, L. Fink, C. Nervi, R. Gobetto, M. U. Schmidt and M. R. Chierotti, Chem.–Eur. J., 2022, 28(6), e202103589 CrossRef CAS PubMed.
- F. T. Martins, P. P. Neves, J. Ellena, G. E. Camí, E. V. Brusau and G. E. Narda, J. Pharm. Sci., 2009, 98, 2336–2344 CrossRef CAS PubMed.
- Y. Kasetti and P. V. Bharatam, Theor. Chem. Acc., 2012, 131, 1–12 Search PubMed.
- S. Agatonovic-Kustrin, B. D. Glass, M. Mangan and J. Smithson, Int. J. Pharm., 2008, 361, 245–250 CrossRef CAS PubMed.
- M. M. De Villiers, R. J. Terblanche, W. Liebenberg, E. Swanepoel, T. G. Dekker and M. Song, J. Pharm. Biomed. Anal., 2005, 38, 435–441 CrossRef CAS PubMed.
- P. Charoenlarp, J. Waikagul, C. Muennoo, S. Srinophakun and D. Kitayaporn, Southeast Asian J. Trop. Med. Public Health, 1993, 24, 712–716 CAS.
- M. Karashima, K. Kimoto, T. Kojima and Y. Ikeda, J. Cryst. Growth, 2014, 390, 30–37 CrossRef CAS.
- World Health Organization, The International Pharmacopoeia, 11th edn, 2022 Search PubMed.
- N. L. Calvo, T. S. Kaufman and R. M. Maggio, J. Pharm. Biomed. Anal., 2016, 122, 157–165 CrossRef CAS PubMed.
- G. R. Desiraju, Chem. Commun., 1997, 1475–1482 RSC.
- G. R. Desiraju, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS.
- V. R. Thalladi, B. S. Goud, V. J. Hoy, F. H. Allen, J. A. K. Howard and G. R. Desiraju, Chem. Commun., 1996, 401 RSC.
- D. J. Berry and J. W. Steed, Adv. Drug Delivery Rev., 2017, 117, 3–24 CrossRef CAS PubMed.
- J. Wounters and L. Quéré, Phamaceutical Salts and Co-crystals, RSC Publishing, Cambridge, 2012 Search PubMed.
- R. D. B. Walsh, M. W. Bradner, S. Fleischman, L. A. Morales, B. Moulton, N. Rodríguez-Hornedo and M. J. Zaworotko, Chem. Commun., 2003, 186–187 RSC.
- N. M. Blaton, O. M. Peeters and C. Deranter, Cryst. Struct. Commun., 1980, 9(1), 181–186 CAS.
- M. Caira, T. Dekker and W. Liebenberg, J. Chem. Crystallogr., 1998, 28(1), 11–15 CrossRef CAS.
- E. V. Brusau, G. E. Camí, G. E. Narda, S. Cuffini, A. P. Ayala and J. Ellena, J. Pharm. Sci., 2008, 97, 542–552 CrossRef CAS PubMed.
- J.-M. Chen, Z.-Z. Wang, C.-B. Wu, S. Li and T.-B. Lu, CrystEngComm, 2012, 14, 6221 RSC.
- J. Chen and T. Lu, Chin. J. Chem., 2013, 31, 635–640 CrossRef CAS.
- K. de Paula, G. E. Camí, E. V. Brusau, G. E. Narda and J. Ellena, J. Pharm. Sci., 2013, 102, 3528–3538 CrossRef CAS PubMed.
- E. L. Gutiérrez, M. S. Souza, L. F. Diniz and J. Ellena, J. Mol. Struct., 2018, 1161, 113–121 CrossRef.
- E. L. Gutiérrez, A. A. Godoy, G. E. Narda and J. Ellena, CrystEngComm, 2020, 22, 6559–6568 RSC.
- E. Saidman, A. K. Chattah, L. Aragón, M. Sancho, G. Camí, C. Garnero and M. Longhi, Eur. J. Pharm. Sci., 2019, 127, 330–338 CrossRef CAS PubMed.
- D. Minda, A. Mioc, C. Banciu, C. Soica, R. Racoviceanu, M. Mioc, I. Macasoi, S. Avram, A. Voicu, A. Motoc and C. Trandafirescu, Processes, 2021, 9, 2185 CrossRef CAS.
- T. Bajaj, C. Singh and G. Das Gupta, J. Drug Delivery Sci. Technol., 2022, 70, 103264 CrossRef CAS.
- Food and Drug Administration (FDA) and U.S. Department of Health and Human Services, Approved Drug Product with Therapeutic Equivalence Evaluations (“Orange Book”), 40th edn, 2020, https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book Search PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Inc., 6th edn, 2007 Search PubMed.
- H. Wan, A. G. Holmén, Y. Wang, W. Lindberg, M. Englund, M. B. Någård and R. A. Thompson, Rapid Commun. Mass Spectrom., 2003, 17, 2639–2648 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362 RSC.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- C. F. Macrae, et al., J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- United States, Pharmacopeia and National Formulary USP 29-NF 24, 2006 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, 1997 Search PubMed.
- L. Zhong and S. F. Parker, R. Soc. Open Sci., 2018, 5, 181363 CrossRef PubMed.
- G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023–1028 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- S. K. Seth, I. Saha, C. Estarellas, A. Frontera, T. Kar and S. Mukhopadhyay, Cryst. Growth Des., 2011, 11, 3250–3265 CrossRef CAS.
- B. B. C. Holanda, R. T. Alarcon, R. B. Guerra, D. Rinaldo, F. C. R. Spazzini, R. A. E. Castro and G. Bannach, J. Anal. Appl. Pyrolysis, 2018, 135, 76–84 CrossRef CAS.
- G. E. Camí, E. V. Brusaua, G. E. Narda and R. M. Maggio, J. Drug Delivery Sci. Technol., 2020, 15, 101344 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2236578. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra07422f |
| This journal is © The Royal Society of Chemistry 2024 |