
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn in-depth physicochemical investigation of drug-loaded core–shell UiO66 nanoMOFs†
Mengli Dinga,
Borja Moreira-Álvarez*b,
Francisco Calderón Celis *b,
Jose Manuel Costa-Fernándezb,
Jorge Ruiz Encinarb and
Ruxandra Gref*a
*b,
Jose Manuel Costa-Fernándezb,
Jorge Ruiz Encinarb and
Ruxandra Gref*a
aInstitut des Sciences Moléculaires d’Orsay, Centre National de la Recherche Scientifique (CNRS), Université Paris-Saclay, 91405 Orsay, France. E-mail: ruxandra.gref@universite-paris-saclay.fr
bDepartment of Physical and Analytical Chemistry, University of Oviedo, Avenida Julian Claveria 8, 33006, Oviedo, Spain. E-mail: borjamoreira@gmail.com; calderonfrancisco@uniovi.es
First published on 5th January 2024
Abstract
Nanosized UiO66 are among the most studied MOF materials. They have been extensively applied in various areas, such as catalysis, gas absorption, electrochemistry, chemical sensing, and biomedical applications. However, the preparation of stable nano-sized UiO66 for drug delivery applications is challenging because of the high tendency of UiO66 to aggregate during storage. To address this issue, we coated UiO66 with oligomers made of crosslinked cyclodextrins. The coated UiO66 exhibited a good stability upon storage for more than three weeks, even for low quantities of coating materials. The resulting core–shell UiO66 were characterized using a set of complementary methods including microscopies, spectroscopies, X-ray diffraction, and thermogravimetric investigations. Size distribution was assessed by orthogonal methods. Cisplatin was loaded in the core–shell nanoparticles, followed by an in-depth analysis by asymmetric flow field-flow fractionation (AF4) hyphenated with inductively coupled plasma-mass spectrometry (ICP-MS). This method combines the extremely high elemental selectivity and ultratrace detection limits of mass spectrometry with the capacity of AF4 to differentiate the diverse populations present in the sample. Free cisplatin and UiO66-associated cisplatin could be well separated by AF4. AF4-ICP-MS/MS analysis provided the exact drug loading, without the need of separating the nanoparticles from their suspension media. These data suggest the potential of AF4-ICP-MS/MS in the optimization of drug delivery systems.
Introduction
Since their first discovery in 1989, metal organic frameworks (MOFs) have attracted extensive attention in various areas, such as catalysis, energy, gas absorption, and biomedicine.1–5 Assembled by coordination between organic linkers and metal ions/clusters, MOFs exhibit versatile supramolecular architectures, both in terms of chemical composition and morphologies. Specifically, a variety of nano-sized MOFs (nanoMOFs) have been widely studied in the drug delivery systems (DDS) area, by virtue of their high capacity to load drugs and release them in a controlled manner, biocompatibility, and biodegradability. Among the most employed families of MOFs are MIL (Materials of the Institute Lavoisier), ZIF (Zeolitic Imidazolate Frameworks), PCNs (Porous Coordination Networks) and UiO (University of Oslo).2,6In particular, UiO66 draws increasing attention, especially since the reported synthesis of defected UiO66.7,8 In 2017, Morris et al. added modulators (acetic, formic, dichloroacetic, or trifluoroacetic acids) during UiO66 preparation in an attempt to improve their physicochemical properties.7 The modulators competed with the organic ligands in coordinating with the metal ions, resulting in the formation of defects in the crystalline structure, which in turn enhanced UiO66's colloidal stability. By controlling both the chemical structure of the modulators and their concentrations, it was possible to modulate the size of the UiO66 within a large range, from less than 50 nm to more than one micron. However, the long term storage stability of UiO66 still needs to be improved.
To circumvent this stability problem, attempts were made to engineer core–shell nanoplatforms by coating UiO66 with chitosan, polydopamine, silica, and other polymers.9–12 For instance, Trushina et al. coated UiO66 with SiO2 resulting in improved colloidal stability in various physiological media.12 However, both the porosity and drug loading ability of UiO66 decreased after coating with SiO2. In other approches, UiO66 were coated with poly(ethylene glycol) (PEG) by click chemistry without affecting their porosity.13 Nevertheless, the synthesis was complex and required the use of large amounts of organic solvents. Therefore, there is a clear need to develop stable core–shell UiO66 nanoMOFs using a simple and green strategy that has no effect on the nanoparticles' porosity.
Due to their well-known biocompatibility and versatility, cyclodextrin (CD) derivatives such as phosphate-CDs (CD-P), polymerized CDs, and CD-based oligomers have been investigated as coating materials to improve nanoMOFs' stability.14–16 Since 2015, our group has been working on the improvement of nanoMOFs stability without affecting their porosity and drug loading ability.14 Functionalized CDs spontaneously anchored onto the surface of MIL100 nanoMOFs. Further coating optimizations were made through the cross-linking of CDs with citric acid first,15 and malic acid later on.16 The resulting CD oligomers could advantageously be functionalized with PEG, and fluorescent molecules. The coated nanoMOFs exhibited good storage stability, and the presence of the coating did not affect porosity and drug loading capacity. However, to our knowledge, the CD-based coatings were only studied on MIL100 and not on other nanoMOFs such as UiO66.
In the last decades, UiO66 nanoMOFs have been widely used as carriers to treat pulmonary diseases, infections, and cancer.17–20 The size of the drug molecules should be smaller than UiO66's windows to enable drug diffusion into the porosity, ensuring an efficient loading. One example of an appropriate drug for loading in UiO66 is cisplatin, the most extensively applied anticancer drug for the treatment of several cancers.21–23 Cisplatin and its prodrug have been successfully loaded within the porosity of UiO66, functionalized or not with amine moieties.18 The presence of cisplatin in the UiO66 was assessed by energy-dispersive X-ray spectroscopy and microwave plasma-atomic emission spectroscopy. Cisplatin loadings reached 4.7 wt%, and 48% of the drug was released within 24 h. However, the spectroscopic techniques used to assess drug loading have poor energy resolution and low sensitivity. In fact, an accurate assessment of cisplatin's loading into UiO66 nanoMOFs would require a very selective and sensitive analytical method.
In this sense, asymmetric flow field-flow fractionation (AF4), in conjunction with more traditional spectroscopic techniques (e.g., UV-Vis, MALS, etc.) has established itself as a powerful analytical tool to characterize nanomaterials,24 and even to assess their drug delivery properties.25,26 Particularly, hyphenated with inductively coupled plasma-mass spectrometry (ICP-MS), AF4-ICP-MS combines the extremely high elemental selectivity and ultratrace detection limits of the mass spectrometry with the capacity of AF4 to differentiate the diverse nanoparticle populations present in the sample.27,28 However, to the best of our knowledge, it has not been applied so far to study drug loading in nanoMOFs.
We aimed here to gain a deep understanding on cisplatin loading into UiO66 nanoMOFs using AF4-ICP-MS/MS. Another novelty here was to coat UiO66 nanoMOFs with CD oligomers by a simple, and organic solvent-free method, to obtain stable, uniform nanoparticles with low toxicity. The coating materials had no effect on UiO66's porosity. Bare and coated UiO66 nanoMOFs were characterized by complementary techniques, such as Transmission Electron Microscopy (TEM), Dynamic Light Scattering (DLS), Nanoparticle Tracking Analysis (NTA), and Tunable Resistive Pulse Sensing (TRPS), to determine their size distribution and morphology. The nanoMOFs' porosity, crystallinity, stability, composition, and toxicity on macrophage J744 cells were investigated. The exact cisplatin incorporation yield and drug distribution within CD-MO@UiO66 were clearly evidenced by AF4-ICP-MS/MS. Noteworthy, the methodology used here avoids the need of separating the nanoMOFs from their suspension media which can lead to artefacts. In a nutshell, we show the ability of AF4-ICP-MS/MS to characterize the cisplatin loaded nanoMOFs throughout the encapsulation process. These investigations set the basis for using AF4-ICP-MS/MS as a diagnostic tool in drug loading and delivery studies.
Materials and methods
Reagents
Zirconyl chloride octahydrate, terephthalic acid, DL-malic acid, sodium phosphate dibasic dodecahydrate, Thiazolyl Blue Tetrazolium Bromide (MTT), cisplatin and absolute ethanol were purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France) without further purification. N,N-dimethylformamide (DMF) was acquired from Alfa Aesar (Kandel, Germany). Acetic acid was purchased from VWR chemicals (Germany). β-CD was obtained from Roquette (France). Milli-Q water and deionized water were obtained from an ELGA water system. Dulbecco's Phosphate Buffered Saline (DPBS) (1X, Gibco) and Dulbecco's Modified Eagle Medium DMEM (1X, Gibco) were used for cell studies. Enzyme Express (1X) TrypLE™, without phenol red (Gibco) was used to detach cells. All products for cell culture studies were sterile.Synthesis of UiO66 nanoMOFs
The synthesis of UiO66 was adapted from a previously reported method using acetic acid as modulator.7 Solution 1 was prepared by dissolving 280 mg zirconyl chloride octahydrate in 40 mL DMF. Solution 2 was prepared by adding 1.5 g terephthalic acid to 30 mL DMF. In a 10 mL vial, 3 mL of solution 1 was mixed with 1 mL of solution 2. Then, acetic acid was added to reach a final concentration of 2.9 M. The reaction mixture was sonicated for 5 min in an ultrasonic bath at room temperature, then heated at 90 °C under gentle magnetic stirring for 18 h. The resulting UiO66 have the general formula Zr6O4(OH)4(C8O4H4)6.7 After three successive washings with DMF and absolute ethanol, the UiO66 nanoMOFs were stored in absolute ethanol until further use.Preparation of CD-MO oligomers
The optimized synthesis of CD-MO oligomers was reported in our previous study.16 Briefly, 1.0 mmol (360 mg) catalyst Na2HPO4·12H2O was added into a 5 mL aqueous solution, followed by mixing with 0.475 mmol (600 mg) β-CD and 2.8 mmol (382 mg) malic acid. After complete solubilization, the solution was concentrated by evaporation (15 min, 160 °C). The obtained dried mixture was heated at 160 °C for 25 min under reduced pressure by connecting the reaction vessel with a suction pump. A yellowish oligomer was recovered and solubilized in 10 mL of deionized water. The mixture was sonicated for 5 min, and then filtered to remove the insoluble fraction. The soluble CD-MO oligomer was dialyzed using a 20 kDa cellulose membrane (Spectrum Laboratories, Rancho Dominguez, USA) and freeze-dried.Preparation of CD-MO@UiO66
The UiO66 ethanolic suspension was centrifuged to discard ethanol and re-dispersed in Milli-Q water. Then, 1 mL of 1 mg mL−1 UiO66 aqueous suspension was added into 1 mL of 0.5 mg mL−1 CD-MO aqueous solution, and the mixture was stirred at room temperature overnight. Finally, CD-MO@UiO66 was centrifuged, and washed with water (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g, 10 min) to remove un-associated CD-MO oligomers. The final nanoMOFs were re-suspended in 1 mL water for further studies.
000 g, 10 min) to remove un-associated CD-MO oligomers. The final nanoMOFs were re-suspended in 1 mL water for further studies.
Preparation of CD-MO@UiO66-cisplatin
The CD-MO@UiO66 suspension was firstly prepared by mixing 1 mL of 1 mg mL−1 UiO66 aqueous suspension with 1 mL of 0.5 mg mL−1 CD-MO oligomers aqueous solution at room temperature overnight. After washing, the CD-MO-coated UiO66 was dispersed in 1 mL water. Then, 1 mL of the resulting 1 mg mL−1 CD-MO@UiO66 was added into 1 mL of a cisplatin solution (2.5 μg mL−1) and the mixture was stirred at room temperature for 24 h. Suspension of CD-MO@UiO66-cisplatin was collected and centrifuged (15000 g, 10 min). The supernatant 1 and pellet 1 were separately collected for Zr/Pt analysis. Pellet 1 was redispersed in water and centrifuge again (15000 g, 10 min). Supernatant 2 and pellet 2 were separately collected again for further analysis.
Characterization of uncoated and CD-MO-coated UiO66
The size and morphology of UiO66 and CD-MO@UiO66 nanoMOFs were characterized by TEM (MET JEOL 1400 (80 kV), Japan). The TEM grids were firstly treated with PELCO easiGlow™ Glow Discharge Cleaning System. Specimens were diluted in water at a final concentration of 50 μg mL−1, dispersed on the grid and excess water was gently wiped out. Average diameters were determined using Image J software based on the observation of at least 50 nanoMOFs.The mean hydrodynamic diameter and polydispersity (PDI) of nanoMOFs were analyzed by DLS (Malvern Panalytical, Nano-ZS, Zetasizer Nano series, Palaiseau, France). Samples were diluted with water at the final concentration of 50 μg mL−1, and the experiment was carried out at 25 °C and an angle of 90°. Experiments were performed in triplicate. Colloidal stability of the uncoated and CD-MO-coated UiO66 was also studied by DLS. To do so, UiO66 and CD-MO@UiO66 nanoMOFs were stored at 4 °C, and analyzed up to 21 days.
The Zeta potential (ZP) of UiO66 and CD-MO@UiO66 in a pH range of 3–9 was determined using a Zetasizer instrument (Malvern Panalytical Nano-ZS, Zetasizer Nano series, Palaiseau, France).
Nanosight (LM10, Malvern Instruments Ltd, Palaiseau, France) was employed to determine both the size distribution and concentrations of nanoMOFs by NTA. The method combines a conventional optical microscope with a laser to illuminate the nanoMOFs in their Brownian motion, and the trajectories are determined using a camera. This enabled to further calculate both the size distribution and the nanoparticle concentration. Results are expressed as the mean of five independent measurements.
TRPS (qNano Gold, Izon, Christchurch, New Zealand) was used to analyze the size distribution of the nanoparticles as they passed through a size-tunable NP200 nanopore. To do so, 1x DPBS was filtered twice with 0.22 μm filters before use as electrolyte. The system was calibrated using 200 nm polystyrene standards at a concentration of 8.2 × 1011 particles per mL and the standard was diluted 1000 times before usage. The stretch of the nanopore (48.85 mm), the voltage (0.46 V), the current (120 nA), pressure (19.82), and particle rate (650 particles per min) were fixed for all the experiments. The freshly prepared nanoparticles were diluted with 1x DPBS to obtain a final concentration of 25 μg mL−1.
The porosity of the UiO66 nanoMOFs before and after coating was determined using an ASAP 2020 (Micromeritics, USA). To measure the Brunauer–Emmett–Teller (BET) surface areas of UiO66 before and after coating with CD-MO oligomers, the samples were first dried at 60 °C overnight to remove free water. After additional degassing at 100 °C overnight, samples were analyzed at −196 °C under high vacuum.
The crystallinity of uncoated and CD-MO-coated UiO66 was investigated by powder X-ray diffraction (PXRD). The experiments were carried out on the MORPHEUS platform at the Laboratoire de Physique des Solides (Université Paris-Saclay, Orsay, France), with a home-made diffraction setup installed on a rotating anode generator (model RUH3R, Rigaku Corp., Japan) at the Cu wavelength (λCuKα = 0.1542 nm) delivered by a multilayer W/Si mirror (Osmic). NanoMOFs powders were filled in cylindric borosilicate capillaries (diameter of 1 mm, WJM-Glass Müller GmbH, Berlin, Germany). Diffraction patterns were collected on a large-area detector MAR345 (marXperts GmBH, Germany) with 150 μm pixel size. The experimental resolution is a Gaussian function with full-width at half-maximum equal to 0.013 Å−1. Extraction of the scattered intensity I as a function of the scattering angle 2θ was obtained from the azimuthal angular integration [0, 2π] of the diffraction patterns with a home-made developed software.
Thermogravimetric analyses (TGA, TGA 4000 Instruments, PerkinElmer, 100–240 V/50–60 Hz) were performed to analyze the composition of UiO66 and to determine the associated amounts of CD-MO oligomers. Samples were first dried at 60 °C in an oven overnight. Approximately 20 mg of samples were accurately weighted and heated from 30 to 600 °C at a heating rate of 3 °C min−1 with an oxygen flow of 20 mL min−1. The association efficiency of CD-MO on UiO66 was calculated based on the dried weight and the mass of residues in CD-MO@UiO66. In addition, the composition of the UiO66, CD-MO oligomers, and CD-MO@UiO66 as dried powders was analyzed by Attenuated Total Reflectance (ATR, PerkinElmer, FT-IR Spectrometer, UATR Two).
Cytotoxicity studies on J744 cells
The cytotoxicity of uncoated and CD-MO-coated UiO66 was investigated using MTT test. Firstly, macrophage J744 cells were seeded in 96-well plates with 10000 cells per well. After incubating at 37 °C for 24 h, cells were treated with various concentrations of both naked and CD-MO-coated UiO66. Nanoparticle suspensions were prepared in sterile water. 20 μL of nanoparticle suspensions were added into each well, to reach final concentrations of 0–0.2 mg mL−1. After 4 h incubation, the DMEM media were discarded from each well and the cells were washed with 200 μL warm PBS, followed by the addition of 200 μL fresh DMEM. Then the cells were incubated overnight. MTT solution was prepared by dissolving MTT powder in 1x DPBS, and filtered (0.22 μm) before usage. The MTT solution (20 μL, 5 mg mL−1) was added into each well and incubated for 1 h. Finally, MTT media was removed, and 150 μL DMSO was added to dissolve formazan crystals. The absorbance was detected with a plate reader (GloMax, Discover Microplate Reader) at the wavelength of 560 nm.
Evaluation of the total metal content
Metal quantification analyses were carried out using a triple quadrupole ICP-MS/MS system equipped with a Micromist nebulizer and a 1.5 mm ID quartz torch injector (Agilent Technologies, Tokyo, Japan). Oxygen was introduced in the reaction cell at 0.35 mL min−1 as cell gas. Zr was acquired in mass-shift mode as 90ZrO+ and Pt was acquired in on-mass mode as 195Pt+ (see ESI† for detailed description of the operational parameters). Determination of total content of Pt and Zr and their corresponding molar ratios was done with external calibration using flow injection analysis (FIA) at 0.7 mL min−1 and a 20 μL injection loop. Pt concentration in calibration solutions was constant (80 ng L−1) while Zr concentration ranged from 0.2 to 20 μg L−1 (Zr/Pt molar ratio ranged from 5 to 500). Pt and Zr ICP-MS standards used for quantification were from Merck (Darmstadt, Germany).Assessment of cisplatin loading in CD-MO@UiO66-cisplatin nanoMOFs
NanoMOFs separation was performed using an AF4 instrument (AF2000 MT Postnova Analytics, Germany). The channel used consisted of a 5 kDa cut-off polyethersulfone (PES) membrane and a 350 μm spacer. Ultrapure water double filtered by 0.1 μm PVDF filters was used as carrier solution. Operational parameters used for the separation are summarized in ESI.† The eluent of the AF4 system was on-line coupled first to the Multiangle light scattering – MALS (PN3621 Postnova Analytics Inc., Germany) detector and then to the ICP-MS/MS instrument. Operation conditions are summarized in SI. Sample recovery calculation and signal integration was done as described elsewhere.29 ICP-MS operational configuration was similar to the previously described for total metal content determination (see ESI†).Results and discussion
Size and morphology characterization of UiO66 nanoMOFs
After the synthesis and purification of UiO66 nanoMOFs schematized in Fig. 1A, their physicochemical properties such as particle size, morphology, and hydrodynamic diameter were assessed. To do so, a set of complementary techniques was required, including TEM, DLS, or NTA, among others.24,30 Firstly, TEM micrographs showed that UiO66 nanoMOFs have homogeneous, round-shape morphologies (Fig. S1A†). The calculated average diameters (103 ± 10 nm) were in agreement with previously reported data.7 | ||
| Fig. 1 Schematic representation of the synthesis of (A) UiO66; (B) CD-MO@UiO66, and (C) CD-MO@UiO66-cisplatin (within the cisplatin structure, H and Cl atoms are represented in blue and green, respectively). | ||
The hydrodynamic diameters and PDI of UiO66 were further analyzed with DLS. As most applications of nanomedicine involve solutions, the hydrodynamic diameter is crucial to correlate nanoparticle sizes with physiological responses.31 As illustrated in Fig. 2A, the mean hydrodynamic diameters of UiO66 were around 130 ± 3 nm, consistent with TEM observations (Fig. S1A†). The low PDI value (0.037 ± 0.024) indicated the narrow UiO66 nanoMOFs size distribution in agreement with TEM investigations. Fig. S2† showed the DLS size and intensity distributions. Despite its universality and simplicity, DLS only allows to study whole populations of NPs. On the opposite, NTA enables to determine the hydrodynamic diameters of individual NPs, as well as the concentration of the NP suspensions, offering in deep quality control of the formulations.32 As shown in Fig. 2A (black line), the average NTA size of UiO66 nanoMOFs was 109 ± 5 nm, in agreement with TEM evaluations.
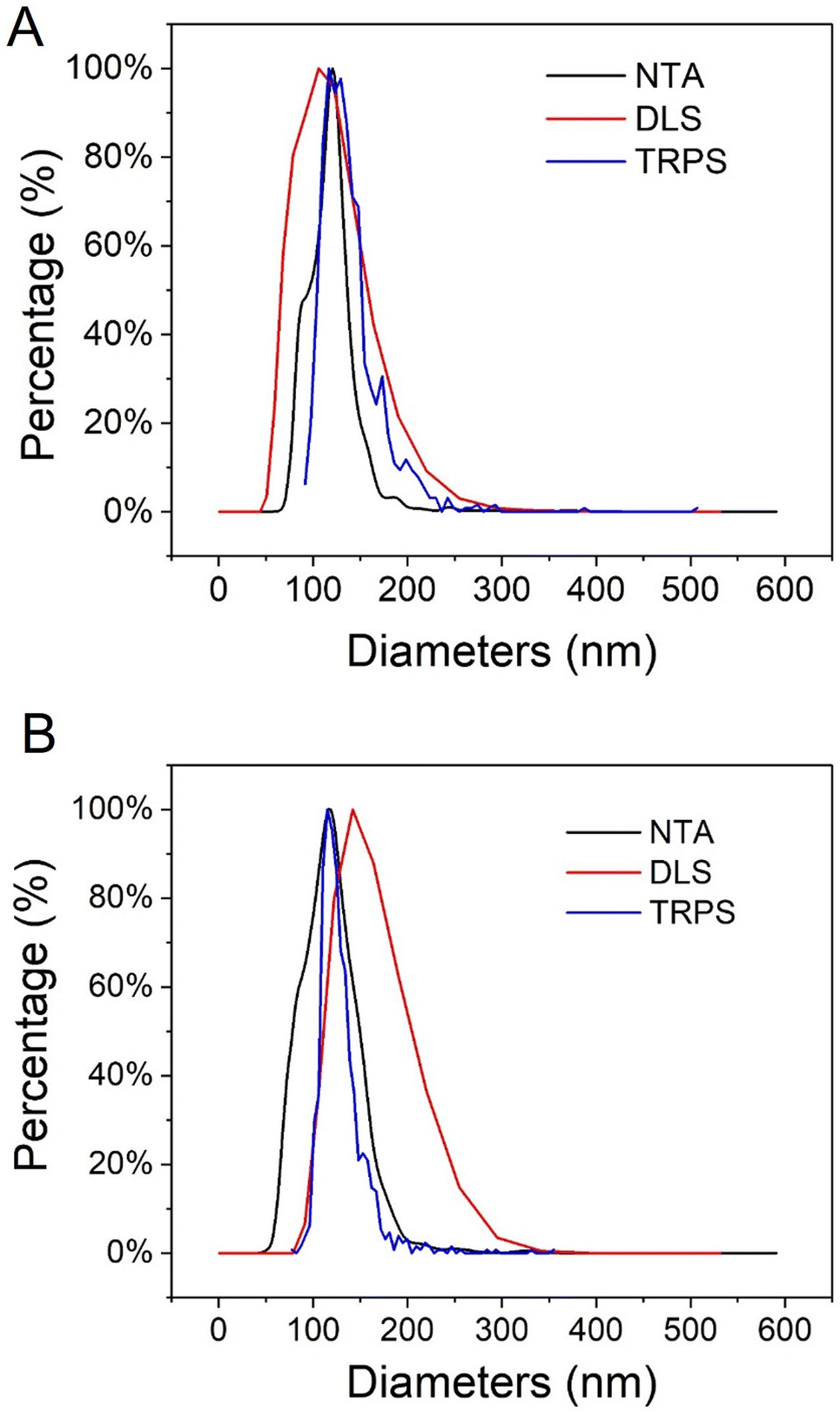 | ||
| Fig. 2 Average hydrodynamic diameters of (A) UiO66 nanoMOFs and (B) CD-MO@UiO66 obtained by DLS (number distribution), NTA and TRPS. | ||
In TRPS, the mean diameter of individual NPs is measured individually as they pass through a size-tunable nanopore. NP size is directly proportional to pore blockade magnitude.33–35 The TRPS size of UiO66 was 105 ± 30 nm (Fig. 2A, blue line). Noteworthy, Fig. 2A shows the good agreement between the different techniques used to determine the size distribution of UiO66 nanoMOFs, highlighting their homogeneous size distribution.32,33,36
Characterization of CD-MO@UiO66
To ensure storage stability, UiO66 nanoMOFs were coated with CD-MO oligomers by incubation in aqueous solutions at room temperature (Fig. 1B). This “green” (organic solvent-free) method has successfully been used to coat CD-base copolymers on MIL 100(Fe) nanoMOFs.16 After coating with CD-MO oligomers, the pH of the resulting CD-MO@UiO66 was 6.5. The coated nanoMOFs were characterized by a set of complementary approaches, as in the case of uncoated nanoMOFs.Investigated by TEM, CD-MO@UiO66 showed similar morphologies and sizes (115 ± 22 nm) as naked UiO66 (103 ± 10 nm) (Fig. S1B†). The hydrodynamic diameter of CD-MO@UiO66 was evaluated as 142 ± 6 nm by DLS, similar to the uncoated UiO66. The mean diameters of CD-MO@UiO66 analyzed by NTA and TRPS were 123 ± 41 nm and 115 ± 25 nm, respectively. Fig. 2B presents a comparison of the size distribution of CD-MO@UiO66 obtained by DLS, NTA and TRPS. There is a good agreement among the data obtained by the three independent methods. Comparison of both coated and uncoated UiO66 suggests that the CD-MO coating did not significantly influence the size of UiO66 nanoMOFs. Moreover, NTA allowed determining the concentration of the nanoMOF suspensions, expressed as particle number per mL. This concentration was similar before and after coating (6.2 × 108 ± 2.0 × 107 and 5.7 × 108 ± 1.41 × 107 particles per mL, respectively). These data suggest that there was no aggregation after the coating process.
The associated amounts of CD-MO oligomers on UiO66 were determined by TGA. As shown in Fig. 3A, there were three weight loss steps within the range of 30–600 °C. The first weight loss step from 30–200 °C corresponds to the evaporation of free and associated water within the UiO66 pores. Then, in the second weight loss step, CD-MO oligomers started to degrade from about 200 °C until complete mass loss at around 600 °C. The amount of associated CD-MO was determined based on the amounts of dried samples and their residues. The association amounts of CD-MO oligomers on UiO66 was 5 wt%.
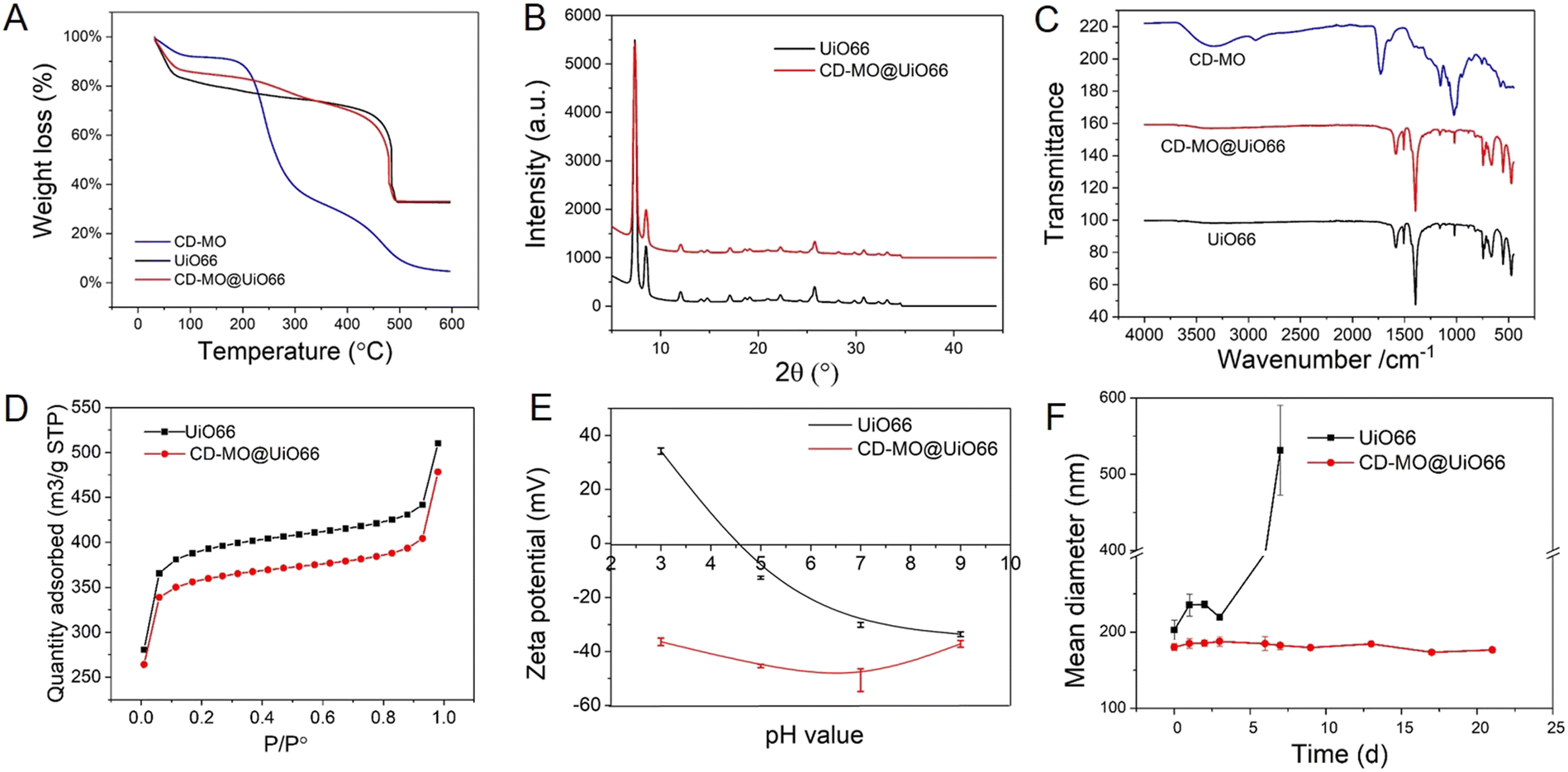 | ||
| Fig. 3 (A) TGA analysis of CD-MO, UiO66, and CD-MO@UiO66; (B) PXRD pattern of UiO66 coated or not with CD-MO oligomers; (C) IR spectra of UiO66, CD-MO, and CD-MO@UiO66; (D) nitrogen adsorption isotherm of UiO66, and CD-MO@UiO66; (E) ZP of UiO66 and CD-MO@UiO66 at pH of 3–9; (F) stability of UiO66 and CD-MO@UiO66 during storage in water at 4 °C within three weeks. The average hydrodynamic diameters were measured by DLS. (black: UiO66, red: CD-MO@UiO66, blue: CD-MO). | ||
The crystalline structure of uncoated and CD-MO-coated UiO66 was investigated by PXRD. Fig. 3B shows similar PXRD patterns of UiO66 and CD-MO@UiO66, suggesting that the CD-MO coating did not affect UiO66's crystalline structure. Similar results were also found in the infrared spectra of bare and coated UiO66 nanoMOFs (Fig. 3C). There were no obvious differences between UiO66 and CD-MO@UiO66, indicating that the CD-MO coatings did not affect the chemical composition of the UiO66.
High nanoMOFs porosity and BET surface areas are critical for drug delivery applications. Here, the porosity of UiO66 coated or not with CD-MO oligomers was investigated by N2 adsorption experiments (Fig. 3D). As calculated, the BET surface area of UiO66 before and after coating with oligomers was 1494 ± 30 m2 g−1 and 1370 ± 42 m2 g−1 respectively, without significant variation (<10%) between them. This suggested that CD-MO coating did not affect the porosity of UiO66.
ZP measurements enabled to investigate the surface charge of UiO66 coated or not with CD-MO oligomers, in a pH range of 3–9. As shown in Fig. 3E, the ZP of UiO66 showed a pH-dependent profile. When the pH increased from 3 to 9, UiO66's ZP decreased from +35 mV to −30 mV. In contrast, the ZP of CD-MO coated UiO66 was always negative whatever the pH. These dramatic ZP differences supported the successful coating of UiO66 with CD-MO. The colloidal stability of uncoated and CD-MO-coated UiO66 was evaluated in aqueous media (pH 6.5) within three weeks. As illustrated in Fig. 3F, UiO66 aggregated rapidly, and their size became larger than 400 nm within one week. In contrast, the size of CD-MO@UiO66 didn't change for up to three weeks, indicating the good colloidal stability of CD-MO@UiO66. This suggested that the oligomer coating could efficiently improve UiO66's colloidal stability.
Therefore, compared with other types of coatings based on silica or dopamine, the coating with CD-MO offers the advantage of simplicity and avoids the need of organic solvents or reactants,9–12 More importantly, CD-MO coatings did not affect the physico chemical properties of the nanoMOFs.
Cytotoxicity study on macrophage J744 cells
As the low toxicity of CD-MO oligomers has been evidenced in our previous study,16 the cytotoxicity of UiO66 and CD-MO@UiO66 were investigated here on macrophage J744 cells. The half-maximal inhibitory concentration (IC50), corresponding to the concentration of a material needed to inhibit a biological process by 50%, is indicative of the nanoparticles' toxicity.As shown in Fig. 4, the cell viability was higher than 85% whatever the UiO66 and CD-MO@UiO66 concentrations within the range 0–200 μg mL−1. No IC50 was obtained in this range, achieved within this concentration, suggesting the low toxicity of both naked and coated UiO66 and with further studies, their potential for biomedical applications.
 | ||
| Fig. 4 Macrophage J744 cells dose-response curves after treatment with nanoparticles (MTT assay). Cell viability was tested after treatment with (A) UiO66; (B) CD-MO@UiO66. Results were exploited as mean ± SD for IC50 values obtained from three or four repeats for each nanoparticle sample. | ||
Characterization of CD-MO@UiO66-cisplatin with ICP-MS/MS
As schematized in Fig. 1C, the anticancer drug cisplatin was loaded within CD-MO@UiO66 by impregnation. The average hydrodynamic diameters of CD-MO@UiO66-cisplatin obtained by DLS were 272 ± 9 nm. They were larger as compared to the unloaded nanoMOFs, most probably because of bridging effects after drug loading. To investigate the drug loading and purification process, FIA-ICP-MS/MS was used to determine the element contents of Zr (UiO66 nanoMOFs) and Pt (cisplatin) through each step of the loading process. Fig. 5 details the cisplatin drug loading and purification process, and the products obtained at each step are indicated by A, B, C, D, E.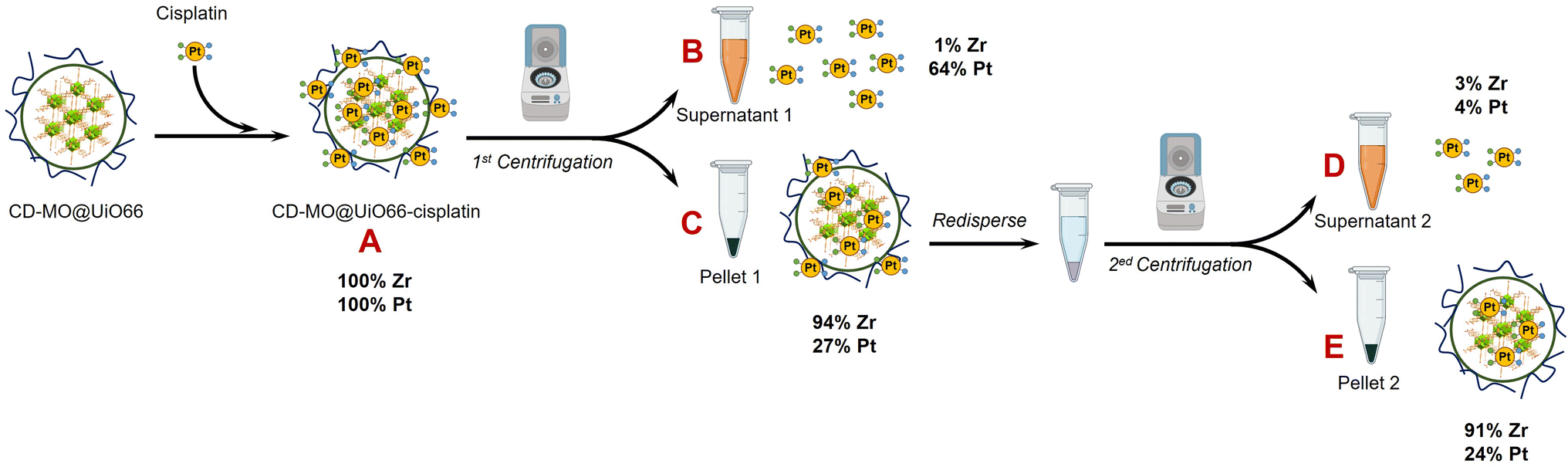 | ||
| Fig. 5 ICP-MS/MS characterization steps during the preparation process of CD-MO@UiO66-cisplatin. Mass balances refer to the mass of Pt and Zr in A. | ||
Firstly, the total amount of Zr and Pt within CD-MO@UiO66-cisplatin was determined. ICP-MS/MS analysis of CD-MO@UiO66-cisplatin (Fig. 5A) resulted in a concentration value of 1.04 ± 0.09 mM of Zr and 3.54 ± 0.13 μM of Pt. These results provided a theoretical ratio of 0.020 cisplatin molecules per nanoMOF cell unit, considering that each nanoMOF cell unit contains 6 atoms of Zr (Table 1A). To explore the amount of the cisplatin actually associated with nanoMOFs and therefore to discriminate the free from the associated cisplatin, CD-MO@UiO66-cisplatin MOFs were purified by centrifugation. Metal contents of Zr and Pt in CD-MO@UiO66-cisplatin obtained herein will be regarded as reference (100%) to which compare subsequent quantifications.
| Step (Fig. 5) | Sample | Cisplatin/nanoMOF cell unit (Zr6) | Encapsulation efficiency (EE, %) |
|---|---|---|---|
| A | CD-MO@UiO66-cisplatin | 2.0 × 10−2 | — |
| C | Pellet 1 | 6.6 × 10−3 | 27 ± 3% |
| E | Pellet 2 | 5.8 × 10−3 | 24 ± 5% |
After a first centrifugation, both Zr and Pt were quantified in supernatant 1 (Fig. 5B) and pellet 1 (Fig. 5C), respectively. ICP-MS/MS quantification revealed that 94 ± 8% of Zr was recovered in the pellet 1, and barely 0.9 ± 0.1% of the Zr in the CD-MO@UiO66-cisplatin nanoMOFs remained in the supernatant. This implies that the nanoMOFs can be efficiently recovered by centrifugation with negligible losses in the supernatant. Nevertheless, the amount of Pt that remains with the nanoMOFs in the pellet 1 is just 27 ± 3%, whereas most of the Pt (64 ± 7%), corresponding to free cisplatin, is found in the supernatant 1. In this way, the molar ratio of Pt/Zr shows a more accurate estimation of the cisplatin drug loading capacity within nanoMOFs, with 0.0066 cisplatins per nanoMOF cell unit (only one out of 151 nanoMOFs cells are occupied with Pt) (Table 1C).
Noteworthy, this value is significantly lower than the theoretical one obtained prior to centrifugation (Table 1A), because of the removal during the centrifugation of all free cisplatin and possibly some loosely adsorbed over the nanoMOFs surface.
However, it cannot be overruled that the centrifugation process may be releasing some of the loaded cisplatin. In order to check this issue, the pellet 1 was redispersed and centrifuged again using the same conditions, and the supernatant 2 (Fig. 5D) and pellet 2 (Fig. 5E) were analyzed. After the second centrifugation, most of the Zr (91 ± 8%) was recovered in the pellet 2, which is consistent with the first centrifugation result.
On the other hand, only 4 ± 1% of the Pt in the supernatant 2 was determined, which hardly corresponded to 15 ± 3% of the Pt present in pellet 1. This means that centrifugation does not seem to release loaded cisplatin. Finally, only 24 ± 5% of the Pt originally added (Fig. 5A) was associated to the nanoMOFs after purification. As most of the Pt is kept in the pellet 2 after the second centrifugation, this Pt can be confidently assumed to correspond to nanoMOF-loaded or tightly associated cisplatin.
As a result, the molar ratio of Pt/Zr determined in the pellet 2 (0.0058 cisplatins per nanoMOF cell unit) (Table 1E), slightly lower than in the pellet 1 due to further purification of free and/or adsorbed cisplatin, shall be a more accurate estimation of the cisplatin loaded into the nanoMOFs. This result highlights the necessity of purification after drug loading process.
AF4-ICP-MS/MS analysis of UiO66 nanoMOFs and free cisplatin
Despite the prowess of ICP-MS/MS in the determination of elemental quantities of Pt and Zr, resulting in an extremely useful tool to evaluate the CD-MO@UiO66-cisplatin drug loading and purification processes, there is one major limitation to be considered. As previously mentioned, Zr and Pt elemental signals determined for CD-MO@UiO66-cisplatin correspond to the total amounts of each element, and they cannot be discriminated or deconvoluted in the different species the element is present. This could lead to inaccurate conclusions regarding the drug loading capacity of the nanoMOFs. Therefore, to have a deeper insight of the species among which Zr and Pt are distributed, we have resorted to the hyphenation of ICP-MS with the separation technique AF4, which has proved extremely useful in the separation of nanoparticles populations.27 Moreover, as we have recently demonstrated, combination of AF4 with online molecular (MALS) and elemental (ICP-MS/MS) detectors is an invaluable tool to characterize nanostructures since it can separate them (AF4), determine their hydrodynamic size (MALS), quantify the elemental composition and determine stoichiometries and molar ratios (ICP-MS/MS) all together in one single analysis.29The optimum AF4-MALS-ICP-MS/MS instrumental conditions are shown in Tables S1 and S2.† CD-MO@UiO66 nanoMOFs and pure cisplatin were separately injected and analyzed, and the fractograms are shown in Fig. 6. AF4-ICP-MS/MS analysis of CD-MO@UiO66 resulted in a single Zr peak at around 5 min. In the case of pure cisplatin, Pt fractogram showed a single peak at a retention time of 6–7 min, clearly separated from the nanoMOFs peak. This means that the free cisplatin and the nanoMOFs present in the same sample can be well separated using AF4, which enables us to characterize better the species distribution and cisplatin loading efficiency.
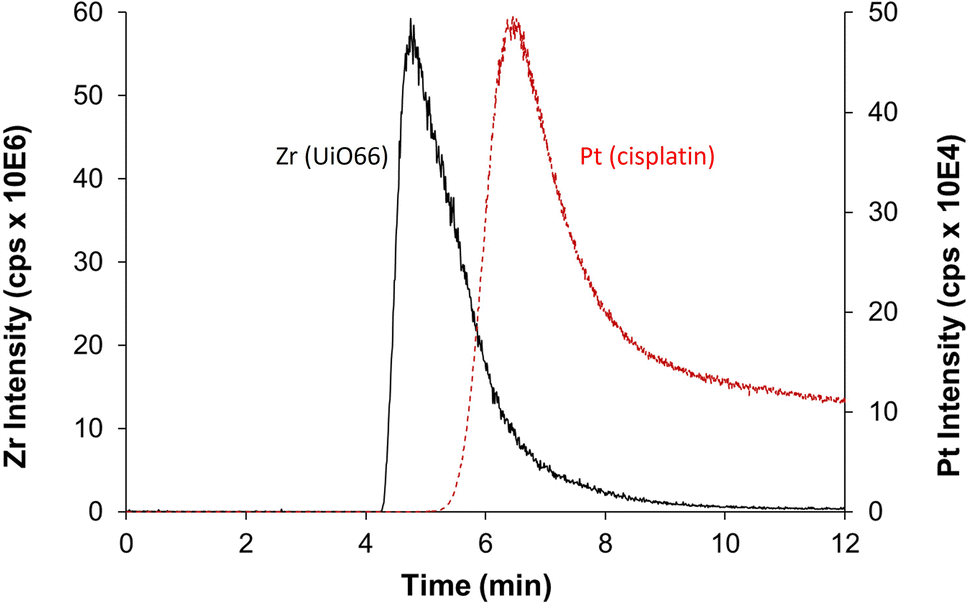 | ||
| Fig. 6 AF4-ICP-MS/MS fractogram of Zr (black, represents for UiO66) in CD-MO@UiO66 sample and Pt (red, represents for cisplatin) in pure cisplatin. | ||
Determination of CD-MO@UiO66-cisplatin drug loading efficiency with AF4-ICP-MS/MS
Collected samples (A, B, C, D, E) indicated in Fig. 5 were individually analyzed with AF4-ICP-MS/MS, and the resulting fractograms are shown in Fig. 7 using the same sample code (A, B, C, D, E). As shown in Fig. 7A, AF4-ICP-MS/MS analysis of loaded CD-MO@UiO66-cisplatin nanoMOFs (Fig. 5A) resulted in a Zr peak similar to the one obtained for CD-MO@UiO66 (Fig. 6). Noteworthy, MALS signal showed a single peak at the same elution time (4–5 min), with a hydrodynamic size population distribution between 160 and 280 nm (Fig. S3†), which was consistent with the DLS result of CD-MO@UiO66-cisplatin. In contrast to Zr, there is not a single Pt peak in the fractogram of CD-MO@UiO66-cisplatin (Fig. 7A). Most of Pt coeluted with Zr, however a Pt shoulder was clearly observed at the retention time of free cisplatin.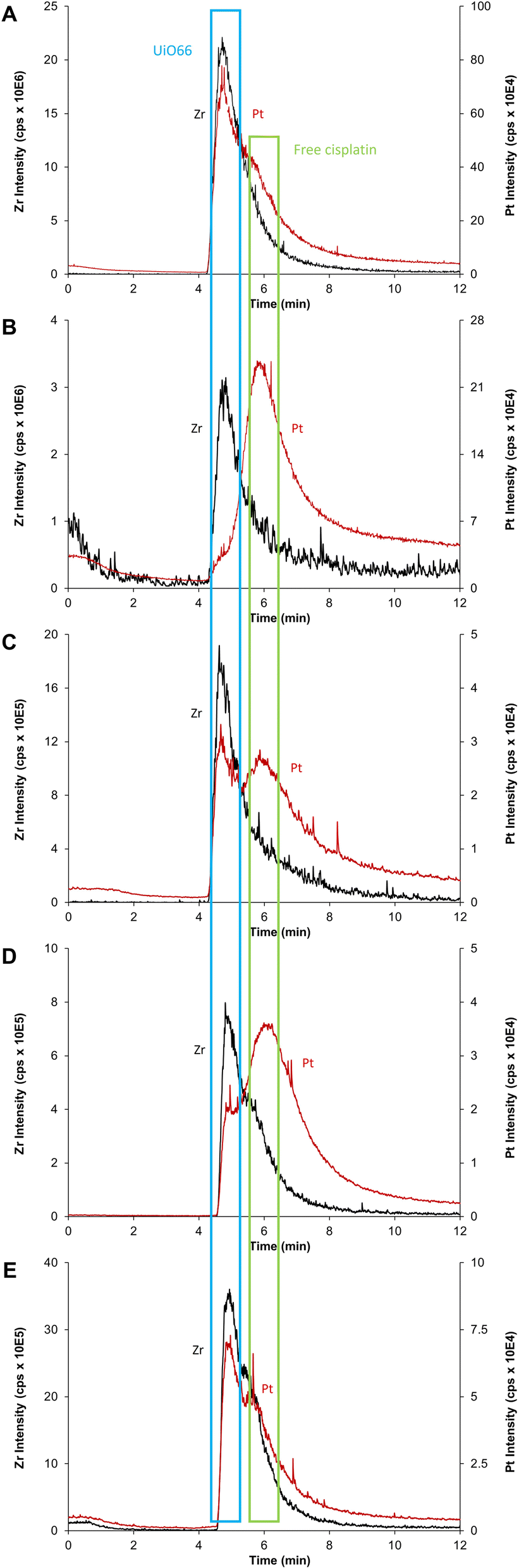 | ||
| Fig. 7 AF4-ICP-MS/MS fractogram of Zr (black, corresponds to UiO66) and Pt (red, corresponds to cisplatin) for (A) CD-MO@UiO66-cisplatin, (B) CD-MO@UiO66-cisplatin supernatant 1, (C) CD-MO@UiO66-cisplatin pellet 1, (D) CD-MO@UiO66-cisplatin supernatant 2, (E) CD-MO@UiO66-cisplatin pellet 2. | ||
After the first centrifugation, the Zr signal in AF4-ICP-MS/MS fractograms obtained for both supernatant 1 (Fig. 5B) and pellet 1 (Fig. 5C) were very similar to the ones obtained for the CD-MO@UiO66. This indicates that the only Zr species eluting is the UiO66 nanoMOFs (Fig. 7B and C). In contrast, the Pt peak profiles obtained for the CD-MO@UiO66-cisplatin, pellet 1 and supernatant 1 clearly differ. Unlike the CD-MO@UiO66-cisplatin profile, in which Pt mainly coeluted with Zr, most of Pt was observed to correspond to free cisplatin (Fig. 7B) in the supernatant 1.
Interestingly, as shown in Fig. 7C, there are two peaks observed in the Pt fractograms of redispersed pellet (Fig. 5C), confirming the coexistence of still some free cisplatin and associated cisplatin after the first centrifugation.
Supernatant 2 (Fig. 5D), obtained after the centrifugation of the resuspended pellet 1, hardly contains either nanoMOFs or cisplatin as demonstrated by the very low Zr and Pt total content determined (3 and 4%, respectively). Its AF4-ICP-MS/MS analysis resulted in a similar Pt peak profile as the supernatant 1, with a very dominant peak of free cisplatin eluting at 6 min (Fig. 7D) as it was previously observed for supernatant 1 (Fig. 5B). Most of the Pt and Zr originally present in pellet 1 was found in the new redispersed pellet 2 instead (Fig. 7E). Unlike in the pellet 1 (Fig. 7C), where the Pt signal at the retention time of the free cisplatin was still significant, the vast majority of the Pt signal of redispersed pellet 2 closely followed the Zr (nanoMOFs) profile.
It appears that while the first centrifugation removed most of the un-associated cisplatin (64% of the Pt originally added), the second centrifugation only removed a small portion (4%) of the Pt present after the first centrifugation (27%) that likely corresponded to loosely adsorbed cisplatin onto the surface of nanoMOFs. In fact, it seems that centrifugation does not promote any significant cisplatin release from the nanoMOFs. Therefore, total metal content by FIA-ICP-MS/MS and AF4-ICP-MS/MS results evidence that two centrifugation processes were required to remove completely the un-associated and loosely adsorbed cisplatin. Therefore, the molar ratio cisplatin/nanoMOFs determined for the pellet 2 after the second centrifugation could be considered as an accurate indicator of the actual cisplatin internally (and stably) loaded within the nanoMOFs (Table 1).
Conclusions
Core–shell CD-MO@UiO66 nanocomposites with good colloidal stability were successfully obtained by coating UiO66 nanoMOFs with CD-MO oligomers using a simple and organic free method. The unique structure of CD-MO oligomers comporting numerous carboxyl groups is favorable for further functionalization with fluorescent moieties or tumor targeting units. The presence of the coating did not affect UiO66's size, morphology, porosity, crystallinity, and chemical composition. CD-MO@UiO66 were loaded with cisplatin. CD-MO@UiO66 exhibited low toxicity towards macrophage cell line. With further studies, cisplatin-loaded CD-MO@UiO66 could find potential applications for cancer treatment.FIA-ICP-MS/MS (total element content) in conjunction with AF4-ICP-MS/MS (elemental speciation) proved to be a powerful tool to determine the real drug loading directly on nanoMOFs suspensions, without the need of separation. Indeed, non-encapsulated free cisplatin and loaded cisplatin could be distinguished within the AF4 separation process. The analysis of various samples at different stages of the encapsulation and purification processes gave a clear picture of the incorporation yields and drug distribution within the nanoMOFs and their suspension medium. AF4-ICP-MS/MS appeared to be a sensitive and elegant strategy to provide a strong control over the different production steps, leading to fully characterized drug-loaded MOFs. It could be an asset in the design of other nanomedicines.
This study sets up the basis of a highly sensitive methodology that could be applied to other types of multifunctional nanoparticles, loaded with drugs and with different types of coatings.
Author contributions
Conceived the study: RG, JMCF and JRE. Performed the experiments: MD, BMA and FCC. Data treatment: MD, RG, BMA, FCC, JMCF and JRE. Wrote the manuscript: MD, RG, BMA, FCC, JMCF and JRE. Funding acquisition: RG, JMCF and JRE. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by a public grant overseen by the French National Research Agency (ANR) as part of the “Investissements d’Avenir” program (Labex NanoSaclay, reference: ANR-10-LABX-0035), by ANR-20-CE19-0020, by Spanish Ministry of Science and Innovation (PID2019-109698GB-I00) and the Principality of Asturias GRUPIN IDI 2021/000081. M.D. acknowledges support from China Scholarship Council (CSC, No. 202108440030). We thank Stéphan Rouzière for help with the PXRD measurement. F.C.C. thank Principality of Asturias for the Margarita Salas Joven postdoctoral grant (AYUD/2021/58459). Authors also wish to thank Agilent Technologies for the technical support.Notes and references
- S. He, L. Wu, X. Li, H. Sun, T. Xiong, J. Liu, C. Huang, H. Xu, H. Sun, W. Chen, R. Gref and J. Zhang, Acta Pharm. Sin. B, 2021, 11, 2362 CrossRef CAS PubMed.
- M. Ding, W. Liu and R. Gref, Adv. Drug Delivery Rev., 2022, 190, 114496 CrossRef CAS PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed.
- Y. Sun, L. Zheng, Y. Yang, X. Qian, T. Fu, X. Li, Z. Yang, H. Yan, C. Cui and W. Tan, Nano-Micro Lett., 2020, 12, 103 CrossRef CAS PubMed.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962 CrossRef CAS.
- V. F. Yusuf, N. I. Malek and S. K. Kailasa, ACS Omega, 2022, 7, 44507 CrossRef CAS PubMed.
- W. Morris, S. Wang, D. Cho, E. Auyeung, P. Li, O. K. Farha and C. A. Mirkin, ACS Appl. Mater. Interfaces, 2017, 9, 33413 CrossRef CAS PubMed.
- J. Xiong, L. Wang, X. Qin and J. Yu, Mater. Lett., 2021, 302, 130427 CrossRef CAS.
- L. Li, S. Han, S. Zhao, X. Li, B. Liu and Y. Liu, RSC Adv., 2020, 10, 45130 RSC.
- H. Chen, Y. Fu, K. Feng, Y. Zhou, X. Wang, H. Huang, Y. Chen, W. Wang, Y. Xu, H. Tian, Y. Mao, J. Wang and Z. Zhang, J. Nanobiotechnol., 2021, 19, 298 CrossRef CAS PubMed.
- N. Rabiee, M. Bagherzadeh, M. Heidarian Haris, A. M. Ghadiri, F. Matloubi Moghaddam, Y. Fatahi, R. Dinarvand, A. Jarahiyan, S. Ahmadi and M. Shokouhimehr, ACS Appl. Mater. Interfaces, 2021, 13, 10796 CrossRef CAS PubMed.
- D. B. Trushina, A. Y. Sapach, O. A. Burachevskaia, P. V. Medvedev, D. N. Khmelenin, T. N. Borodina, M. A. Soldatov and V. V. Butova, Pharm, 2022, 14, 1325 CAS.
- I. Abanades Lazaro, S. Haddad, S. Sacca, C. Orellana-Tavra, D. Fairen-Jimenez and R. S. Forgan, Chem, 2017, 2, 561 CAS.
- V. Agostoni, P. Horcajada, M. Noiray, M. Malanga, A. Aykac, L. Jicsinszky, A. Vargas-Berenguel, N. Semiramoth, S. Daoud-Mahammed, V. Nicolas, C. Martineau, F. Taulelle, J. Vigneron, A. Etcheberry, C. Serre and R. Gref, Sci. Rep., 2015, 5, 7925 CrossRef CAS PubMed.
- J. Qiu, X. Li, K. Steenkeste, N. Barroca-Aubry, C. Aymes-Chodur, P. Roger, J. M. Casas-Solvas, A. Vargas-Berenguel, C. Rihouey, L. Picton and R. Gref, Int. J. Pharm., 2020, 581, 119281 CrossRef CAS PubMed.
- M. Ding, J. Qiu, S. Rouziere, C. Rihouey, L. Picton and R. Gref, Int. J. Mol. Sci., 2023, 24, 1757 CrossRef CAS PubMed.
- B. M. Jarai, Z. Stillman, L. Attia, G. E. Decker, E. D. Bloch and C. A. Fromen, ACS Appl. Mater. Interfaces, 2020, 12, 38989 CrossRef CAS PubMed.
- K. A. Mocniak, I. Kubajewska, D. E. M. Spillane, G. R. Williams and R. E. Morris, RSC Adv., 2015, 5, 83648 RSC.
- N. Rakhshani, N. Hassanzadeh Nemati, A. R. Saadatabadi and S. K. Sadrnezhaad, J. Drug Delivery Sci. Technol., 2021, 66, 102881 CrossRef CAS.
- H. Chen, C. Qiu, Y. Jiang, X. Liao, D. Wu, M. Shen and T. Ding, Food Sci. Hum. Wellness, 2022, 11, 269 CrossRef CAS.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364 CrossRef CAS PubMed.
- L. Qi, Q. Luo, Y. Zhang, F. Jia, Y. Zhao and F. Wang, Chem. Res. Toxicol., 2019, 32, 1469 Search PubMed.
- A. M. Florea and D. Busselberg, Cancers, 2011, 3, 1351 Search PubMed.
- B. Moreira-Alvarez, L. Cid-Barrio, H. S. Ferreira, J. M. Costa-Fernández and J. R. Encinar, J. Anal. At. Spectrom., 2020, 35, 1518 RSC.
- J. Bian, N. Gobalasingham, A. Purchel and J. Lin, Molecules, 2023, 28, 4169 CrossRef CAS PubMed.
- B. Roda, V. Marassi, A. Zattoni, F. Borghi, R. Anand, V. Agostoni, R. Gref, P. Reschiglian and S. Monti, Anal. Bioanal. Chem., 2018, 410, 5245 CrossRef CAS PubMed.
- M. Menendez-Miranda, M. T. Fernandez-Arguelles, J. M. Costa-Fernandez, J. R. Encinar and A. Sanz-Medel, Anal. Chim. Acta, 2014, 839, 8 CrossRef CAS PubMed.
- D. Bouzas-Ramos, J. I. Garcia-Alonso, J. M. Costa-Fernandez and J. Ruiz Encinar, Anal. Chem., 2019, 91, 3567 CrossRef CAS PubMed.
- B. Moreira-Alvarez, A. L. Larraga-Urdaz, A. Fuentes-Cervantes, M. L. Fernandez-Sanchez, J. M. Costa-Fernandez and J. R. Encinar, Talanta, 2023, 256, 124309 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871 RSC.
- C. M. Maguire, M. Rosslein, P. Wick and A. Prina-Mello, Sci. Technol. Adv. Mater., 2018, 19, 732 CrossRef CAS PubMed.
- T. Bourguignon, A. A. Torrano, L. Houel-Renault, A. Machelart, P. Brodin and R. Gref, Int. J. Pharm., 2021, 610, 121202 CrossRef CAS PubMed.
- F. Caputo, R. Vogel, J. Savage, G. Vella, A. Law, G. Della Camera, G. Hannon, B. Peacock, D. Mehn, J. Ponti, O. Geiss, D. Aubert, A. Prina-Mello and L. Calzolai, J. Colloid Interface Sci., 2021, 588, 401 CrossRef CAS PubMed.
- G. S. Roberts, S. Yu, Q. Zeng, L. C. Chan, W. Anderson, A. H. Colby, M. W. Grinstaff, S. Reid and R. Vogel, Biosens. Bioelectron., 2012, 31, 17 CrossRef CAS PubMed.
- M. Chaupard, M. Frutos and R. Gref, Part. Part. Syst. Charact., 2021, 38, 2100022 CrossRef CAS.
- E. Pancani, J. Mathurin, S. Bilent, M.-F. Bernet-Camard, A. Dazzi, A. Deniset-Besseau and R. Gref, Part. Part. Syst. Charact., 2018, 35, 1700457 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07098k |
| This journal is © The Royal Society of Chemistry 2024 |