
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetric dicyanobenzothiadiazole (DCBT) dyes with a 1.5 eV excited state reduction potential range†
Qing Yun Li a,
Ethan C. Lamberta,
Ravinder Kaura,
Nathan I. Hammera and
Jared H. Delcamp*ab
a,
Ethan C. Lamberta,
Ravinder Kaura,
Nathan I. Hammera and
Jared H. Delcamp*ab
aDepartment of Chemistry and Biochemistry, University of Mississippi, University, 322 Coulter Hall, MS 38677, USA. E-mail: delcamp@olemiss.edu
bMaterials and Manufacturing Directorate, Air Force Research Laboratory, 2230 Tenth Street, Wright-Patterson AFB, OH 45433, USA
First published on 21st February 2024
Abstract
Strong molecular photooxidants are important in many disciplines including organic synthesis and renewable energy. In these fields, strongly oxidizing chromophores are employed to drive various transformations from challenging bond formations to energy storage systems. A range of photooxidant strengths are needed to drive these processes. A series of 8 symmetrically bisarylated 5,6-dicyano[2,1,3]benzothiadiazole (DCBT) dyes were studied for their tunability toward breadth of light absorption and photooxidant strength. The dye oxidation strength and light absorption tunability is the result of appending various aryl substituents on the periphery of the DCBT core which shows remarkable tunability of the final chromophore. The dyes are studied via steady-state absorption and emission, time-correlated single photon counting, computational analysis, and cyclic voltammetry. In changing the peripheral aryl substituents via electronics, sterics, and π-conjugation length, a series of dyes are arrived at with a dramatic 1.5 eV range in oxidizing strength and >200 nm (0.95 eV) absorption maxima tunability. Furthermore, two dyes in the series exhibit strong oxidizing strength while still approaching red light absorbance (>650 nm onset) which provides unique opportunities for the use of lower energy light to affect chemical transformations. Ultimately, this series provides options for photooxidations that allow for energetic tuning and selectivity for a given chemical transformation.
Introduction
Strong photooxidants are of interest in multiple fields such as sustainable synthetic chemistry1–9 and solar-to-fuel generation.10–18 In these systems, a strong photooxidant is often used to create greater driving forces in order to achieve higher efficiencies and to access new reactions.4,19–23 Depending on the photooxidant used, selectivity towards certain processes can also be increased.23–26 Molecular photooxidants are often characterized by two important features, oxidizing strength as well as excited state (ES) lifetime. Thus, literature known photooxidants are commonly metal-based, such as [Ru(bpy)3]2+, given that organic photosensitizers often have short-lived excited state lifetimes in the nanosecond regime.27 However, organic dyes exhibiting much longer ES lifetimes in the microsecond regime are becoming more common such as with the widely studied acridinium based photooxidants.8,19,23 More recently, a series of benzo[c][1,2,5]thiadiazole-5,6-dicarbonitrile (DCBT) based dyes have been synthesized and studied for their photooxidizing abilities to reveal a dye (QL11) of strong oxidizing strength as well as a long lived ES lifetime (16 μs measured by transient absorption spectroscopy).28The exceptionally withdrawn nature of the DCBT core makes it an ideal addition to a strong photooxidant systems since this core can be used in photoinduced charge transfer systems to oxidize weakly donating groups appended to it.29 While the literature known DCBT dyes showcased symmetric and asymmetric moieties, the symmetric photosensitizers exhibited greater oxidizing strength as well as longer lived excited state lifetimes in comparison to their asymmetric counterparts,28 making symmetric DCBT dyes of greater interest and the focus of this work. Furthermore, the ease of synthetic access to a symmetric, doubly aryl substituted derivative allows for rapidly accessed dyes, resulting in an array of photooxidants of varying oxidation strengths.28,29
In this report, a series of six new symmetrical DCBT based dyes were synthesized along with 3 known dyes (Fig. 1). All 9 dyes were studied in order to assess the effect of the varying aryl groups on the oxidizing ability of the molecule. As previously shown, the energetics of a DCBT dye is highly tunable through the usage of various aryl substituents.28 A range of electronic effects are probed through derivatives containing a cyano group, an alkoxy group, thiophene, and multiple amine groups with varying donation strength. A tolyl derivative is also synthesized to test the effect of increased steric hindrance inducing a twisted π-system. Furthermore, a derivative featuring an extra phenyl spacer is also synthesized to see the effects of extended π-conjugation.
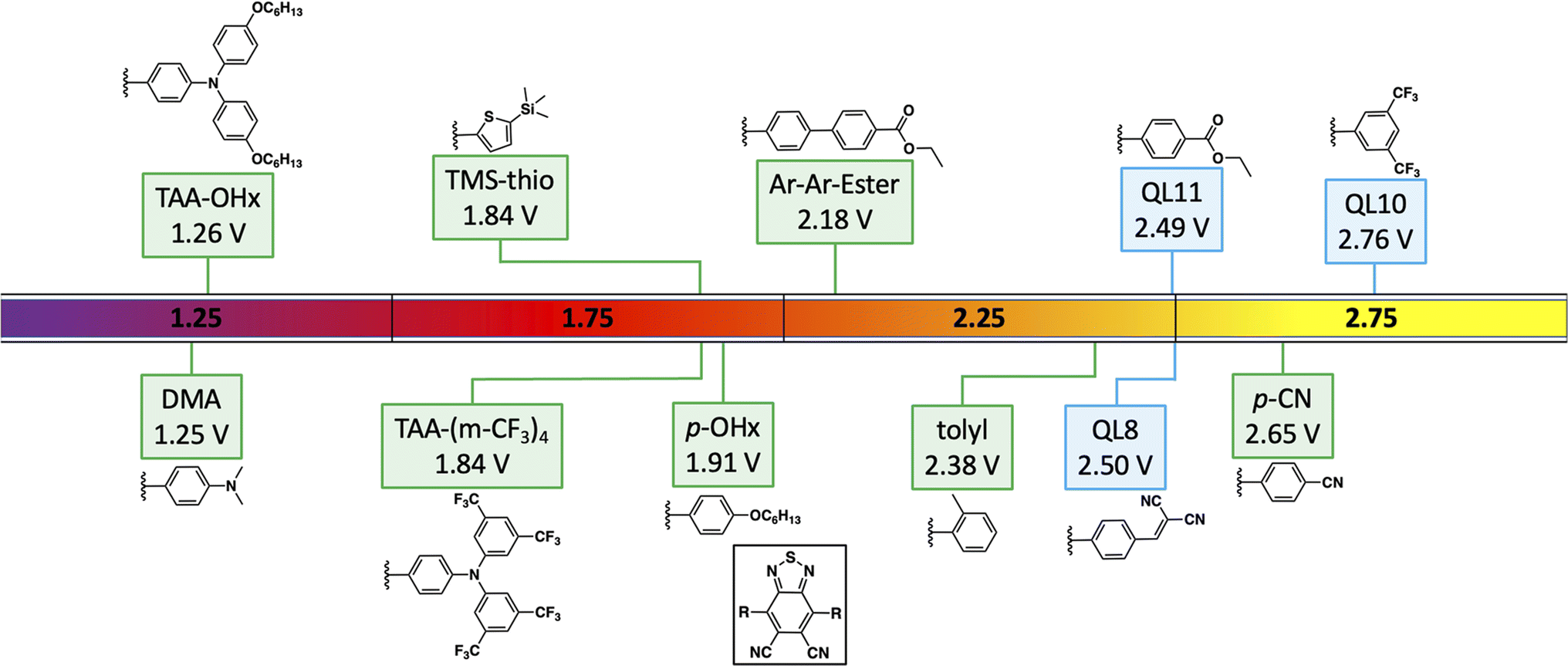 | ||
| Fig. 1 Previously reported (aryl)2DCBT dyes28,29 (with QL codes) and the new dyes presented in this work. Potential values are versus normal hydrogen electrode (NHE). | ||
Experimental section
General information
All commercially obtained reagents were used as received. Benzo[c][1,2,5]thiadiazole-5,6-dicarbonitrile (1), 4-bromo-N,N-bis(4-(hexyloxy)phenyl)aniline (TAA-(OHx)-Br), and (5-bromothiophen-2-yl)trimethylsilane (TMS-thio-Br) show in Scheme 1 were prepared following literature precedent.30–32 Thin-layer chromatography (TLC) was conducted with Sorbtech silica XHL TLC plates and visualized with UV light. Flash column chromatography was performed with Sorbent Tech P60, 40–63 μm (230–400 mesh). Flash column chromatography was performed using a Teledyne CombiFlash Rf + system. The silica gel cartridges were purchased from Luknova SuperSep (FC003012, 50 μm). 1H and 13C{1H} NMR spectra were recorded on either a Bruker Avance-400 (400 MHz) or Bruker Avance-300 (300 MHz) instrument and are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm, (CD3)2CO at 2.05 ppm). Data reported as s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad, ap = apparent, dd = doublet of doublets; coupling constant(s) in Hz. UV spectra were measured with a Cary 5000 UV-Vis-NIR spectrometer. Emission spectra were measured with a Horiba FluoroMax SpectroFluorimeter. Curves were fitted with the LOESS function in Igor which allows for easier data interpretation and comparisons. Molar absorptivities were measured with an AvaSpec-ULS2048-USB2-50 Spectrometer (Pine Research part RRAVSP3), and the light source was a UV/Vis/NIR Light Source (Pine Research part RRAVSP). These two systems were used in conjunction with the AvaSoft8 software program. Cyclic voltammetry curves were measured with a C–H Instruments electrochemical analyzer CHI600E. Tetrabutylammonium hexafluorophosphate (0.1 M in dichloromethane) was used as the electrolyte. The working electrode is glassy carbon, the pseudo-reference electrode is a silver wire, and the counter electrode is platinum. A scan rate of 100 mV s−1 was used. Ferrocenium/ferrocene is used as an internal standard (taken as 0.70 V vs. normal hydrogen electrode (NHE) in dichloromethane (DCM)).33,34 For electrospray ionization (ESI) high resolution mass spectrometry (HRMS), a quadruple-TOF system was used to obtain the data both in positive and negative modes with a Waters Synapt HDMS. IR experiments were recorded on a Bruker Alpha FT-IR spectrometer.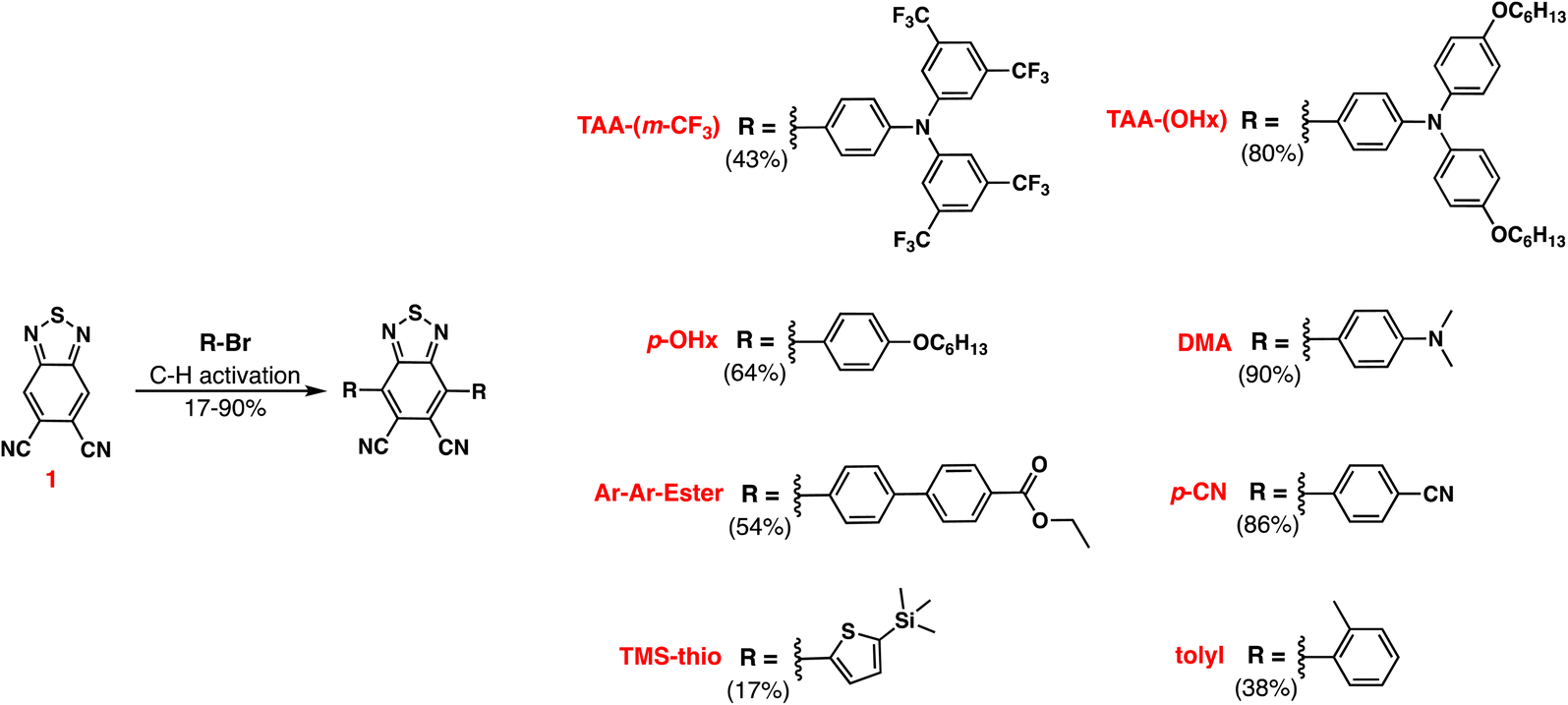 | ||
| Scheme 1 Synthetic route to the DCBT dyes. | ||
Computational approach
Molecules were drawn in ChemDraw (19.1.0.5) and saved as an MDL Molfile. Molecular geometries of the considered species were then optimized with the MMFF94 force field via Avogadro (1.2.0). Dihedral angles about non-cyclic single bonds were set to between 0 and 90 degrees manually to avoid local minima conformations. Accurate geometry optimization were performed sequentially by DFT using Gaussian 16 (ref. 35) with the M06-2X36 functional applying the following basis sets: first 3–21g, second 6-31G(d,p),37 and finally 6-311G(d,p).37 Time dependent-DFT (TD-DFT) computations were performed with optimized geometries with the M06-2X functional and 6-311G(d,p) basis set to compute the vertical transition energies and oscillator strengths. This functional and basis set were previously found to have good agreement with experimental findings for DCBT dyes.28Time-correlated single photon counting (TCSPC)
Emission lifetime curves were obtained using the 375 nm, 405 nm, or 485 nm line of an LDH series 405B pulsed diode laser (pulse width approx. 100 ps) as the excitation source and emission was detected using a PicoQuant PDM series single photon avalanche diode (time resolution approx. 50 ps) and a TimeHarp 260 time-correlated single photon counter (25 ps resolution). Appropriate filters were placed before the sample and detector. Time-resolved emission kinetics were best modeled by a single exponential function. Fluorescene lifetime values were measured in dichloromethane (5 × 10−4 M).Synthetic protocols
Results and discussion
As previously shown, the strongly electron withdrawing nature of the DCBT core characterizes what are typically electron deficient substituents as electron donating in comparison to the core.29 In studying this new series of dyes, computational analysis with density functional theory (DFT) was undertaken in order to better understand these electronic comparisons. Geometry optimizations were performed at the M06-2X/6-311G(d,p) level of theory36,37 with the Gaussian 16 software package.35 For the entire series of dyes, the highest occupied molecular orbital (HOMO) is delocalized across the entire dye, spanning from the aryl substituents to the DCBT core (Fig. 2 and S1–S7, ESI†). Likewise, the lowest unoccupied molecular orbital (LUMO) is observed to be positioned primarily on the DCBT core. Modest LUMO presence is observed on any other portion of the dyes across the entire series. This suggests that all of the aryl substituents are serving as electron donating groups in comparison to the DCBT core. The entire dye series exhibits intramolecular charge transfer (ICT) behavior, specifically as a donor–acceptor–donor system (D–A–D), for the HOMO to LUMO transition that changes regions of the molecules the orbitals are located on.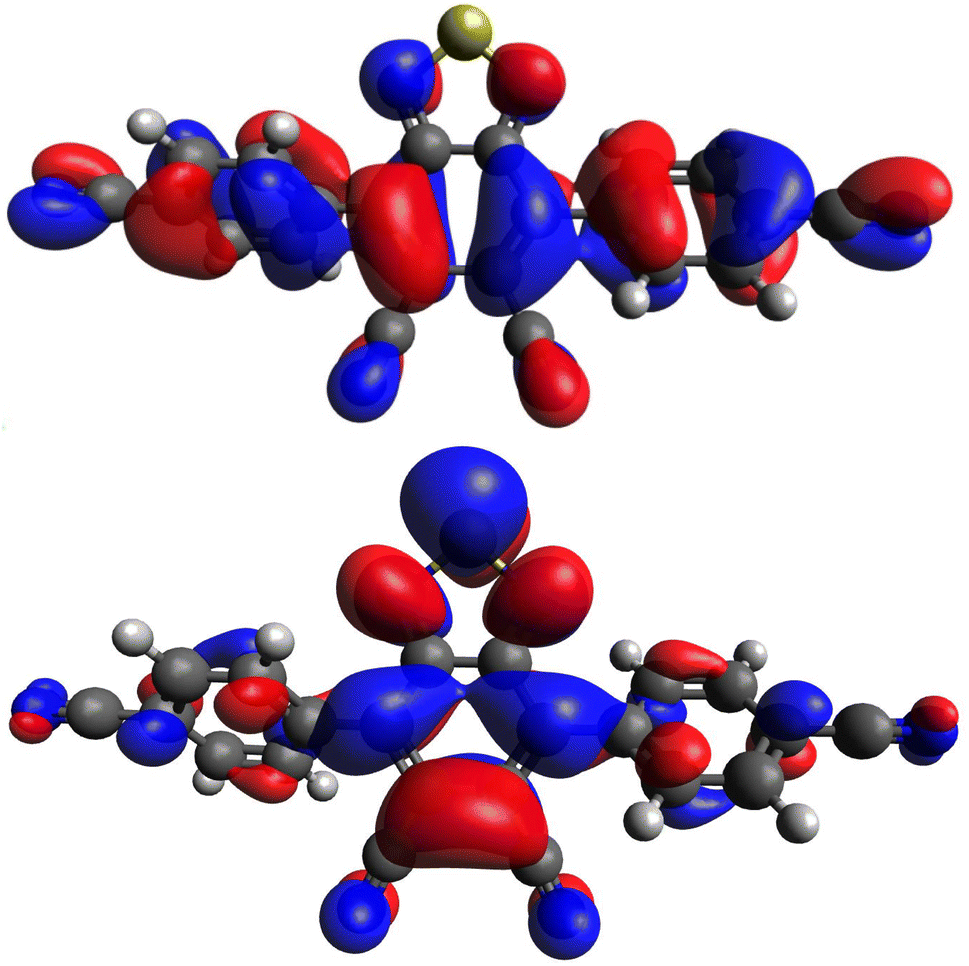 | ||
| Fig. 2 Computationally derived HOMO (top) and LUMO (bottom) orbitals of (p-CN)2DCBT using the M06-2X functional and 6-311G(d,p) basis set. Iso values are set to 0.02. Orbital images for the remaining dyes are in the ESI (Fig. S1–S7, ESI†). | ||
Time-dependent (TD)-DFT calculations were carried out at the M06-2X/6-311G(d,p) level of theory to better understand the orbital transitions within the series (Table 1). Calculations showed that the S0 to S1 transition is largely made up of the HOMO to LUMO transition in all cases (Tables 1 and S1, ESI†). The oscillator strengths for these transitions ranged from 0.2 to 0.7 which is to be expected from ICT systems. Solvatochromatic effects were studied by comparing the absorption spectra of (p-OHx)2DCBT in solvents of varying polarity to yield a trend of red-shifting absorption as the polarity of the solvent decreased (Fig. S19†). This solvent polarity dependence further suggests the ICT behavior of this series. In comparing the HOMO and LUMO energies, it is seen that the HOMO and LUMO energies range over 2.5 eVs and 1.1 eVs respectively, suggesting that the appended groups have a greater effect on the HOMO than the LUMO.
| Dyes | HOMO (eV) | LUMO (eV) | Orbitals | % Cont. | Vertical transition (nm | eV) | Oscillator strength |
|---|---|---|---|---|---|---|
| p-CN | −8.65 | −3.23 | H → L | 96% | 347 | 3.57 | 0.4667 |
| TMS-thio | −7.41 | −2.68 | H → L | 98% | 421 | 2.95 | 0.5452 |
| TAA-OHx | −6.13 | −2.29 | H → L | 91% | 495 | 2.51 | 0.7089 |
| H-2 → L | 6% | |||||
| Ar-Ar-ester | −7.79 | −2.75 | H → L | 90% | 374 | 3.32 | 0.7356 |
| H-2 → L | 2% | |||||
| H-4 → L | 4% | |||||
| Tolyl | −8.15 | −2.60 | H → L | 97% | 343 | 3.61 | 0.2192 |
| DMA | −6.50 | −2.18 | H → L | 96% | 456 | 2.72 | 0.5018 |
| TAA-(m-CF3)4 | −7.58 | −3.04 | H → L | 87% | 406 | 3.05 | 0.7081 |
| H-2 → L | 8% | |||||
| p-OHx | −7.38 | −2.47 | H → L | 97% | 394 | 3.15 | 0.3991 |
Having observed intriguing tunable energetics computationally, syntheses of the target dyes was attempted. The DCBT dyes can be accessed through a single symmetric C–H activation cross-coupling using the DCBT core (intermediate 1)30 and the respective aryl bromide group for each chromophore to yield the final dyes in 17–90% yield (Scheme 1). Through the entire series, one of two C–H activation conditions were used to achieve the end product.29
Steady-state absorption and emission spectroscopy were used to study the optical properties of the dyes (Fig. 3, Table 2). The absorption maximum spectral range spans from 363 to 567 nm in the order of tolyl = p-CN < Ar-Ar-Ester < p-OHx < TAA-(m-CF3)4 < TMS-thio < DMA < TAA-OHx in dichloromethane. Interestingly, (tolyl)2DCBT and (p-CN)2DCBT have the same absorption maxima despite having greatly different electronic properties in their aryl substituents which suggests sterics can have a significant influence on the energy of the charge transfer event. Despite the sterics of the tolyl group, a longer wavelength λabsmax is observed as part of a broad peak at 363 nm indicative of a ICT S0 to S1 transition for (tolyl)2DCBT in comparison to the DCBT core which has sharper transitions from ∼325 to 350 nm (Fig. S18†). (Ar2Ester)2DCBT and (p-OHx)2DCBT follow with the third and fourth highest energy λabsmax values, respectively. (Ar2Ester)2DCBT absorbs at a lower wavelength in comparison to (p-OHx)2DCBT despite its extended conjugation with the extra phenyl group due to the strong electron withdrawing nature of the ester groups. Next in the series, (TAA-(m-CF3)4)2DCBT and (TMS-thio)2DCBT have very similar λabsmax values at 456 and 466 nm, respectively, suggesting that a withdrawn amine donor has similar electronic effects to thiophene. Finally, the two electron-rich amines, (DMA)2DCBT and (TAA-OHx)2DCBT exhibit absorption at the longest wavelengths of the series at 542 and 567 nm, respectively. The emission maxima of the dyes follow a similar trend to the absorption maxima with minor differences. While (tolyl)2DCBT and (p-CN)2DCBT shared the same absorption maxima, (tolyl)2DCBT emits at a longer wavelength than (p-CN)2DCBT. This is likely due to the sterics of the tolyl substituent that increases reorganization energy, resulting in a larger Stokes shift. (TMS-thio)2DCBT and (TAA-(m-CF3)4)2DCBT are observed to emit at essentially the same wavelength. Likewise, the same is seen with (DMA)2DCBT and (TAA-OHx)2DCBT. The absorption and emission curve intercept (E(0–0)) values for the series ranges from 405 to 633 nm (Fig. S8–S16, ESI†) which follows the same trend as λabsmax with the exception of (p-CN)2DCBT absorbing at a lower wavelength than (tolyl)2DCBT. Additionally, the dyes exhibit molar absorptivities ranging from 3200 to 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 M−1 cm−1 (Table 2; Fig. S17, ESI†).
600 M−1 cm−1 (Table 2; Fig. S17, ESI†).
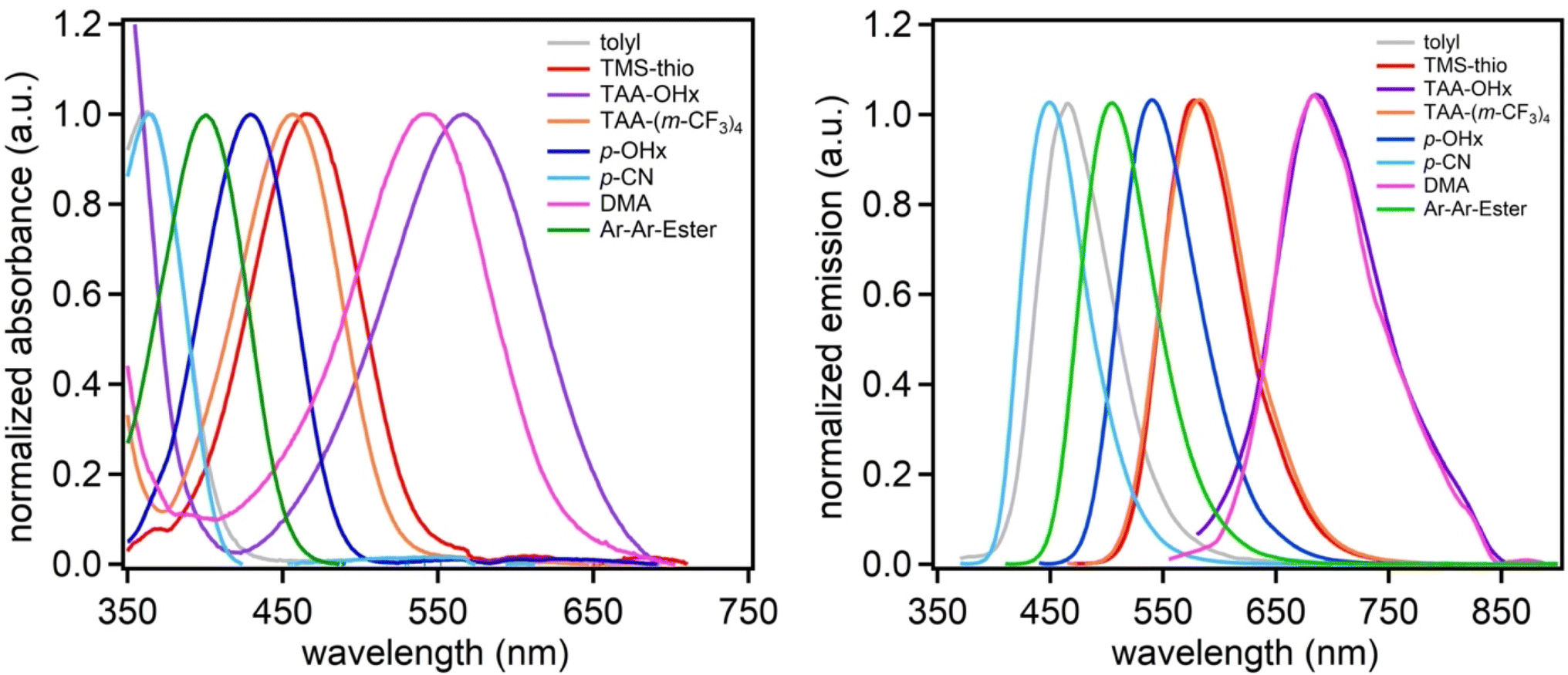 | ||
| Fig. 3 Normalized absorption and emission curves in dichloromethane under argon. All absorption spectra were taken at 1 × 10−5 M except for (tolyl)2DCBT which was taken at 1 × 10−4 M due to low signal. All emission spectra were taken at 1 × 10−5 M except (TAA-OHx)2DCBT which was taken at 1 × 10−4 M due to low signal. Dyes are coded by their aryl substituents. | ||
| Dye | λabsmax (nm) | ε (M−1 cm−1) | λemismaxa (nm) | E(0–0)b (nm) | E(S/S–)c (V) | E(S*/S–)d (V) | τemise (ns) |
|---|---|---|---|---|---|---|---|
| a When excited at λabsmax.b Found via the intercept from the normalized absorption and emission curves (Fig. S8–S16).c Found via cyclic voltammetry using a glassy carbon working electrode, platinum counter electrode, silver wire pseudo-reference electrode, 0.1 M tetrabutylammonium hexafluorophosphate as electrolyte in dichloromethane, and ferrocene as an internal standard. Value reported versus NHE by addition of +0.7 V versus ferrocene.33,34d Found via the equation E(S*/S–) = E(S/S–) + E(0–0); where E(0–0) is converted from nm to eV by the equation eV = 1240/nm.e Obtained via fluorescence lifetime measurements in DCM (5 × 10−4 M). | |||||||
| p-CN | 363 | 11400 |
451 | 405 | −0.41 | 2.65 | 2.9 |
| TMS-thio | 466 | 7700 | 581 | 527 | −0.51 | 1.84 | 9.2 |
| TAA-OHx | 567 | 12300 |
690 | 633 | −0.70 | 1.26 | 6.3* |
| Ar-Ar-ester | 401 | 13600 |
509 | 452 | −0.56 | 2.18 | 4.8 |
| Tolyl | 363 | 3200 | 468 | 412 | −0.63 | 2.38 | 3.9* |
| DMA | 542 | 10600 |
685 | 623 | −0.74 | 1.25 | 1.9 |
| TAA-(m-CF3)4 | 456 | 12000 |
583 | 519 | −0.55 | 1.84 | 7.7 |
| p-OHx | 429 | 9100 | 543 | 484 | −0.65 | 1.91 | 9.2 |
| DCBT | 332 | 13000 |
370 | 340 | −0.61 | 2.74 | — |
Electrochemical data was collected through cyclic voltammetry (CV) to obtain ground state reduction potentials (E(S/S–)) of the dyes (Fig. 4). The dyes begin with neutral charge at 0.0 V versus NHE and are eventually reduced by 1 electron to give the dye radical anion as the CV sweep travels in the negative direction. After reduction, the potential sweep direction is switched and an electron is then removed from the radical anion dye species to give a reversible redox process. The reduction values range from −0.41 to −0.74 V versus normal hydrogen electrode (NHE). These values are then used to calculate the excited state reduction potential (E(S*/S–)) using the equation E(S*/S–) = E(S/S–) + E(0–0) (Fig. 5). The excited state reduction potentials ranged over 1.4 V, from 1.25 to 2.65 V vs. NHE. However, the ground state reductions range over ∼0.3 V. This affirms the tunability of the E(S*/S–) through the selection of the aryl substituents on the DCBT core and shows the wide range of photooxidants that can be generated with this molecular series. While (DMA)2DCBT and (TAA-OHx)2DCBT exhibit the weakest photooxidizing strengths of the series, both dyes absorb at much lower energies (greater than 540 nm) in comparison to the rest of the series which could aid in the selectivity of various chemical transformations with the use of lower energy light.
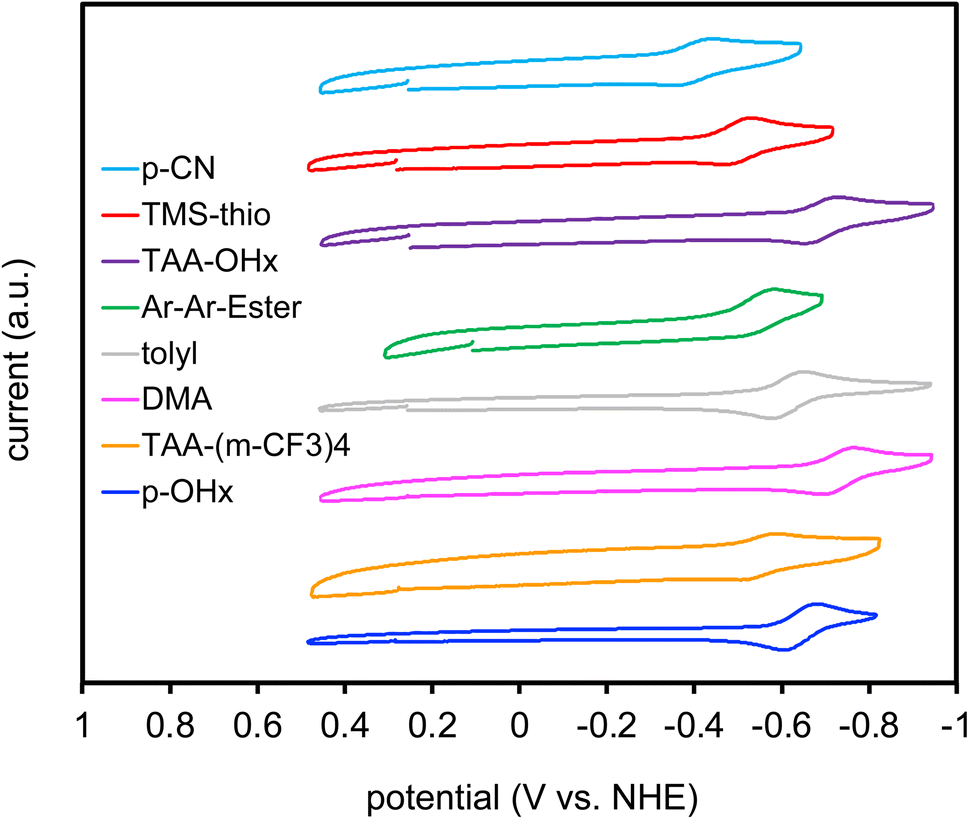 | ||
| Fig. 4 Cyclic voltammograms of the DCBT dyes. Dyes are coded by their aryl substituents. | ||
 | ||
| Fig. 5 Energy level diagram of the DCBT and QL dyes with E(S/S–) values as the top values and E(S*/S–) as the bottom values. Dyes are coded by their aryl substituents. | ||
The obtained E(S*/S–) value range shows that this class of DCBT photosensitizer is capable of being tuned to match or exceed the oxidizing abilities of other recent strong organic photooxidants such as the phenanthrenequinone derivatives that exhibited excited state reduction potentials ranging from 1.40 V to 1.73 V versus saturated calomel electrode (SCE) (or 1.64 V and 1.97 V versus NHE).39 The popular acridinium dye class has been widely studied in the literature due to the strong oxidizing potentials ranging from 2.56 V to 3.05 V versus SCE (2.80 V to 3.29 V versus NHE) for the most powerful “super oxidants”.40 While the oxidizing potentials of the acridinium class surpass that presented in this work, the DCBT motif shows a large range of tunability (1.51 eV range) which can be matched to a very wide range of substrate potentials for chemical transformations.
Time-correlated single photon counting (TCSPC) was undertaken to obtain excited state lifetimes of the series. Lifetimes ranged from 1.9 to 9.2 ns (Table 2). (p-OHx)2DCBT and (TMS-thio)2DCBT are observed to have the longest fluorescence lifetimes of the series. Interestingly, a general trend of increasing fluorescence lifetimes as absorption maxima increases is observed for the majority of the dyes in the series (Fig. 6). This observation goes against typical trends according to the energy gap law.41–43 It is also important to note that (DMA)2DCBT and (TAA-OHx)2DCBT differs from the rest of the series and do not follow this trend. The reason for the deviation from the observed trend for these two derivatives is not apparent, but these two dyes both employ the strongest aryl donor groups with the least positive E(S*/S–) values by more than 0.5 V. Despite (DMA)2DCBT and (TAA-OHx)2DCBT having the least positive E(S*/S–) values, it is important to note that these two photooxidants are still stronger than some commonly known chemical oxidants such as “Magic Blue” and I2. Furthermore, the oxidizing strength of these two dyes are mediated by low energy light (>540 nm absorption maxima; >600 nm absorption onset) which differs in comparison to other literature known photooxidants with much higher photoexcitation energy requirements (≤∼450 nm).44 Thus, these materials may be able to drive reactions requiring strong oxidants without the use of higher energy light which is incompatible with some functionalities. As a preliminary demonstration of the thermodynamic and kinetic competence of these dyes to drive photochemical transformations, three reactions based on p-methoxystyrene derived substrates were undertaken showing up to 95% consumption of the starting material using 450 nm light and (p-OHx)2DCBT to give products known in the literature (up to 29% yield with 5 turnovers) with several additional species (30–40%) (Fig. S21†).23,45
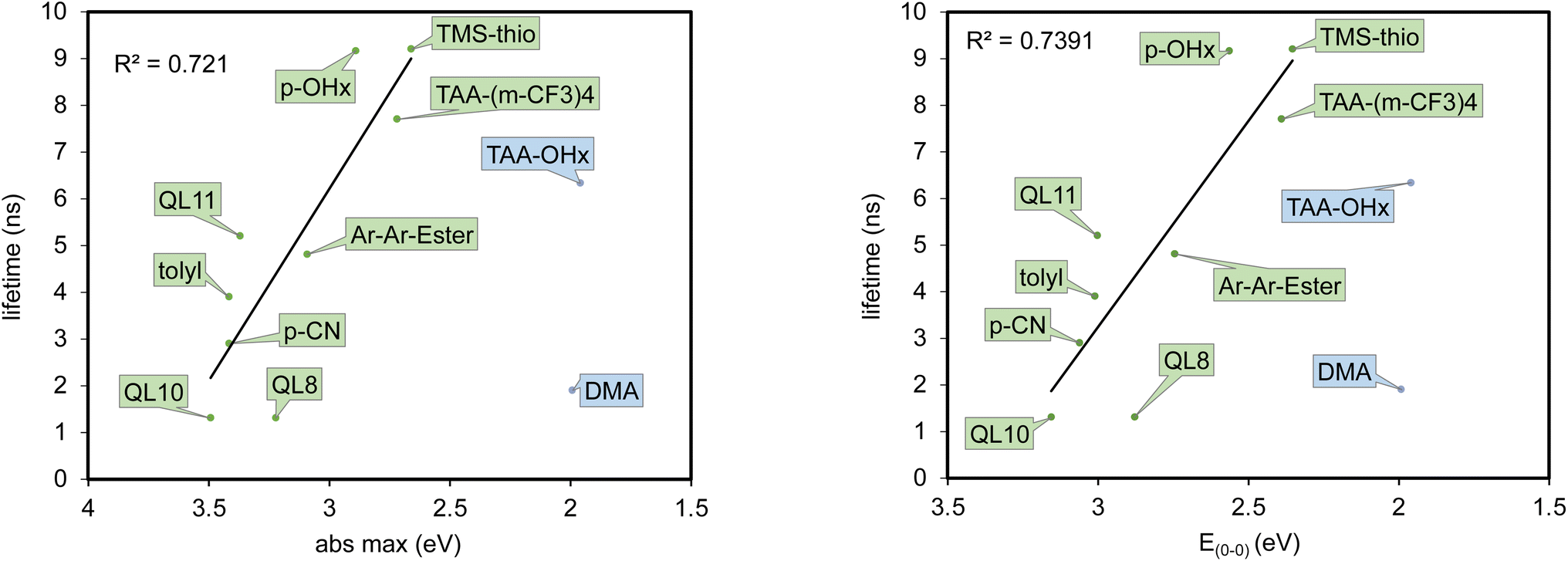 | ||
| Fig. 6 Fluorescence lifetimes of DCBT dyes plotted with respect to λabsmax (left) and E(0–0) (right). Trend line and R2 value only applies to the green dataset. Dyes are coded by their aryl substituents. | ||
Conclusions
A series of DCBT dyes were synthesized and characterized computationally, electrochemically, and photophysically. Computations reveal this series to be intramolecular charge transfer systems, more specifically D–A–D systems. Electrochemical measurements showed E(S*/S–) values ranging from 1.25 to 2.76 V, revealing an excited state reduction driving force range of 1.5 eV. This shows incredible energetic tunability for this class of molecules, enabling the tuning of the photooxidant to a substrate for more selective, milder reactions in some cases. Steady state absorption and emission spectroscopy was undertaken to characterize this series of dyes as visible light absorbers and emitters. Namely, (TAA-OHx)2DCBT and (DMA)2DCBT are lower energy visible light absorbers which allows for reaction selectivity when used in catalysis. Beyond selectivity, stronger photooxidants are necessary in order to access new chemical transformations. This was recently demonstrated with a water soluble version of QL11 named QL11-OEG; which is QL11 with oligomeric ethylene glycol chains.28 The strong organic photooxidant QL11-OEG was shown to drive a water oxidation system which typically employs metal oxidants. The dyes put forward in this study have the potential to drive new reaction systems with tunable properties to the reaction system needs. Finally, an unexpected relationship is discovered between the excited state fluorescence lifetime and absorption maxima. This series of symmetric DCBT dyes shows the tunability of this class of photosensitizers (over a 1.5 eV range of excited state reduction potentials) as well as the suitability of these dyes for various catalytic oxidative processes. Future directions for this work include further expanding upon the energetic range of these dyes as well as further understanding and probing their excited state lifetimes via transient absorption spectroscopy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Q. Y. L., R. K., and J. H. D. thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences for supporting this work under award DE-SC0019131. E. C. L. and N. I. H. thank the National Science Foundation (NSF) for supporting this research with award NSF OIA-1757220 and CHE-1954922.References
- K. Ohkubo, K. Suga and S. Fukuzumi, Solvent-Free Selective Photocatalytic Oxidation of Benzyl Alcohol to Benzaldehyde by Molecular Oxygen Using 9-Phenyl-10-Methylacridinium, Chem. Commun., 2006, 19, 2018–2020 RSC.
- M. Uygur, J. H. Kuhlmann, M. C. Pérez-Aguilar, D. G. Piekarski and O. García Mancheño, Metal- and Additive-Free C–H Oxygenation of Alkylarenes by Visible-Light Photoredox Catalysis, Green Chem., 2021, 23, 3392–3399 RSC.
- D. Steuernagel and H.-A. Wagenknecht, Photocatalytic Synthesis of Acetals and Ketals from Aldehydes and Silylenolethers without the Use of Acids, Chem.–Eur. J., 2023, 29, e202203767 CrossRef CAS PubMed.
- P. P. Singh, J. Singh and V. Srivastava, Visible-Light Acridinium-Based Organophotoredox Catalysis in Late-Stage Synthetic Applications, RSC Adv., 2023, 13, 10958–10986 RSC.
- Z. Luo, C. Lu, G. Histand and D. Lin, One-Step Visible Light Photoredox-Catalyzed Purine C8 Alkoxylation with Alcohol, J. Org. Chem., 2022, 87, 11558–11564 CrossRef CAS PubMed.
- X. Kan, J.-C. Wang, Z. Chen, J.-Q. Du, J.-L. Kan, W.-Y. Li and Y.-B. Dong, Synthesis of Metal-Free Chiral Covalent Organic Framework for Visible-Light-Mediated Enantioselective Photooxidation in Water, J. Am. Chem. Soc., 2022, 144, 6681–6686 CrossRef CAS PubMed.
- E. Skolia, P. L. Gkizis, N. F. Nikitas and C. G. Kokotos, Photochemical Aerobic Oxidation of Sulfides to Sulfoxides: The Crucial Role of Wavelength Irradiation, Green Chem., 2022, 24, 4108–4118 RSC.
- B. Zilate, C. Fischer and C. Sparr, Design and Application of Aminoacridinium Organophotoredox Catalysts, Chem. Commun., 2020, 56, 1767–1775 RSC.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- A. B. Muñoz-García, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Dye-Sensitized Solar Cells Strike Back, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC.
- D. Zhang, M. Stojanovic, Y. Ren, Y. Cao, F. T. Eickemeyer, E. Socie, N. Vlachopoulos, J.-E. Moser, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, A Molecular Photosensitizer Achieves a Voc of 1.24 V Enabling Highly Efficient and Stable Dye-Sensitized Solar Cells with Copper(II/I)-Based Electrolyte, Nat. Commun., 2021, 12, 1777 CrossRef CAS PubMed.
- Y. H. Hong, Y.-M. Lee, W. Nam and S. Fukuzumi, Molecular Photocatalytic Water Splitting by Mimicking Photosystems I and II, J. Am. Chem. Soc., 2022, 144, 695–700 CrossRef CAS PubMed.
- D. Antón-García, J. Warnan and E. Reisner, A Diketopyrrolopyrrole Dye-Based Dyad on a Porous TiO2 Photoanode for Solar-Driven Water Oxidation, Chem. Sci., 2020, 11, 12769–12776 RSC.
- X. M. C. Ta, R. Daiyan, T. K. A. Nguyen, R. Amal, T. Tran-Phu and A. Tricoli, Alternatives to Water Photooxidation for Photoelectrochemical Solar Energy Conversion and Green H2 Production, Adv. Energy Mater., 2022, 12, 2201358 CrossRef CAS.
- Q. Li, Y. Ouyang, H. Li, L. Wang and J. Zeng, Photocatalytic Conversion of Methane: Recent Advancements and Prospects, Angew. Chem., Int. Ed., 2022, 61, e202108069 CrossRef CAS PubMed.
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Artificial Photosynthesis: From Nanosecond Electron Transfer to Catalytic Water Oxidation, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed.
- R. R. Rodrigues, A. Peddapuram, A. L. Dorris, N. I. Hammer and J. H. Delcamp, Thienopyrroledione-Based Photosensitizers as Strong Photoinduced Oxidants: Oxidation of Fe(Bpy)32+ in a >1.3 V Dye-Sensitized Solar Cell, ACS Appl. Energy Mater., 2019, 2, 5547–5556 CrossRef CAS.
- R. R. Rodrigues, H. Cheema and J. H. Delcamp, A High-Voltage Molecular-Engineered Organic Sensitizer–Iron Redox Shuttle Pair: 1.4 V DSSC and 3.3 V SSM-DSSC Devices, Angew. Chem., Int. Ed., 2018, 57, 5472–5476 CrossRef CAS PubMed.
- Y. Lee and M. S. Kwon, Emerging Organic Photoredox Catalysts for Organic Transformations, Eur. J. Org Chem., 2020, 2020, 6028–6043 CrossRef CAS.
- J. Moon, Y. K. Moon, D. D. Park, S. Choi, Y. You and E. J. Cho, Visible-Light-Induced Trifluoromethylation of Unactivated Alkenes with Tri(9-Anthryl)Borane as an Organophotocatalyst, J. Org. Chem., 2019, 84, 12925–12932 CrossRef CAS PubMed.
- T. Bortolato, S. Cuadros, G. Simionato and L. Dell'Amico, The Advent and Development of Organophotoredox Catalysis, Chem. Commun., 2022, 58, 1263–1283 RSC.
- A. Tlili and S. Lakhdar, Acridinium Salts and Cyanoarenes as Powerful Photocatalysts: Opportunities in Organic Synthesis, Angew. Chem., 2021, 133, 19678–19701 CrossRef.
- N. A. Romero and D. A. Nicewicz, Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- G. Han, G. Li, J. Huang, C. Han, C. Turro and Y. Sun, Two-Photon-Absorbing Ruthenium Complexes Enable near Infrared Light-Driven Photocatalysis, Nat. Commun., 2022, 13, 2288 CrossRef CAS PubMed.
- L. Mei, J. M. Veleta and T. L. Gianetti, Helical Carbenium Ion: A Versatile Organic Photoredox Catalyst for Red-Light-Mediated Reactions, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS PubMed.
- R. Obertík, J. Chudoba, J. Šturala, J. Tarábek, L. Ludvíková, T. Slanina, B. König and R. Cibulka, Highly Chemoselective Catalytic Photooxidations by Using Solvent as a Sacrificial Electron Acceptor, Chem.–Eur. J., 2022, 28, e202202487 CrossRef PubMed.
- S. P. Pitre, C. D. McTiernan, W. Vine, R. DiPucchio, M. Grenier and J. C. Scaiano, Visible-Light Actinometry and Intermittent Illumination as Convenient Tools to Study Ru(Bpy)3Cl2 Mediated Photoredox Transformations, Sci. Rep., 2015, 5, 16397 CrossRef CAS PubMed.
- Q. Y. Li, L. A. Hunt, K. H. Wijesinghe, C. Curiac, A. Williams, A. Dass, N. I. Hammer and J. H. Delcamp, Dicyanobenzothiadiazole (DCBT) Organic Dye as a Visible Light Absorbing Strong Photoinduced Oxidant with a 16 Microsecond Long-Lived Excited State, Adv. Energy Mater., 2023, 13, 2203102 CrossRef CAS.
- J. Zhang, T. C. Parker, W. Chen, L. Williams, V. N. Khrustalev, E. V. Jucov, S. Barlow, T. V. Timofeeva and S. R. Marder, C–H-Activated Direct Arylation of Strong Benzothiadiazole and Quinoxaline-Based Electron Acceptors, J. Org. Chem., 2016, 81, 360–370 CrossRef CAS PubMed.
- C. Burmester and R. Faust, A Reliable Route to 1,2-Diamino-4,5-Phthalodinitrile, Synthesis, 2008, 1179–1181 CAS.
- J. H. Delcamp, A. Yella, M. K. Nazeeruddin and M. Grätzel, Modulating dye E(S+/S*) with Efficient Heterocyclic Nitrogen Containing Acceptors for DSCs, Chem. Commun., 2012, 48, 2295–2297 RSC.
- E. Zhu, J. Hai, Z. Wang, B. Ni, Y. Jiang, L. Bian, F. Zhang and W. Tang, Two-Dimensional Polyfluorenes Bearing Thienylenevinylene π-Bridge-Acceptor Side Chains for Photovoltaic Solar Cells, J. Phys. Chem. C, 2013, 117, 24700–24709 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- R. C. Weast and M. J. Astle, CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, The CRC Press, Boca Raton, Fla., 63rd edn, 1982, pp. 1982–1983 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, Self-consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- F. Mo, D. Qiu, Y. Jiang, Y. Zhang and J. Wang, A Base-Free, One-Pot Diazotization/Cross-Coupling of Anilines with Arylboronic Acids, Tetrahedron Lett., 2011, 52, 518–522 CrossRef CAS.
- J. Talvitie, I. Alanko, A. Lenarda, N. Durandin, N. Tkachenko, M. Nieger and J. Helaja, Electron-Deficient Phenanthrenequinone Derivative for Photoactivated Hydrogen Atom Transfer Mediated Oxidation of Secondary Alcohols, ChemPhotoChem, 2023, 7, e202300107 CrossRef CAS.
- J. Žurauskas, S. Boháčová, S. Wu, V. Butera, S. Schmid, M. Domański, T. Slanina and J. P. Barham, Electron-Poor Acridones and Acridiniums as Super Photooxidants in Molecular Photoelectrochemistry by Unusual Mechanisms, Angew. Chem., Int. Ed., 2023, 62, e202307550 CrossRef PubMed.
- S. J. A. Jang, Simple Generalization of the Energy Gap Law for Nonradiative Processes, J. Chem. Phys., 2021, 155, 164106 CrossRef CAS PubMed.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, Application of the Energy Gap Law to the Decay of Charge-Transfer Excited States, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- R. Englman and J. Jortner, The Energy Gap Law for Radiationless Transitions in Large Molecules, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. S. Hamilton and D. A. Nicewicz, Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational data, spectroscopic and electrochemical data, and NMR spectra. See DOI: https://doi.org/10.1039/d3ra06575h |
| This journal is © The Royal Society of Chemistry 2024 |