
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of potential drug candidates to treat gastritis and associated oxidative stress based on some novel 2-aryl-1H-naphtho[2,3-d]imidazole: synthesis, in vitro and in silico analysis
Amina Sultanaa,
Aneela Wahaba,
Ghulam Fareed *b,
Hamna Rafiqb,
Khalid Mohammed Khanc,
Mehreen Lateefd,
Nazia Fareeda,
Shafqat Hussaine and
Sikander Khan Sherwanif
*b,
Hamna Rafiqb,
Khalid Mohammed Khanc,
Mehreen Lateefd,
Nazia Fareeda,
Shafqat Hussaine and
Sikander Khan Sherwanif
aDepartment of Chemistry, Federal Urdu University of Arts, Science and Technology, Gulshan-e-Iqbal, Karachi, Pakistan
bPharmaceutical Research Centre, PCSIR Laboratories Complex, Karachi, Pakistan. E-mail: fareedchm@yahoo.com
cH. E. J. Research Institute of Chemistry, International Centre for Chemical and Biological Sciences, University of Karachi, Pakistan
dDepartment of Biochemistry, Bahria University Medical and Dental College, Bahria University, Karachi, Pakistan
eDepartment of Chemistry, University of Baltistan, Skardu, Gilgit-Baltistan 1600, Pakistan
fDepartment of Microbiology, Federal Urdu University of Arts, Science and Technology, Gulshan-e-Iqbal, Karachi, Pakistan
First published on 2nd January 2024
Abstract
To identify potential scaffolds to treat gastritis and oxidative stress, 2-aryl-1H-naphtho[2,3-d]imidazole derivatives (1–15) were synthesized. The synthesis was conveniently carried out by condensing 2,3-diaminonaphthalene with variously substituted aldehydes to yield 15 new 2-aryl-1H-naphtho[2,3-d]imidazole derivatives. Structures of all synthesized compounds were elucidated using MS and NMR spectroscopic techniques. Compounds containing an imidazole moiety have continued to spark interest in the field of medicinal chemistry due to their unique properties. In continuation of this statement, to further explore the biological potential of these types of compounds, newly synthesized imidazole derivatives were evaluated for their inhibitory potential against urease and antioxidant activities. Compounds 4 and 11 were identified as the most potent urease inhibitors in the series, with IC50 values of 34.2 ± 0.72 and 42.43 ± 0.65 μM, respectively. Compounds 1, 3, 6, 11, and 15, with EC50 values in the range of 37–75 μg ml−1, showed significant antioxidant activity. Molecular docking studies of the selected synthesized compounds 3, 4, 9, and 11 were also performed to determine their binding interaction with the jack bean urease. Through docking studies, it was revealed that all the compounds that showed good inhibitory potential against urease fit well within the protein's binding pocket. Furthermore, ADME analysis was carried out to explore the drug-likeness properties of the compounds. The findings of the present work revealed that compounds 4 and 11 could be better options to treat gastritis and associated oxidative stress.
Introduction
The term “gastritis” primarily refers to abnormal stomach lining inflammation. Many people with gastritis do not have any symptoms, but others may experience upper abdominal pain or discomfort, nausea, and vomiting.1 Helicobacter pylori (H. pylori) is a Gram-negative bacterium that causes gastric inflammation, leading to peptic ulcers. The infection mainly originates from developing a defense mechanism that helps H. pylori survive in the stomach's acidic environment and improves its capacity to spread illness.2 A particular adaptability is the activity of the enzyme urease, which breaks down urea and produces ammonia. Ammonia then neutralizes stomach acid, enabling H. pylori to persist and colonize the gastric mucosa (Fig. 1). Urease is a nickel-containing enzyme responsible for urea's catalytic degradation to ammonia and carbon dioxide. Bacterial urease is a virulence factor implicated in the pathogenesis of various gastrointestinal infections, including peptic ulcers. Bacterial urease from Helicobacter pylori is essential for its survival, as the released ammonia serves to protect the bacteria from the surrounding gastric acid. Inhibitions of urease are therefore highly sought after and considered important in medicinal chemistry.3,4 | ||
| Fig. 1 Possible mechanism of naphthaleneimidazoles to treat gastritis. | ||
In addition, the other main feature of H. pylori is its capability to adhere to the gastric epithelium, which is achieved through receptor-mediated adhesion and initiates apoptosis (cellular damage).5,6 The generation of the reactive oxygen species (ROS) in the presence of H. pylori induces the inflammatory pathway. The oxidation is mediated by the overproduction of nitric oxide, a reactive oxygen species produced by inducible nitric oxide synthase (iNOS). The activity of iNOS is a characteristic factor of H. pylori infection, which causes gastritis.7–9 The compounds containing inhibitory activity towards urease and antioxidant properties, which scavenge and deactivate free radicals, may protect the body from infections and associated cellular damages.10,11 Several FDA-approved medications are available to treat gastritis. However, primary antibiotic resistance is the main factor affecting their results. They are also known to initiate respiratory infections (Fig. 2).
 | ||
| Fig. 2 Imidazole ring-containing FDA-approved drugs to treat gastritis (ulcer). | ||
Benzimidazole, also known as 1H-benzimidazole and 1,3-benzodiazole, is a heterocyclic pharmacophore composed of a benzene ring fused with a five-membered imidazole ring.12 Numerous studies have been conducted in recent years that have shown intriguing results on the chemistry, structure–activity relationship, and biological activities of several benzimidazole-based compounds.13 Researchers are interested in the biological activity of synthetic imidazole 5,6-dimethylbenzimidazole since it is a breakdown product of vitamin B12, and some of its derivatives have vitamin B12-like action.14 Benzimidazole and its derivatives, both natural and synthetic, reveal a broad spectrum of biological activities.15 The naturally occurring benzimidazole moiety of vitamin B12 has been known to improve CNS function.16
Similarly, kealiiquinone, a benzimidazole-based alkaloid, has exhibited anticancer properties.17 Benzimidazole derivatives continue to spark an interest in the field of medicinal chemistry. Consequently, a wide assortment of derivatives of benzimidazole have been reported for their favorable physiological and pharmacological properties, such as enzyme inhibition,18–20 antimicrobial, cytotoxic, antidiabetic,21 antiasthmatic, antileukemic,22 antihypertensive and antihepatitis B virus,23 antileishmanial,2 anti-HIV and cardiotonic, anti-inflammatory and analgesic,25,26 diuretic,27 antitumor and antiasthmatic,26 anthelmintic and antihistaminic24 properties. Benzimidazole derivatives have shown tremendous potential in remedying infections, obesity, epilepsy, and ulcers.28,29
Thus, to further explore the biological potential of the naphthaleneimidazole scaffold to treat gastritis, a library of variously substituted 2-aryl-1H-naphtho[2,3-d]imidazole derivatives (1–15) were synthesized (Fig. 3). Structures of all synthesized compounds were elucidated using EIMS, IR, and NMR spectroscopic techniques. All of the synthesized derivatives were evaluated for the said activities to identify lead molecules with dual potential.
 | ||
| Fig. 3 Enzyme inhibition activity (IC50 of compounds) of urease. | ||
Results and discussion
Chemistry
A series of 2-aryl-1H-naphtho[2,3-d]imidazole (1–15) derivatives was synthesized via the facile condensation of 2,3-diaminonaphthalene with appropriate aromatic aldehydes using sodium metabisulfite (Na2S2O5) in N,N-dimethyl formamide (DMF). The solid product obtained after adding cold water was filtered, washed with water, crystallized from ethanol, and finally dried in air. The chemical structure of these synthesized derivatives was determined using EIMS, IR, and NMR spectroscopies (Scheme 1).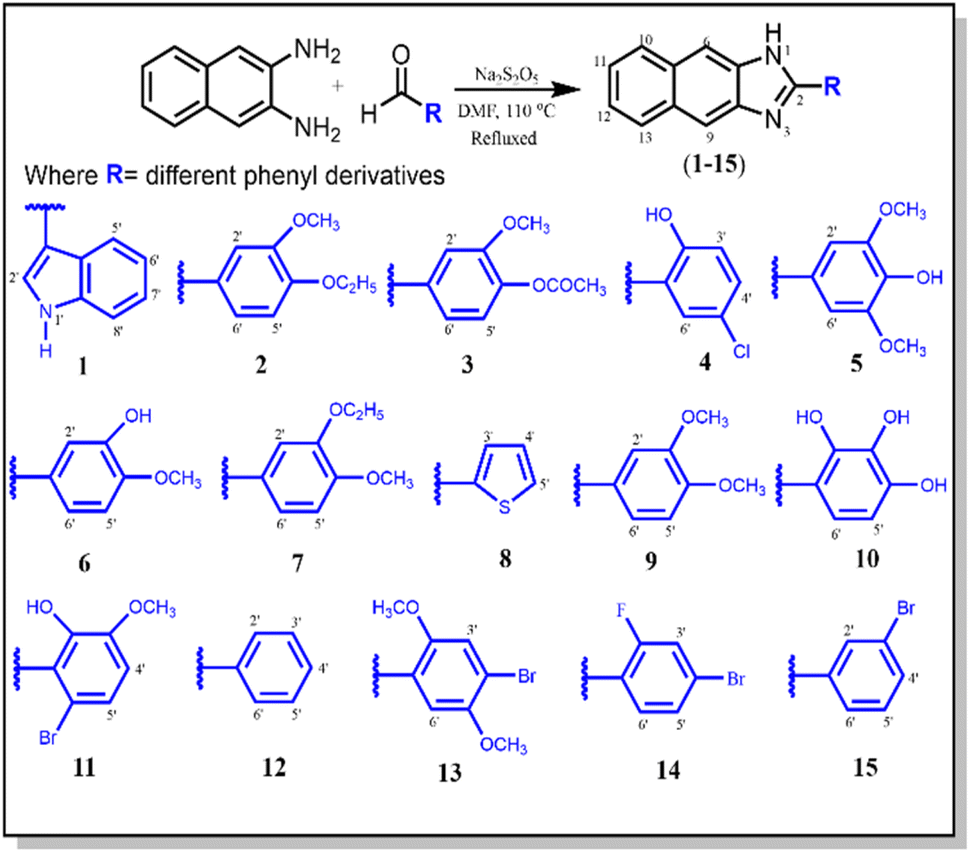 | ||
| Scheme 1 Synthesis of 2-aryl-1H-naphtho[2,3-d]imidazole derivatives (1–15). | ||
The IR spectra for all compounds (1–15) exhibited strong absorption bands for –NH in the 3245–3501 cm−1 range and for –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– at 1461–2895 cm−1. The proton NMR of these compounds displayed a singlet of the –NH proton in between 9.99 and 13.71 ppm, and a signal of CN was observed in the 146.1–152.3 ppm range in the 13C NMR spectra. They all furnished satisfactory CHN elemental analysis.30,31
N– at 1461–2895 cm−1. The proton NMR of these compounds displayed a singlet of the –NH proton in between 9.99 and 13.71 ppm, and a signal of CN was observed in the 146.1–152.3 ppm range in the 13C NMR spectra. They all furnished satisfactory CHN elemental analysis.30,31
The molecular formula C19H16N2O2 of the representative compound 9 was determined using EIMS analysis, which showed the [M+] ion peak at m/z 304(100). The proton NMR of compound 9 indicated –NH as a singlet at δ 10.61, aromatic protons were observed in the δ 8.06–7.20 range, and the two singlets at δ 3.90 and 3.86 confirmed the two –OCH3 groups on the 4 and 3 positions of the phenyl ring. In the IR spectra, the absorption bands at 3384 and 1687 cm−1 revealed –NH and –CN–, respectively, whereas the –OCH3 group and aromatic ring were confirmed by absorption bands at 1211 and 3124 cm−1, respectively.32 The carbon atoms were determined using 13C NMR spectroscopy, which showed the presence of the quaternary carbon of the –CN group and the phenyl ring at δ 151.0 and 123.6, respectively. The signals at δ 57.5 and 56.9 indicated the presence of two –OCH3 carbons. In addition, the signals in between δ 137.9 and 109.9 defined the aromatic moiety.31,33 Based on the above spectral analysis, the new synthesized compound 9 was found to be 2-(3,4-dimethoxyphenyl)-1H-naphtho[2,3-d]imidazole. Likewise, the other synthesized compounds (1–15) were characterized as described in the experimental section.
In vitro urease inhibitory activity
The synthesized naphthaleneimidazoles (1–15) were screened for their in vitro urease inhibitory activities. Only seven compounds exhibited varying degrees of urease inhibition. Among the series, 4 and 11 were found to be the most active with IC50 values of 34.2 ± 0.72 and 42.43 ± 0.65 μM, respectively, as compared to the standard inhibitor thiourea (IC50 = 21.7 ± 0.12 μM). The activity may result from the 2-hydroxyl and 5-chloro groups substituted on the phenyl ring in both the compounds, 4 and 11, although compound 11 also contains a 3-methoxy substituent on the same phenyl ring. Compound 3, having 3-methoxy and 4-acetoxy substituents, showed >2-fold decreased urease inhibition with an IC50 value of 56.3 ± 0.78 μM. Compounds 1, 2, 8, and 9 exhibited relatively low activities, having IC50 values of 92.3 ± 0.20, 73.5 ± 0.65, 86.4 ± 0.78 and 67.14 ± 0.67 μM, respectively. Other compounds in the series were inactive (Table 1). The potential inhibition activity associated with IC50 is presented in Fig. 3. Compound 4 showed promising urease inhibition activity as compared to compound 11. It may be due to the more electron-withdrawing inductive effect of the chloro group compared to the bromo group in compound 11, which is less electronegative and larger than the chloro group.34| Compound | Urease inhibition IC50 ± SDa | Antioxidant activity | |
|---|---|---|---|
| % Inhibition ± SDa | EC50 (μg ml−1) | ||
| a SD = standard deviation; NA = not applicable. | |||
| 1 | 92.3 ± 0.20 | 71.1 ± 0.01 | 37.5 |
| 2 | 73.5 ± 0.65 | 43 ± 0.01 | — |
| 3 | 56.3 ± 0.78 | 67.4 ± 0.01 | 75 |
| 4 | 34.2 ± 0.72 | 29 ± 0.01 | — |
| 5 | NA | 12 ± 0.01 | — |
| 6 | NA | 71 ± 0.01 | 37.5 |
| 7 | NA | 57.8 ± 0.01 | 100 |
| 8 | 86.4 ± 0.78 | 32 ± 0.01 | — |
| 9 | 67.14 ± 0.67 | 57.8 ± 0.02 | 100 |
| 10 | NA | 57.9 ± 0.02 | 100 |
| 11 | 42.43 ± 0.65 | 71.1 ± 0.01 | 37.5 |
| 12 | NA | 43 ± 0.01 | — |
| 13 | NA | 57.7 ± 0.01 | 100 |
| 14 | NA | 23 ± 0.01 | — |
| 15 | NA | 69.3 ± 0.01 | 75 |
| Thiourea | 21.7 ± 0.12 | — | — |
| Acetohydroxamic acid | 37.0 | ||
| Ascorbic acid | — | 80 | 8.3 |
In vitro antioxidant activity
The synthesized derivatives (1–15) were also screened for their in vitro antioxidant activity against 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical and ascorbic acid as a standard. Five compounds, 1, 3, 6, 11, and 15, showed significant % inhibition 71.1, 67.4, 71, 71, and 69.3% with EC50 = 37.5, 75, 37.5, 37.5, and 75 μg ml−1, respectively. The presence of 2-indole may be responsible for the good activity of compound 1. However, the presence of the 4-acetoxy and 3-hydroxyl groups in compounds 3 and 6, respectively, accompanied by the methoxy group, can be attributed to their significant activity. The considerable properties of compounds 11 and 15 can be attributed to the bromo substitution in both rings. Four compounds, 7, 9, 10, and 13, exhibited moderate antioxidant activity with the % inhibition of 57.8, 57.8, 57.9, and 57.7, respectively, having EC50 = 100 μg ml−1 for each compound (Table 1). The potential antioxidant activity of the compounds is presented in Fig. 4. The steric crowding of the ortho-substituted methoxy (–OCH3) group in compound 11 enhances its antioxidant activity because it is an electron-donating group. An electron donor can increase the electron cloud density of the benzene ring, decrease the dissociation energy of the phenolic hydroxyl bond, and then enhance its free radical scavenging ability. Compound 4 showed no remarkable antioxidant activity because of the absence of an electron-donating group at the ortho position.35 | ||
| Fig. 4 Antioxidant activity of compounds. | ||
Drug-likeness evaluation
The compounds that potentially inhibit urease and possess antioxidant properties (3, 4, 9, and 11) were assessed for drug-likeness evaluation by various parameters (Table 2). The predicted properties are summarized in Table 3. All compounds contain molecular weights that were within the acceptable limit, i.e., <500 Da. The rotatable bonds within the molecules indicate their flexibility. The permeability through the cell membrane is log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P ≤ 5, indicating good membrane permeability, as the logp values of all of the compounds ranged from 3.63 to 3.95, indicating excellent lipophilicity. Lipophilicity is an important property of the molecule that plays a role in determining how it functions inside the body. The total polar surface area (TPSA) values were calculated to analyze the absorption and membrane permeability profiles, and the outcome demonstrated an average TPSA value of 54.6 Å2, proposing their absorption through the intestine (<140 Å2). It has been shown that the TPSA value and the number of rotatable bonds can indicate that a molecule is orally active. All compounds contain high GI absorption, and none of the molecules violated the Lipinski rule of five.
P ≤ 5, indicating good membrane permeability, as the logp values of all of the compounds ranged from 3.63 to 3.95, indicating excellent lipophilicity. Lipophilicity is an important property of the molecule that plays a role in determining how it functions inside the body. The total polar surface area (TPSA) values were calculated to analyze the absorption and membrane permeability profiles, and the outcome demonstrated an average TPSA value of 54.6 Å2, proposing their absorption through the intestine (<140 Å2). It has been shown that the TPSA value and the number of rotatable bonds can indicate that a molecule is orally active. All compounds contain high GI absorption, and none of the molecules violated the Lipinski rule of five.
| Compounds | 3 | 4 | 9 | 11 |
| MW | 332.35 | 294.74 | 304.34 | 369.21 |
| Heavy atoms | 25 | 21 | 23 | 23 |
| Aromatic heavy atoms | 19 | 19 | 19 | 19 |
| Rotatable bonds | 4 | 1 | 3 | 2 |
| H-bond acceptors | 4 | 2 | 3 | 3 |
| H-bond donors | 1 | 2 | 1 | 2 |
| TPSA | 64.21 | 48.91 | 47.14 | 58.14 |
| ilogP |
2.54 | 2.59 | 2.69 | 2.54 |
| Consensus logP |
3.63 | 3.97 | 3.71 | 3.95 |
| ESOL logS |
−4.56 | −4.98 | −4.64 | −5.34 |
| Ali logS |
−4.79 | −5.04 | −4.66 | −5.28 |
| GI absorption | High | High | High | High |
| BBB permeant | Yes | Yes | Yes | Yes |
| Lipinski violations | 0 | 0 | 0 | 0 |
| Ghose violations | 0 | 0 | 0 | 0 |
| Veber violations | 0 | 0 | 0 | 0 |
| Egan violations | 0 | 0 | 0 | 0 |
| Muegge violations | 0 | 0 | 0 | 0 |
| Bioavailability score | 0.55 | 0.55 | 0.55 | 0.55 |
| Compounds | Binding energy |
|---|---|
| 3 | −7.48 |
| 4 | −6.86 |
| 9 | −7.14 |
| 11 | −6.99 |
| Thiourea | −3.04 |
| Omeprazole | −6.94 |
| Lansoprazole | −7.20 |
| Rabeprazole | −6.69 |
Molecular docking studies
To gain insight regarding the binding affinity and intermolecular interactions of the synthetic analogs (3, 4, 9, and 11) with the target jack bean urease enzyme (PDB ID 4H9M), molecular docking was performed using AutoDock Vina integrated with USCF Chimera software. The active site of urease contains hydrophilic residues, including His 593, His 594, Arg 439, His 519, Glu 493, Asp 494, Leu 595, and hydrophobic residues Gly 550 and Ala 440. Additionally, the urease enzyme contained a modified tyrosine residue KCX-220 and two nickel ions in their active site, which account for a crucial role by binding essential amino acids with ligands and augmenting urease activity. The docking conformations of all analogs in comparison with standards, i.e., thiourea, omeprazole, lansoprazole, and rabeprazole, were established within the active site. The docking results are analyzed using the docking scores, binding modes, and interaction of ligands with the functional residues of urease. The standard inhibitor, i.e., thiourea, was shown to have a −3.04 kcal mol−1 binding affinity (Table 3).Based on the docking calculations of naphthaleneimidazoles, the docking scores of all compounds against the urease were >6.0 kcal mol−1. Other standards, including omeprazole, lansoprazole, and rabeprazole, show docking scores of −6.94, −7.2, and −6.69 kcal mol−1, respectively. The docking poses of all standards are presented in Fig. 5.
 | ||
| Fig. 5 Docking pose of standards: (a) thiourea, (b) omeprazole, (c) lansoprazole, (d) rabeprazole. | ||
The docking results revealed that all compounds interact with the active site of urease (Fig. 6). In compound 3, the conventional hydrogen bonding was found with the nonpolar aliphatic amino acid Ala 440 at 2.1 Å. On the other hand, two H-bonds are formed between the O2 of compound 3 and the NH group of Arg 609. CME 592 was also shown to make hydrophobic contact. The docking results of compound 4 show the formation of an H-bond between the NH group and two oxygen atoms of Asp 494 at a distance of 2.8 Å. However, it was also found that compound 4 makes interactions with Ni 901 and 902 at a distance of 2.7 Å and 3.2 Å, respectively. The presence of chloride in compound 4 enhanced the electronegativity and the stability to bind with urease. Some hydrophobic contacts were also observed with Ala 6636, Met 637, and Ala 440. In compound 9, H-bonds were shown between the 3-OH group at the para position of the extended phenyl ring with the NH2 group of the Arg 609 residue. Ala 440 and His 593 made hydrophobic interactions, while Arg 493 was shown to interact through cation–π interaction.
 | ||
| Fig. 6 Docking pose of naphthaleneimidazole compounds: (a) compound 3, (b) compound 4, (c) compound 9, (d) compound 11. | ||
Compound 11 also makes an H-bond with Asp 494 at a distance of 3.1 Å. It shows π–π interaction with His 593 and a hydrophobic bond with Ala 440. Results of the docking studies revealed the binding interactions of benzimidazole compounds with urease. It is observed that such a class of pharmacophore is mainly dependent on phenyl ring substituents. Therefore, the outcomes of the present study will be useful to design a new target against the urease enzyme to treat gastritis.
Experimental
General
All the reagents and solvents used were purchased from Sigma-Aldrich and used without further purification. The Gallen Kamp melting point apparatus was used to determine the melting points that were uncorrected. EIMS spectra were measured using a Jeol JMS-600H machine. NMR spectra were recorded on Avance AV-300, 400, and 500 MHz machines in deuterated dimethyl sulfoxide (DMSO-d6) with trimethylsilane (TMS) as an internal standard. The progress of the reaction was monitored on pre-coated silica gel glass TLC plates (Kieselgel 60, 254, E. Merck, Germany). IR spectra were recorded using a JASCO-302-A spectrophotometer. The elemental (CHN) analysis was performed using a Carlo Erba Strumentazione-Model-1106, Italy.Urease inhibition assay
The urease inhibitory activity of all synthesized derivatives was determined using the indophenol method.36 The results were obtained after measuring the ammonia production during the reaction. The reaction mixture of 25 μl of urease enzyme solution, 55 μl of buffer (0.01 M K2HPO4·3H2O, 1 mM EDTA, and 0.01 M LiCl, pH 8.2), and 100 mM urea were incubated in 96-well plates having 5 μl of test compounds (1 mM) for 15 min at 30 °C. Briefly, 45 μl each of phenol reagent (1% w/v phenol and 0.005% w/v sodium nitroprusside) and 70 μl of alkali reagent (0.5% w/v NaOH and 0.1% active chloride NaOCl) were added to each well. The increasing absorbance was measured at 630 nm after 50 min through a microplate reader (Molecular Device, USA). All reactions were performed in triplicate in a final volume of 200 μl. The results, i.e., change in absorbance per minute, were processed on SoftMax Pro software (Molecular Device, USA). Thiourea was used as a standard for comparison. Percentage inhibition was calculated from the formula:| % Inhibition = 100 − (ODtestwell/ODcontrol) × 100 |
Antioxidant activity assay
The antioxidant activity of the compounds (1–15) was determined by using the procedure described by Lee et al.37 The stable radical solution of 1,1-diphenyl-2-picrylhydrazyl (DPPH) was prepared in ethanol (300 μM). A 10 μl volume of the test samples and 90 μl solution of the stable radical (DPPH) was added in 96-well microtiter plates, and incubated at 37 °C for 30 minutes. The absorbance was measured at 515 nm using a spectrophotometer. The percent inhibition of radicals by the treatment of the test sample was found by comparison with DMSO as the negative control. Ascorbic acid was used as the standard.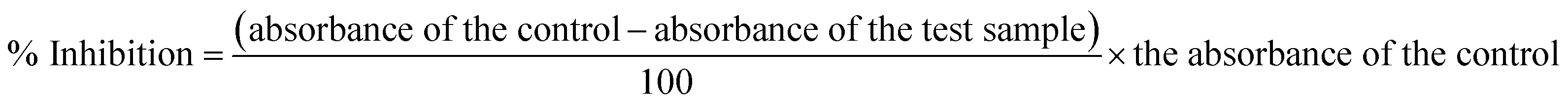
The calculated EC50 value denotes the concentration (in μg ml−1) of the sample required to scavenge 50% of DPPH.
In silico study
 | ||
| Fig. 7 Optimized 3D structure of compound (a) 3, (b) 4, (c) 9, (d) 11. | ||
The molecular structures of the compounds were acquired from ChemDraw and PDB files were obtained from Chimera software for docking. The docking parameters used for the ligand simulation are: exhaustiveness = 10; center_x = 19.82, center_y = −58.26 and center_z = −25.99; size_x = 25.25, size_y = 25 and size_z =25. Throughout the docking experiment, the protein structures were kept rigid, while the torsions or degrees of freedom for the ligands were allowed full rotations. Twenty conformational modes were obtained. The 3D images were visualized by using USCF Chimera software.41
General procedure for the synthesis of compounds (1–15)
In a typical reaction (Scheme 1), sodium metabisulfite (Na2S2O5) was mixed with a stirring solution of 2,3-diaminonaphthalene (3.12 mmol, 0.50 g) and substituted aromatic aldehydes (3.16 mmol, 0.51 g) in DMF (15 ml). The reaction mixture was then refluxed at 110 °C for 4 h. The reaction progress was monitored using TLC. After the completion of the reaction, the reaction contents were cooled at room temperature. Then, cold distilled water was added with vigorous shaking until the formation of precipitates. It was kept aside in an ice bath to settle down the precipitates. Subsequently, the solid product was filtered, washed with distilled water, and crystallized from ethanol to afford pure 2,3-diaminonaphthaleneimidazole derivatives (1–15).N); 1H-NMR: (500 MHz, DMSO-d6): δ 13.71 (1H, bs, –NH), 12.21 (1H, s, H-1′), 8.45 (1H, br.d, J = 6.0 Hz, H-5′), 8.44 (1H, s, H-2′), 8.13 (2H, s, H-6/9), 8.08 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.60 (1H, br.d, J = 6.0 Hz, H-8′), 7.44 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 7.30 (2H, m, H-6′/7′); 13C-NMR: (125 MHz, DMSO-d6): δ 153.1, 137.8(2×), 134.5, 126.7(2×),126.1(2×), 125.6, 123.9, 122.3(2×), 120.9, 118.7, 118.6, 114.9, 110.7, 103.8; EI-MS: m/z (rel. abund.%), 283 (M+, 44), 282 (10), 149 (15), 141 (10), 140 (11), 135 (38), 115 (9), 71 (34), 44 (100); anal. calcd for C19H13N3 (283.11): C, 80.54; H, 4.62; N, 14.83; found: C, 80.52, H, 4.61; N, 14.85.N), 1124 (OCH3); 1H-NMR (400 MHz, DMSO-d6): δ 13.22 (1H, bs, –NH), 8.16 (2H, s, H-6/9), 8.07 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.87 (2H, dd, J = 8.4, 2.0 Hz, H-2′/6′), 7.43 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 7.23 (1H, d, J = 8.4 Hz, H-5′), 4.14 (2H, q, J = 7.2 Hz, –OCH2), 3.92 (3H, s, –OCH3), 1.37 (3H, t, J = 7.2 Hz, –CH3); 13C-NMR: (100 MHz, DMSO-d6): δ 152.7, 151.8, 149.4, 137.8, 126.5(2×), 126.3(2×), 123.9, 123.6, 122.5(2×), 114.6(2×), 112.0, 110.6, 63.8, 55.7, 13.4; EI-MS: m/z (rel. abund.%), 318 (M+, 100%), 303 (6), 289 (57), 288 (15), 275 (13), 273 (7), 261 (31), 260 (13), 168 (5), 141 (7), 140 (18); anal. calcd for C20H18N2O2 (318.14): C, 75.45; H, 5.70; N, 8.80; found: C, 75.43; H, 5.69; N, 8.78.N); 1H-NMR (400 MHz, DMSO-d6): δ 10.81 (1H, s, –NH), 8.22 (2H, s, H-6/9), 8.08 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 8.02 (1H, d, J = 2.0 Hz, H-2′), 7.89 (1H, dd, J = 8.4, 2.0 Hz, H-6′), 7.44 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 7.40 (1H, d, J = 8.4 Hz, H-5′), 3.94 (3H, s, OCH3), 2.31 (3H, s, –COCH3); 13C-NMR: (100 MHz, DMSO-d6): δ 169.1 (–OCO), 150.9 (CN), 147.7, 138.5(2×), 137.8, 127.6, 126.7(2×), 126.5(2×), 123.4, 122.1(2×), 119.2, 114.2(2×), 110.9, 55.6, 19.9; EI-MS: m/z (rel. abund.%), 332 (M+, 87), 298 (6), 291 (56), 290 (100), 289 (54), 275 (23), 192 (5), 141 (8), 140 (17); anal. calcd for C20H16N2O3 (332.12); C, 72.28; H, 4.85; N, 8.43; found: C, 72.23; H, 4.84; N, 8.44.N); 1H-NMR (400 MHz, DMSO-d6): δ 13.30 (1H, s, –NH), 8.25 (1H, d, J = 2.4 Hz, H-6′), 8.09 (2H, s, H-6/9), 8.06 (2H, m, H-10/13), 7.49 (2H, m, H-11/12), 7.48 (1H, dd, J = 8.8, 2.4 Hz, H-4′), 7.10 (1H, d, J = 8.8 Hz, H-3′); 13C-NMR: (100 MHz, DMSO-d6): δ 152.1, 151.9, 137.2(2×), 130.6, 127.6, 126.5, 125.8(2×), 125.7(2×), 123.0(2×), 117.1(2×), 116.8 EI-MS: m/z (rel. abund.%), 294 (M+, 100), 258 (10), 140 (26), 111 (8), 76 (9); anal. calcd for C17H11ClN2O (294.06); C, 69.28; H, 3.76; N, 9.50; found: C, 69.26; H, 3.77; N, 9.49.N), 1084 (OCH3); 1H-NMR (400 MHz, DMSO-d6): δ 10.99 (1H, s, –NH), 8.17 (2H, s, H-6/9), 8.07 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.63 (2H, s, H-2′/6′), 7.44 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 3.92 (6H, s, 2OCH3); 13C-NMR: (100 MHz, DMSO-d6): δ 152.1, 147.9(2×), 136.9(2×), 136.8, 125.8, 127.1(4×), 121.2, 113.9(2×), 106.4, 56.3(2×) EI-MS: m/z (rel. abund.%), 320 (M+, 100), 319 (15), 304 (7), 289 (10), 273 (9), 140 (11), 115 (8); anal. calcd for C19H16N2O3 (320.12); C, 71.24; H, 5.03; N, 8.74; found: 71.22; H, 5.02; N, 8.75.N), 1089 (OCH3); 1H-NMR (400 MHz, DMSO-d6): δ 10.55 (1H, s, –NH), 8.12 (2H, s, H-6/9), 8.05 (2H, dd, J = 6.4, Hz, 3.2, H-10/13), 7.73 (2H, m, H-2′/6′), 7.43 (2H, dd, J = 6.4, 3.2, Hz, H-11/12), 7.21 (1H, d, J = 8.4 Hz, H-5′), 3.89 (3H, s, –OCH3); 13C-NMR: (100 MHz, DMSO-d6): δ 151.9, 146.5, 137.2(2×), 137.1, 127.6(4×), 125.3, 123.6, 122.2(2×), 114.2, 114.1(2×), 112.2, 55.9; EI-MS: m/z (rel. abund.%), 290 (M+, 100), 289 (5), 275 (8), 274 (35), 247 (19), 141 (7), 140 (10), 123 (11), 108 (9); anal. calcd for C18H14N2O2 (290.11); C, 74.47; H, 4.86; N, 9.65; found: C, 74.45; H, 4.87; N, 9.63.N), 1105 (OCH3),1H-NMR (300 MHz, DMSO-d6): δ 9.99 (1H, s, –NH), 8.16 (2H, s, H-6/9), 8.05 (2H, dd, J = 6.4, 3.2, Hz, H-10/13), 7.86 (2H, br. s, H-2′/6′), 7.45 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 7.25 (1H, d, J = 8.8 Hz, H-5′), 4.14 (2H, q, J = 6.9 Hz, –OCH2), 4.19 (3H, s, –OCH3), 1.37 (3H, t, J = 6.9 Hz, –CH3); 13C-NMR: (75 MHz, DMSO-d6): δ 151.9, 148.7, 146.4, 136.8(2×), 126.3(4×), 124.5, 122.0, 121.8(2×), 116.1(2×), 113.1, 111.3, 34.6, 56.1, 15.1; EI-MS: m/z (rel. abund.%), 318 (M+, 100), 317 (5), 287 (5), 273 (7), 258 (12), 243 (9), 230 (10), 168 (6), 152 (11), 141 (9), 140 (43), 123 (11), 115 (13); anal. calcd for C20H18N2O2 (318.14); C, 75.45; H, 5.70; N, 8.80; found: C, 75.44; H, 5.71; N, 8.79.N); 1H-NMR (300 MHz, DMSO-d6): δ 11.06 (1H, s, –NH), 8.04 (2H, s, H-6/9), 8.00 (3H, m, Ar–H) 7.84 (1H, d, J = 4.2 Hz, H-5′), 7.38 (2H, dd, J = 6.4, 3.2, Hz, H-11/12), 7.28 (1H, t, J = 4.2 Hz, H-4′); 13C-NMR: (75 MHz, DMSO-d6): δ 146.1, 142.6, 136.4(2×), 128.0, 127.9, 126.7, 125.9(2×), 125.7(2×), 124.2(2×), 115.5(2×), EI-MS: m/z (rel. abund.%), 250 (M+, 100), 249 (5), 141 (18), 140 (22), 114 (43), 109 (15), 97 (13), 83 (14), 64 (57); anal. calcd for C15H10N2S (250.06); C, 71.97; H, 4.03; N, 11.19; found: C, 71.96; H, 4.04; N, 11.20.N), 1211 (OCH3); 1H-NMR (300 MHz, DMSO-d6): δ 10.61 (1H, s, –NH), 8.06 (2H, s, H-6/9), 8.01 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.85 (1H, br. s, H-2′), 7.82 (1H, d, J = 8.0 Hz, H-6′), 7.38 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 7.20 (1H, d, J = 8.0 Hz, H-5′), 3.90, (3H, s, –OCH3), 3.86 (3H, s, –OCH3); 13C-NMR: (75 MHz, DMSO-d6): δ 151.0, 148.3, 147.8, 137.7(2×), 127.1(2×), 127.0(2×), 123.9, 123.6, 123.1(2×), 116.0, 113.1, 109.9, 57.1, 56.9; EI-MS: m/z (rel. abund.%), 304 (M+, 100), 303 (12), 289 (15), 273 (9), 261 (32), 242 (7), 218 (31), 192 (15), 168 (10), 163 (11), 141 (21) 140 (95), 138 (12), 121 (8), 113 (40), 91 (5), 75 (13); anal. calcd for C19H16N2O2 (304.12); C, 74.98; H, 5.30; N, 9.20; found: C, 74.96; H, 5.31; N, 9.22.N), 1059 (OCH3); 1H-NMR (400 MHz, DMSO-d6): δ 10.82 (1H, s, –OH), 10.65 (1H, s, –OH), 10.27 (1H, s, –OH), 8.70 (2H, s, H-6/9), 8.03 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.42 (2H, dd, J = 6.4, 3.2 H-11/12), 7.09 (1H, d, J = 8.4 Hz, H-6′), 6.51 (1H, d, J = 8.4 Hz, H-5′); 13C-NMR: (100 MHz, DMSO-d6): δ 153.0, 149.2, 144.6, 136.3(2×), 131.9, 126.2(4×), 121.5, 121.2(2×), 115.0(2×), 107.6, EI-MS: m/z (rel. abund.%), 292 (M+, 100) 262 (12), 168 (9), 151 (5), 140 (14), 126 (13), 115 (23), 79 (12); anal. calcd for C17H12N2O3 (292.08); C, 69.86; H, 4.14; N, 9.58; found: C, 69.85; H, 4.14; N, 9.57.N), 1135 (OCH3); 1H-NMR (400 MHz, DMSO-d6): δ 11.05 (1H, s,–NH), 8.12 (2H, s, H-6/9), 8.03 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.40 (2H, dd, J = 6.4, 3.2 Hz, H-11/12), 7.21 (1H, d, J = 8.0 Hz, H-5′), 7.09 (1H, d, J = 8.0 Hz, H-4′), 3.86 (3H, s, –OCH3); 13C-NMR: (100 MHz, DMSO-d6): δ 152.3, 147.6, 143.4, 137.1, 128.1, 127.3(4×), 123.7(2×), 122.9, 120.7(2×), 114.9(2×), 55.9; EI-MS: m/z (rel. abund.%), 368 (M+, 50), 367 (20), 289 (5), 259 (7), 243 (15), 229 (8), 169 (6), 152 (13), 141 (9), 140 (47), 76 (10); anal. calcd for C18H13Br N2O2 (368.02); C, 58.56; H, 3.55; N, 7.59; found: C, 57.69; H, 3.57; N, 7.58.N); 1H-NMR (400 MHz, DMSO-d6): δ 12.05 (1H, s, –NH) 8.29 (2H, dd, J = 7.2, 3.6 Hz, H-2′/6′), 8.21 (2H, s, H-6/9), 8.08 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.68 (3H, m, H-3′/4′/5′), 7.44 (2H, dd, J = 6.4, 3.2 Hz, H-11/12); 13C-NMR: (100 MHz, DMSO-d6): δ 150.6, 138.5–109.8 (CAr); EI-MS: m/z (rel. abund.%), 244 (M+, 72), 243 (6), 141 (10), 140 (28), 114 (29), 104 (5), 89 (6), 77 (99); anal. calcd for C17H12N2 (244.10); C, 83.58; H, 4.95; N, 11.47; found: C, 83.61; H, 4.91; N, 11.45.N), 1029 (OCH3); 1H-NMR (400 MHz, DMSO-d6): δ 12.15 (1H, s, –NH), 8.17 (2H, s, H-6/9), 8.04 (3H, m, H-10/13/6′), 7.58 (1H, s, H-3′), 7.42 (2H, m, H-11/12), 4.05 (3H, s, –OCH3), 3.94 (3H, s, –OCH3); 13C-NMR: (100 MHz, DMSO-d6): δ 150.7, 148.7, 145.6, 135.5, 125.8(6×), 120.1, 115.1, 113.7(2×), 111.2, 55.5, 54.8; EI-MS: m/z (rel. abund.%), 384 (M+, 2, 100), 381 (23), 382 (96), 367 (20), 352 (38), 335 (6), 242 (9), 217 (31), 168 (5), 141 (6), 140 (26), 74 (7); anal. calcd for C19H15 Br N2O2 (382.03); C, 59.55; H, 3.95; N, 7.31; found: C, 59.59; H, 3.97; N, 7.35.N); 1H-NMR (300 MHz, DMSO-d6): δ 10.49 (1H, br s, –NH), 8.28 (1H, d, J = 8.4 Hz, H-6′), 8.22 (2H, s, H-6/9), 8.02 (2H, m, H-10/13), 7.88 (1H, d, J = 2.1 Hz, H-3′), 7.68 (1H, dd, J = 8.4, 2.1 Hz, H-5′), 7.39 (2H, m, H-11/12); 13C-NMR: (75 MHz, DMSO-d6): δ 158.1, 150.7, 131.2(2×), 129.3, 125.9(2×), 125.7, 125.6(2×), 123.5(2×), 123.1, 122.6, 120.2, 111.7(2×) EI-MS: m/z (rel. abund.%), 340 (M+, 90), 187 (5), 172 (7), 156 (6), 141 (8), 140 (23), 80 (4), 78 (72), 63 (70); anal. calcd for C17H10Br F N2 (340.00); C, 59.85; H, 2.95; N, 8.21; found: C, 59.83; H, 2.99; N, 8.18.N); 1H-NMR (300 MHz, DMSO-d6): δ 11.88 (1H, s, –NH), 8.48 (1H, br. s, H-2′), 8.29 (1H, d, J = 7.8 Hz, H-6′), 8.21 (2H, s, H-6/9), 8.08 (2H, dd, J = 6.4, 3.2 Hz, H-10/13), 7.83 (1H, d, J = 7.8 Hz, H-4′), 7.62 (1H, t, J = 7.8 Hz, H-5′), 7.44 (2H, dd, J = 6.4, 3.2 Hz, H-11/12); 13C-NMR: (75 MHz, DMSO-d6): δ 151.9, 137.8(2×), 132.1, 130.0, 129.7, 127.0, 126.8(2×), 126.7, 126.5(2×), 123.0(2×), 120.5, 116.2(2×); EI-MS: m/z (rel. abund.%), 322 (M+, 100), 321 (3), 243 (20), 214 (10); 183 (9), 162 (18), 155 (8), 141 (7), 140 (31), 121 (19), 114 (12), 75 (5); anal. calcd for C17H11BrN2 (322.01); C, 63.18; H, 3.43; N, 8.67; found: C, 63.20; H, 3.45; N, 8.65.Conclusions
A series of 2-aryl-1H-naphtho[2,3-d]imidazole derivatives (1–15) was designed and synthesized as structural analogs of well-known biologically active benzimidazoles. The synthetic strategy employed used a simple and facile condensation reaction between 2,3-diaminonaphthalene and various aryl/hetero aryl aldehydes. Structures of the synthesized compounds were elucidated using MS and NMR spectroscopic techniques. To explore the potential of 2,3-naphthaleneimidazole as a biologically active scaffold, enzyme inhibition assays were carried out against urease. Compounds 4 and 11 were identified as the most active urease inhibitors (IC50 = 34.2 ± 0.72 μM and 42.43 ± 0.65 μM, respectively). Compounds 1, 3, 6, 11, and 15 exhibited excellent antioxidant activity. This trend indicates the selectivity of 2-aryl-1H-naphtho[2,3-d]imidazole derivatives towards remedying various infections, and the most active urease inhibitor and antioxidant compounds 4 and 11 can be developed further to produce more potent drug candidates from this class of compounds.Author contributions
Amina Sultana: synthesis, experiments, and writing original draft preparation. Shafqat Hussain: assistance in synthesis. Aneela Wahab: spectroscopic characterization of compounds. Khalid Mohammed Khan: synthesis scheme, editing and reviewing of an article. Mehreen Lateef, Sikander Khan Sherwani and Hamna Rafiq: biological activities, writing of results and prediction of urease mechanism. Ghulam Fareed and Nazia Fareed: in silico studies, conceptualization, writing, editing and reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the Pakistan Academy of Science, 3-Constitution Avenue, G-5/2, Islamabad-44000, Pakistan, under PAS Project No. 111.References
- C. Röcken, J. Cancer Res. Clin. Oncol., 2023, 149, 467–481 CrossRef PubMed.
- I. Gullo, F. Carneiro, C. Oliveira and G. M. Almeida, Pathobiology, 2017, 85, 50–63 CrossRef PubMed.
- P. A. Chalk, A. D. Roberts and W. M. Blows, Microbiology, 1994, 140, 2085–2092 CrossRef CAS PubMed.
- P. Kafarski and M. Talma, J. Adv. Res., 2018, 13, 101–112 CrossRef CAS.
- F. Wang, W. Meng, B. Wang and L. Qiao, Cancer Lett., 2014, 345, 196–202 CrossRef CAS PubMed.
- S. Kayali, M. Manfredi, F. Gaiani, L. Bianchi, B. Bizzarri, G. Leandro, F. Di Mario and G. L. De' Angelis, Acta Biomed., 2018, 89, 72–76 Search PubMed.
- A. J. P. O. de Almeida, J. C. P. L. de Oliveira, L. V. da Silva Pontes, J. F. de Souza Júnior, T. A. F. Gonçalves, S. H. Dantas, M. S. de Almeida Feitosa, A. O. Silva and I. A. de Medeiros, Oxid. Med. Cell. Longevity, 2022, 2022, 1225578 Search PubMed.
- D. M. Hardbower, T. de Sablet, R. Chaturvedi and K. T. Wilson, Gut Microbes, 2013, 4, 475–481 CrossRef PubMed.
- U. Jain, K. Saxena and N. Chauhan, Helicobacter, 2021, 26(3), e12796 CrossRef CAS PubMed.
- A. Ulfig and L. I. Leichert, Cell. Mol. Life Sci., 2021, 78, 385–414 CrossRef CAS.
- K. Yahiro, Y. Akazawa, M. Nakano, H. Suzuki, J. Hisatune, H. Isomoto, J. Sap, M. Noda, J. Moss and T. Hirayama, Cell Death Discovery, 2015, 1, 15035 CrossRef CAS PubMed.
- M. Alamgir, D. St, C. Black and N. Kumar, Top. Heterocycl. Chem., 2007, 87–118 CAS.
- P. F. Asobo, H. Wahe, J. T. Mbafor, A. E. Nkengfack, Z. T. Fomum, E. F. Sopbue and D. Döpp, J. Chem. Soc., Perkin Trans. 1, 2001, 457–461 RSC.
- A. S. Alpan, S. Zencir, I. Zupkó, G. Coban, B. Réthy, H. S. Gunes and Z. Topcu, J. Enzyme Inhib. Med. Chem., 2009, 24, 844–849 CrossRef CAS PubMed.
- J. G. Walker, A. J. Mackinnon, P. Batterham, A. F. Jorm, I. Hickie, A. McCarthy, M. Fenech and H. Christensen, Br. J. Psychiatry, 2010, 197, 45–54 CrossRef PubMed.
- B. M. Mehta, D. V. Rege and R. S. Satoskar, Am. J. Clin. Nutr., 1964, 15, 77–84 CrossRef CAS PubMed.
- M. Alkaloid, I. Kawasaki, N. Taguchi, T. Yamamoto, M. Yamashita and S. Ohta, Tetrahedron Lett., 1995, 36, 8251–8254 CrossRef.
- R. G. Khalifah, J. I. Rogers and J. Mukherjee, Biochemistry, 1987, 26, 7057–7063 CrossRef CAS PubMed.
- W. Zhang, Y. Ramamoorthy, T. Kilicarslan, H. Nolte, R. F. Tyndale and E. M. Sellers, Drug Metab. Dispos., 2002, 30, 314–318 CrossRef CAS PubMed.
- D. W. Woolley, J. Biol. Chem., 1944, 152, 225–232 CrossRef CAS.
- R. Vinodkumar, S. D. Vaidya, B. V. S. Kumar, U. N. Bhise, S. B. Bhirud and U. C. Mashelkar, Arkivoc, 2008, 2008, 37–49 Search PubMed.
- Z. A. Al-Mudaris, A. S. A. Majid, D. Ji, B. A. Al-Mudarris, S.-H. Chen, P.-H. Liang, H. Osman, S. K. K. J. Din and A. M. S. A. Majid, PLoS One, 2013, 8, e80983 CrossRef PubMed.
- K. F. Ansari and C. Lal, J. Chem. Sci., 2009, 121, 1017–1025 CrossRef CAS.
- S. R. Brishty, M. J. Hossain, M. U. Khandaker, M. R. I. Faruque, H. Osman and S. M. A. Rahman, Front. Pharmacol, 2021, 12, 762807 CrossRef CAS.
- S. M. Sondhi, N. Singh, A. Kumar, O. Lozach and L. Meijer, Bioorg. Med. Chem., 2006, 14, 3758–3765 CrossRef CAS.
- S. Geetha and K. Vijayakumar, Res. J. Pharm. Technol., 2020, 13, 3383 CrossRef.
- Y. Radha, A. Manjula, B. M. Reddy and B. V. Rao, ChemInform, 2012, 43(16) DOI:10.1002/chin.201216122.
- L. Luca, Curr. Med. Chem., 2006, 13, 1–23 Search PubMed.
- B. Narasimhan, D. Sharma and P. Kumar, Med. Chem. Res., 2010, 20, 1119–1140 CrossRef.
- I. Fryšová, J. Slouka and J. Hlavác, Arkivoc, 2006, 2006, 207–212 Search PubMed.
- A. Voskiene, B. Sapijanskaite, V. Mickevicius, K. Kantminiene, M. Stasevych, O. Komarovska-Porokhnyavets, R. Musyanovych and V. Novikov, ChemInform, 2011, 42(38) DOI:10.1002/chin.201138155.
- A. Mobinikhaledi, M. Zendehdel, P. Safari, A. Hamta and S. M. Shariatzadeh, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 165–170 CrossRef CAS.
- G. Navarrete-Vázquez, H. Moreno-Diaz, F. Aguirre-Crespo, I. León-Rivera, R. Villalobos-Molina, O. Muñoz-Muñiz and S. Estrada-Soto, Bioorg. Med. Chem. Lett., 2006, 16, 4169–4173 CrossRef PubMed.
- M. Rashid, H. Rafique, S. Roshan, S. Shamas, Z. Iqbal, Z. Ashraf, Q. Abbas, M. Hassan, Z. U. R. Qureshi and M. H. H. B. Asad, BioMed Res. Int., 2020, 2020, 1–11 CrossRef.
- J. Chen, J. Yang, L. Ma, J. Li, N. Shahzad and C. K. Kim, Sci. Rep., 2020, 10, 2611 CrossRef CAS PubMed.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971–974 CrossRef CAS.
- S. K. Lee, Z. H. Mbwambo, H. Chung, L. Luyengi, E. J. C. Gamez, R. G. Mehta, A. D. Kinghorn and J. M. Pezzuto, Comb. Chem. High Throughput Screening, 1998, 1, 35–46 CrossRef CAS.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |