
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegration of network pharmacology, molecular docking, and simulations to evaluate phytochemicals from Drymaria cordata against cervical cancer†
Kunal Bhattacharya ab,
Bhargab Chandra Natha,
Ekbal Ahmeda,
Pukar Khanalc,
Nongmaithem Randhoni Chanuad,
Satyendra Dekaa,
Dibyajyoti Dasae and
Amit Kumar Shrivastava*f
ab,
Bhargab Chandra Natha,
Ekbal Ahmeda,
Pukar Khanalc,
Nongmaithem Randhoni Chanuad,
Satyendra Dekaa,
Dibyajyoti Dasae and
Amit Kumar Shrivastava*f
aPratiksha Institute of Pharmaceutical Sciences, Guwahati, Assam 781026, India
bRoyal School of Pharmacy, The Assam Royal Global University, Assam 781035, India
cDepartment of Pharmacology and Toxicology, KLE College of Pharmacy, KLE Academy of Higher Education and Research (KAHER), Belagavi 590010, India
dFaculty of Pharmaceutical Science, Assam Downtown University, Assam 781026, India
eDepartment of Pharmaceutical Sciences, Dibrugarh University, Dibrugarh 786004, India
fDepartment of Pharmacology, Universal College of Medical Sciences, and Teaching Hospital, Bhairahawa, Rupandehi, 32900, Nepal. E-mail: sr.akshri.ucms.np@gmail.com
First published on 30th January 2024
Abstract
Introduction: Cervical cancer is prevalent among women worldwide. It is a type of cancer that occurs in the cells of the cervix, the lower part of the uterus. Mostly, it is observed in developing nations due to limited access to screening tools. Natural products with anticancer properties and fewer side effects have gained attention. Therefore, this study evaluates the potential of Drymaria cordata as a natural source for treating cervical cancer. Methodology: Phytocompounds present in Drymaria cordata were screened for their molecular properties and drug-likeness. The selected compounds were studied using systems biology tools such as network pharmacology, molecular docking, and molecular dynamics simulations, including MMGBSA studies. Results: Through network pharmacology, molecular docking, and molecular dynamics simulations, quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside was identified as a hit compound targeting HRAS and VEGFA proteins. These proteins were found to be responsible for the maximum number of pathway modulations in cervical cancer. Conclusion: Drymaria cordata exhibits potential for treating cervical cancer due to the presence of quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside. Further validation of these findings through in vitro and in vivo studies is required.
1. Introduction
Cervical cancer continues to present a substantial global health challenge, representing a serious risk to the health of women worldwide.1,2 According to the World Health Organization (WHO), cervical cancer ranks as the fourth most prevalent cancer in women globally, with 604![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases and 342000 fatalities reported in 2020.3 Despite recent advancements in the field of diagnosis and treatment, there remains a persistent demand for the development of innovative, efficacious, and safer therapeutic alternatives for this malignancy.4
000 new cases and 342000 fatalities reported in 2020.3 Despite recent advancements in the field of diagnosis and treatment, there remains a persistent demand for the development of innovative, efficacious, and safer therapeutic alternatives for this malignancy.4
In recent times, there has been a renewed enthusiasm for investigating the potential of natural products as a reservoir of bioactive molecules with anticancer effects. This renewed interest can be attributed to the wide range of structural variations found in natural products and their historical significance in drug discovery.5,6 Drymaria cordata, commonly referred to as chickweed or “Calabar woman's eye,” is a plant species extensively utilized for ethnomedicinal purposes across diverse cultural contexts. The utilization of this botanical remedy is prevalent in treating various medical conditions, including peptic ulcers, female sterility, headaches, glomerulonephritis, sleeping disorders, convulsions, and febrile illnesses in children. It is commonly incorporated as a constituent in numerous regional polyherbal formulations, addressing both primary and secondary ailments such as colds, headaches, coryza, bronchitis, leprosy, tumors, etc. Alkaloids, flavonoids, tannins, saponins, phenols, and terpenoids are some of the secondary plant metabolites found in this plant, and they have been shown to have a wide range of medicinal effects, including antibacterial, analgesic, antipyretic, expectorant, anxiolytic, anti-diabetic, sinusitis, and cytotoxic effects.7,8
The utilization of traditional knowledge can be a valuable resource for contemporary scientific research. In light of its potential significance, we have conducted a computational analysis to explore the phytochemical components of Drymaria cordata and their potential efficacy in combating cervical cancer. The present investigation is conducted using sophisticated computational techniques such as molecular docking, network pharmacology, and molecular dynamics (MD) simulations. Network pharmacology provides a comprehensive viewpoint, enabling an investigation of the complex network of interactions among the discovered phytochemicals and various targets within the framework of cervical cancer-related pathways.9,10
Identifying small molecule-biomolecular target binding affinity and interaction types requires molecular docking.11 Molecular docking was used to determine the binding patterns of selected phytochemicals from Drymaria cordata with cervical cancer-associated molecular targets. These analyses revealed how these phytochemicals may affect cancer-related biological processes. On the other hand, molecular dynamics simulations allowed us to investigate the dynamic behavior and stability of ligand-receptor complexes. Docked complexes were simulated extensively to learn more about the conformational changes, flexibility, and residence durations of the ligands within their binding sites. Understanding the structural basis of binding and evaluating the accuracy of the docking predictions greatly benefits from such data.12 The utilization of advanced computational techniques serves as the fundamental basis of our study, as we strive to make a valuable contribution to the continuously expanding array of approaches targeted at tackling the global issue posed by cervical cancer.
2. Materials and methods
2.1. Identification of phytoconstituents of Drymaria cordata
The phytochemical constituents of Drymaria cordata were identified through the utilization of several databases, including Traditional Chinese Medicine Systems Pharmacology (TCMSP) (https://tcmsp-e.com/tcmsp.php)13 and Traditional Chinese Medicine Information Database (TCMID) (https://bidd.group/TCMID/),14 which are recognized sources in the field of Traditional Chinese Medicine. Additionally, a comprehensive review of existing literature pertaining to Drymaria cordata was conducted to gather relevant information. The 3D structures of the identified phytochemicals were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). For structures that were not available in the chemical databases, they were drawn using ChemDraw (https://revvitysignals.com/products/research/chemdraw).2.2. Screening of molecular properties and toxicity prediction of phytochemicals
To determine the molecular properties and drug-likeness score of each compound, MolSoft L.L.C (https://www.molsoft.com/)15 was utilized by querying each SMILE. The server predicts an overall drug-likeness score using Molsoft's chemical fingerprints. The training set for this mode consisted of 5K marketed drugs from WDI (positives) and 10K carefully selected non-drug compounds (negatives). Additionally, the toxicity of the phytochemicals was predicted using Protox II (https://tox-new.charite.de/protox_II/).16 Protox-II utilizes computer-based models trained with authentic data obtained from in vitro or in vivo experiments. These models predict the toxicological properties of both actual and virtual compounds. The determination of the acute toxicity class and various endpoints for a given compound is achieved through the utilization of trained machine-learning algorithms that assess chemical similarities to known toxic compounds.2.3. Prediction of cervical cancer target genes
To identify genes associated with cervical cancer, we utilized the DisGeNET database (https://www.disgenet.org/search).17 Target genes were restricted to Homo sapiens, and duplicates were eliminated to ensure accuracy and prevent redundancy. This approach aimed to provide a comprehensive understanding of genes potentially involved in the pathogenesis of cervical cancer.2.4. Target prediction of Drymaria cordata phytochemicals
To identify the target genes of the phytochemicals present in Drymaria cordata, this study utilized three different databases: SwissTargetPrediction (http://www.swisstargetprediction.ch/),18 Way2Drug database (http://www.way2drug.com/),19 and Similarity Ensemble Approach (SEA) (https://sea.bkslab.org/).20 By employing this multi-database approach, we aimed to provide a more comprehensive understanding of the potential target genes of the phytochemicals in Drymaria cordata.To identify target genes of Drymaria cordata phytochemicals potentially related to cervical cancer, we imported the predicted target genes of both the phytochemicals and cervical cancer into VENNY 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/). By utilizing a Venn diagram, we identified common potential target genes between the drug and the disease.
2.5. Protein–protein network and enrichment analysis
To conduct a comprehensive investigation into the potential modulatory impacts of phytochemicals from Drymaria cordata on proteins associated with cervical cancer, the discovered target genes were subjected to enrichment analysis using the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/)21 with a confidence score of 0.9. This analysis identified gene ontology terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways potentially modulated by the phytochemicals in Homo sapiens at a false discovery rate of 5%. For protein–protein interaction assessment, established interactions from curated databases and experimentally determined interactions were considered. Predicted interactions were also included based on factors such as gene neighborhood, gene fusions, gene co-occurrence, text mining, co-expression, and protein homology. It's important to note that these evaluations assumed the statistical background of the entire genome. Disconnected nodes were excluded from the network, and hub genes were identified by selecting the top 10 genes under “Degree,” “Closeness,” and “Betweenness,” with calculation of the topological features of each node in the network.2.6. Compound-protein-pathway network construction and analysis
Before constructing the network, all KEGG disease records were screened for cancer relevance using keywords such as “melanoma,” “sarcoma,” “cancer,” “glioma,” “leukemia,” and “carcinoma.” The disease-protein-phytochemical network was then built using Cytoscape v3.9.1 (ref. 22) in a directed manner, and each node in the network was assessed and ranked based on its “degree” value.2.7. Molecular docking
The objective of molecular docking is to elucidate the binding affinities of the compound 5,4′-dihydroxy-7-methoxyflavone-6-C (2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside with VEGF (Vascular Endothelial Growth Factor) and HRAS (HRas proto-oncogene) proteins, and Quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside with VEGF, CTNNB1 (Catenin B 1), and HRAS proteins. Docking analyses were conducted using Autodock v4.2.6 (ref. 23) for the compound of interest.The pre-established co-crystallized X-ray structure, sourced from the RCSB PDB (VEGF, PDB ID: 4QAF; CTNNB1, PDB ID: 7AFW and HRAS, PDB ID: 7JII), facilitated the determination of protein binding cavities. Residue locations within a 3 Å radius were computed utilizing the co-crystallized ligand. Chimera software (https://www.cgl.ucsf.edu/chimera/)24 was used to remove co-crystallized ligands during the cavity selection phase, followed by energy minimization using steepest descent and conjugate gradient methods. After this step, the non-polar hydrogens of the receptor and target compound were merged, and the data for both were saved in the pdbqt format.
The molecular docking process was carried out inside a grid box with the following dimensions: 18 × 15 × 17 Å for 4QAF, 10 × 12 × 14 Å for 7AFW, and 17 × 13 × 18 Å for 7JII, with a 0.3 Å spacing. Protein–ligand complex docking studies were conducted using the Lamarckian Genetic Algorithm (LGA) framework. Three independent molecular docking experiments were performed, each comprising 50 solutions. The population size for each experiment was set at 500, with a total of 2500000 evaluations undertaken. The maximum number of generations allowed for each experiment was 27, while all other parameters were kept at their default settings.
Following the completion of the docking procedure, RMSD clustering maps were produced using re-clustering, employing clustering tolerances of 0.25, 0.50, and 1. The objective was to choose the most favorable cluster, characterized by the lowest energy score and maximum population. The efficiency of docking studies was further validated by removing the co-crystallized ligands from the receptor protein and re-docking at the same place, calculating the binding energy and RMSD with X-ray structures having the co-crystallized ligands.
2.8. Molecular dynamics simulation
The complexes of 4QAF + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside and 7JII + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside underwent molecular dynamics simulations using the Desmond software25 for 100 nanoseconds.The first step in constructing protein and ligand complexes for molecular dynamics simulation involved docking studies. Molecular docking studies, conducted in static conditions, accurately predict ligand binding states. Molecular dynamics (MD) simulations involve integrating Newton's classical equation of motion26,27 to compute the temporal evolution of atomic displacements. Docking techniques provide a fixed depiction of a molecule's binding conformation within the active region of a protein. Simulations establish predictions about ligand binding states under physiological conditions.28,29
The Protein Preparation Wizard performed preliminary processing, including complex optimization and minimization, on the protein–ligand complex. Each system was developed using the System Builder tool. The solvent model chosen was the Transferable Intermolecular Interaction Potential 3 Points (SPC), with an orthorhombic box. The simulation used the OPLS 2005 force field,30 and counter ions were added for charge neutrality. A solution containing 0.15 M sodium chloride (NaCl) replicated physiological conditions. The NPT ensemble was used throughout the simulation, maintaining a temperature of 300 K and a pressure of 1 atm.
Before the simulation, the models underwent a relaxation process.31 Trajectories were recorded at regular 100 picosecond intervals for evaluation. Simulation stability was assessed by comparing various metrics, including root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent accessible surface area (SASA), and hydrogen bonds formed between the protein and ligand throughout the simulation duration.
2.9. Binding free energy analysis
The binding free energy of the complex was determined using the molecular mechanics and generalized Born surface area approach (MM-GBSA).32,33 The MM-GBSA free energy of binding was quantified in the last fifty frames of the simulation trajectory using the Python script ‘thermal_mmgsba.py'. The binding free energy of MM-GBSA (kcal mol−1) was determined by aggregating various energy modules, namely coulombic, van der Waals, covalent, self-contact, hydrogen bond, lipophilic, and solvation of ligand and protein, using the principle of additivity. The value of ΔG bind can be found by employing the following equation:| ΔGbind = ΔGMM + ΔGsolv − ΔGSA |
3. Results and discussion
3.1. Identification and screening of Drymaria cordata phytochemicals
According to literature and database studies, a total of 22 compounds (Table S1†) have been identified in Drymaria cordata. However, only 6 compounds-stigmasterol, 24-ethyl-cholesta-5,22E-dien-3β-O-β-D-glucopyranosyle, 5,4′-dihydroxy-7-methoxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside, quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside, 5,7,3′,4′-tetrahydroxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside, and Cassiaoccidentalin A – were selected for further studies based on high drug likeness scores (Table S1†) and toxicity analysis (Table S2†).3.2. Identification of cervical cancer targets
Previously recorded targets for cervical cancer (UMLS CUI: C4048328) were identified using the DisGeNET database. In total, 1817 cervical cancer hits were found in the primary search. Using SwissTarget prediction, Way2Drug database, and Similarity Ensemble Approach, 59 target genes of the 6 selected ligands were identified. A Venn diagram (Fig. S1†) was constructed to find potential cervical cancer target genes, revealing 59 common targets of Drymaria cordata against cervical cancer. These 59 targets, presented in Fig. S1,† were considered as the cervical cancer targets of Drymaria cordata phytochemicals and subjected to further analysis.3.3. Protein–protein interaction and gene-enrichment analysis
The interaction network of 59 proteins exhibited 53 different edges with an average node degree of 1.8, an average local clustering coefficient of 0.284, an expected number of edges of 25, and a protein–protein interaction enrichment p-value of 5.51 × 10−7 (Fig. S2a†). There are 24 disconnected nodes (DAPK1, EPHA3, CA13, BCHE, ST6GAL1, CAT, KRT17, NR1H2, CD83, ERAP1, CD38, CLU, LGALS9, G6PD, SMN2, ABCG2, ABCB1, EPHB6, CA12, DHCR24, HMOX1, PLAU, ALP1, and ABCC4). Additionally, AR, ESR1, IL6, VEGFA, CTNNB1, HRAS, and TNF were identified as major hub genes (Fig. S2†). The detailed scores for each protein, including average short path length, betweenness centrality, clustering coefficient, closeness centrality, eccentricity, stress, degree, neighbourhood connectivity, number of undirected edges, topological coefficient, edge count, indegree, outdegree, and radiality, are summarized in Fig. S3.†The network of hub genes was further explored for gene ontology and pathway enrichment analysis. The analysis revealed the regulation of 34 different cellular components, with a major emphasis on adherence junction (GO:0005912), protein–DNA complex (GO:0032993), membrane raft (GO:0045121), and membrane microdomain (GO:0098857). Genes CTNNB1 (Catenin B 1), TNF (Tumor Necrosis Factor), VEGFA (Vascular Endothelial Growth Factor A), and ESR1 (Estrogen Receptor 1) were identified as major modulators (Fig. S4a†). Similarly, the study identified 45 significant molecular functions, with a notable focus on the regulation of β-catenin binding (GO:0008013), cytokine activity (GO:0005125), and cytokine receptor binding (GO:0005126). Genes AR (Androgen Receptor), ESR1, CTNNB1, IL6 (Interleukin 6), TNF, and VEGFA played major roles in governing these molecular functions, as depicted in Fig. S4b.† Furthermore, the analysis encompassed a comprehensive exploration of 1277 distinct biological processes. Among the enriched processes, gland development (GO:0048732), epithelial cell proliferation (GO:0050673), positive regulation of DNA-binding transcription factor activity (GO:0051091), and regulation of DNA-binding transcription factor activity (GO:0051090) were found to be majorly modulated by the genes VEGFA, AR, TNF, IL6, HRAS, ESR1, and CTNNB1 (Fig. S4c†).
3.4. KEGG pathway analysis
A total of 74 KEGG pathways were identified to be regulated by the hub genes CTNNB1, TNF, VEGFA, ESR1, AR, IL6, and HRAS, of which 12 pathways were associated with different types of cancers. Among these, 6 genes (CTNNB1, AR, IL6, ESR1, HRAS, and VEGFA) were modulated in the ‘Pathways in cancer’ (hsa05200) at a strength of 1.51 with a false discovery rate of 7.54 × 10−7 (Fig. S5 and S6†).3.5. Compound-protein-pathway network
The compound-target-pathway network (Fig. 1) was constructed based on significantly enriched pathways using Cytoscape to understand the pharmacological mechanism of Drymaria cordata phytocompounds against cervical cancer and other types of cancers. In the network, zero scores were observed for clustering coefficient, self-loops, stress, and betweenness centrality. Among all the selected phytocompounds, the maximum number of hub genes were modulated by quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside (HRAS, IL6, TNF, AR, and VEGFA) and 5,4′-dihydroxy-7-methoxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside (TNF, AR, CTNNB1, VEGFA, and HRAS), while the maximum number of pathways were modulated by HRAS, CTNNB1, and VEGFA.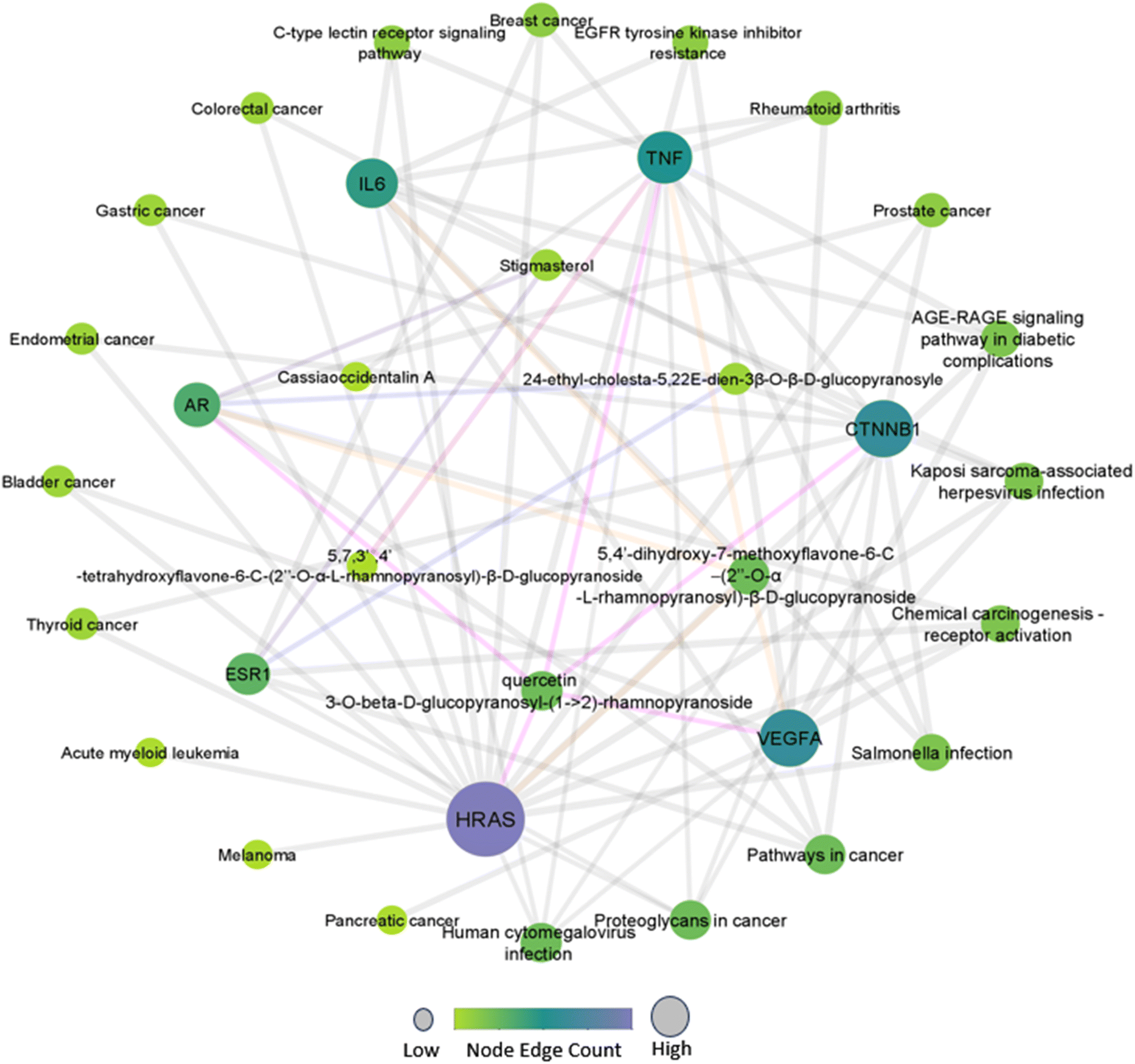 | ||
| Fig. 1 Interaction network of phytocompounds with hub genes and regulated pathways. | ||
3.6. Molecular docking studies
All binding energy scores were calculated from the best cluster (95%) within an RMSD of 0.25 Å. The docked complex between VEGFA (VEGF) and 5,4′-dihydroxy-7-methoxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside showed a low binding affinity (ΔG – 5.0 kcal mol−1). During the interaction, it exhibited conventional hydrogen bonds with Thr16, Tyr18, Asn48, Lys70, and Thr71 (Fig. 2a). Quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside showed higher binding affinity (ΔG – 7.6 kcal mol−1) with hydrogen bonds formed with Thr16 and Val85 residues, and carbon hydrogen bonds with Gly15, and pi–alkyl interactions with Lys70 residues (Fig. 2b). The docking algorithm and scoring were validated with the co-crystallized ligand OMA, showing similar binding energy (ΔG – 7.1 kcal mol−1) and an RMSD value of 0.745 Å for the redocked OMA and crystal structure with OMA (Fig. 2c and d), validating the docking study.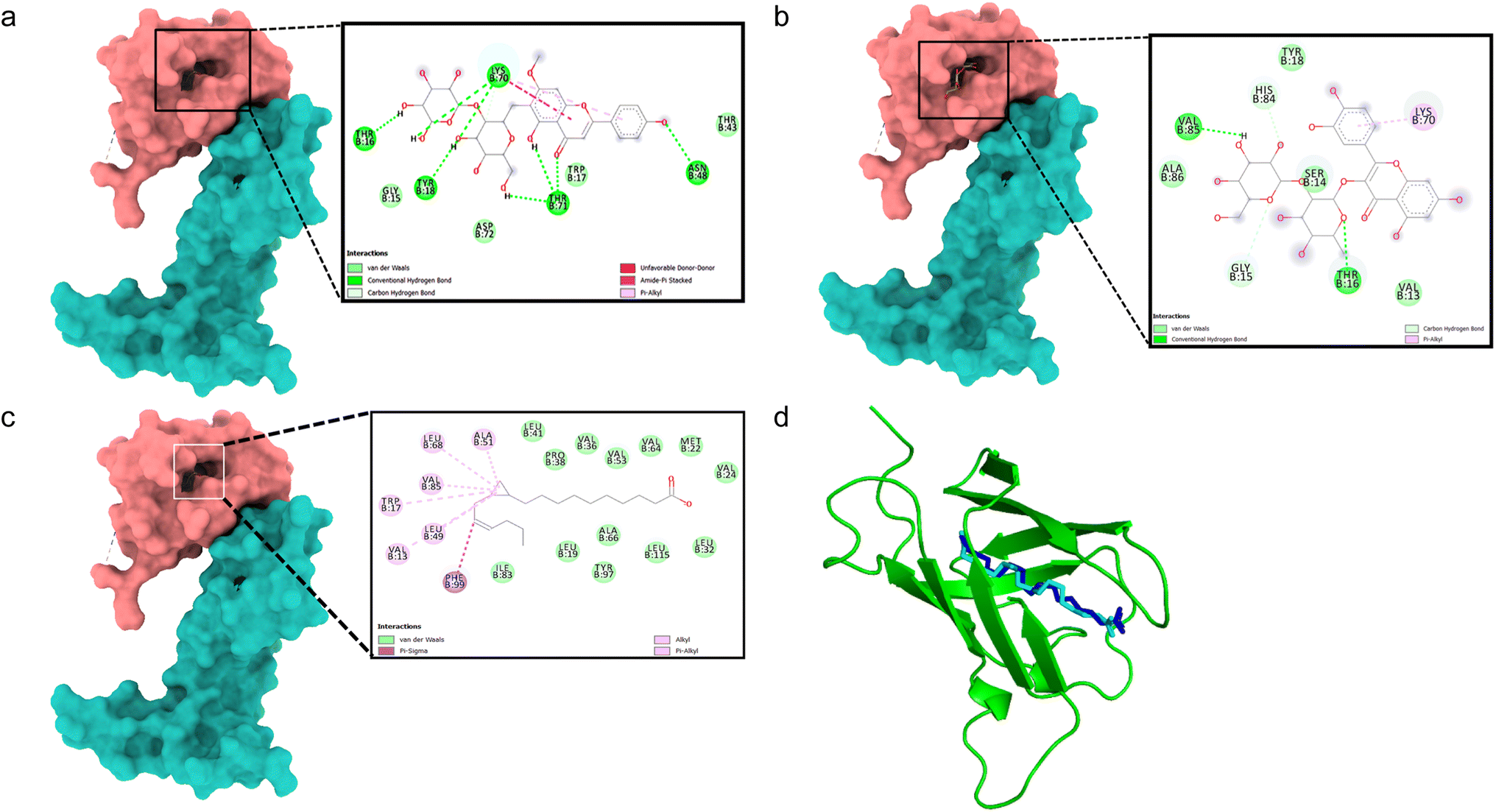 | ||
| Fig. 2 Molecular surface view of the 4QAF with (a) 5,4′-dihydroxy-7-methoxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside and (b) quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside (c) OMA bound in deep cavity. 2D interaction is exhibiting the interactions between ligands and proteins. (d) Is exhibiting superimposed structure of redocked co-crystallized ligand (cyan) with protein and the crystal structure (blue) for docking validation at the binding cavity. | ||
Docking between CTNNB1 and quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside showed low binding affinity (ΔG – 5.71 kcal mol−1) with conventional hydrogen bonds formed with Asn206, Asp207, and Lys242 residues (Fig. 3a). The validation with the co-crystallized ligand R90 revealed a similar binding energy (ΔG – 5.9 kcal mol−1) and an RMSD value of 0.070 Å for the redocked R90 and crystal structure with R90 (Fig. 3b and c), validating the docking study.
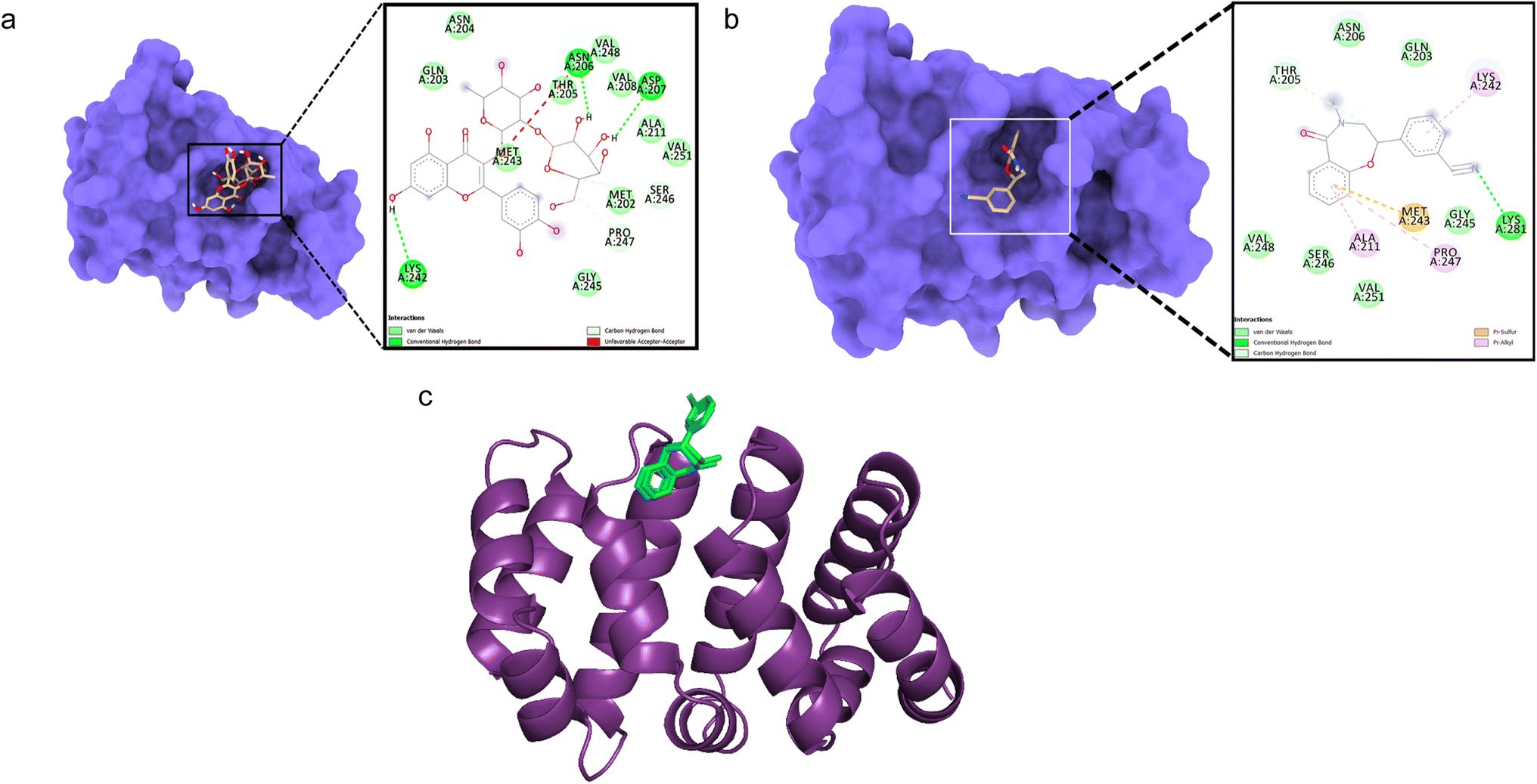 | ||
| Fig. 3 Molecular surface view of the 7AFW with (a) quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside and (b) R90 bound in deep cavity. 2D interactions is exhibiting the interactions between ligand and protein and dotted lines exhibiting interactions. (c) Is exhibiting superimposed structure of redocked co-crystallized ligand (green) with protein and the crystal structure (cyan) for docking validation at the binding cavity. | ||
Docking between HRAS and 5,4′-dihydroxy-7-methoxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside showed a considerable binding affinity (ΔG – 5.7 kcal mol−1). During the interaction, it exhibited conventional hydrogen bonds with Lys117, pi–alkyl interactions with Leu120, pi–cation interaction with Asp30 and Lys147, and pi–pi interaction with Phe28 residues (Fig. 4a). Quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside showed higher binding affinity (ΔG – 8.4 kcal mol−1) with conventional hydrogen bonds formed with Asp30, Asp119, Ala146, and Lys147 residues, pi–alkyl interactions with Ala18, Lys117, Ala146 residues, pi–cation with Lys117, and pi–pi interaction with Phe28 residues (Fig. 4b). The validation with the co-crystallized ligand GDP revealed a similar binding energy (ΔG – 10.4 kcal mol−1) and an RMSD value of 0.532 Å for the redocked GDP and crystal structure with GDP (Fig. 4c and d), validating the docking study.
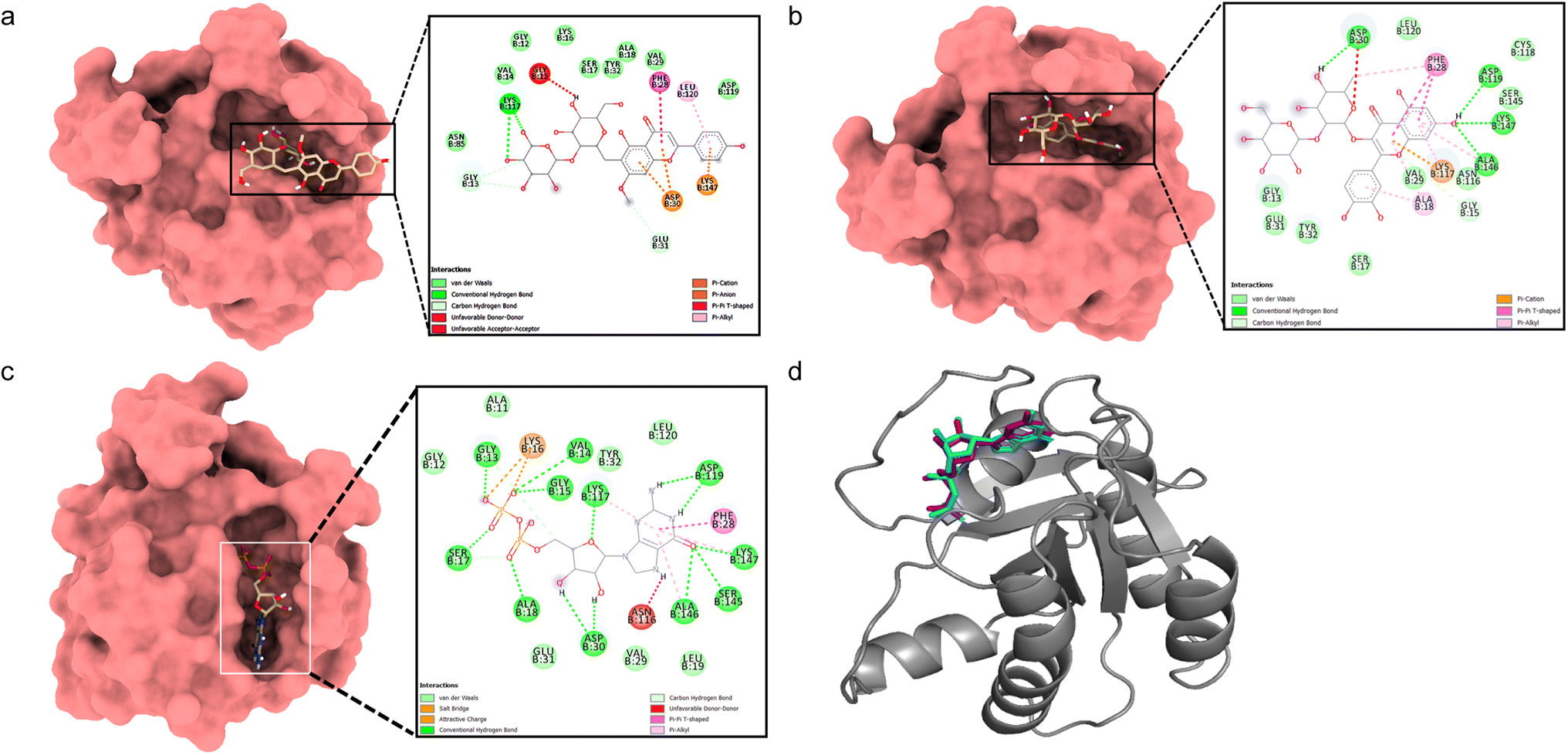 | ||
| Fig. 4 Molecular surface view of the 7JII with (a) 5,4′-dihydroxy-7-methoxyflavone-6-C-(2′′-O-α-L-rhamnopyranosyl)-β-D-glucopyranoside and (b) quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside (c) GDP bound in deep cavity. 2D interaction exhibiting the interactions between ligands and proteins and dotted lines exhibiting interactions. (d) Is exhibiting superimposed structure of redocked co-crystallized ligand (purple) with protein and the crystal structure (cyan) for docking validation at the binding cavity. | ||
3.7. Molecular dynamics simulation
The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) are the major indicators for defining the qualitative stability of the docked ligand–protein complex. The high RMSD values represent the high deviation in structural changes compared to the initial structure at the starting point, showing less stability of the ligand–protein complex. The result of the present study revealed that the co-crystal of VEGF showed good stability and compactness. Also, fluctuation was observed from 0.8 to 6.4 Å at 20–60 nanoseconds. Similarly, quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside with protein VEGF complex represents initial fluctuation from 0.8–7.2 Å at approximately 15 nanoseconds after that constant stability was observed. The RMSD value of the co-crystal of HRAS represents minor fluctuation between 1.2 to 3.2 Å up to 20 ns, after that it was stable. Similarly, the ligand–protein complex showed good stability throughout the simulation time. The initial minor fluctuation was observed from 1–1.75 Å (Fig. 5). The RMSF values of the co-crystal structure were maintained within the range of 1–4 Å for VEGF and 0.5–2 Å for HRAS. In the co-crystal structure of VEGF, significant fluctuation was observed from residue 89–95 (Glu13, Val14, Val15, Lys16, Phe17, Met18, Asp19). Similarly, the co-crystal structure of HRAS exhibited fluctuation in residue numbers 60–63 (Gln61, Glu62, Glu63, Tyr64). The RMSF values were in the range of 0.8–3.2 Å for the VEGF-quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside complex and 0.4–2 Å for the HRAS-quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside complex (Fig. 6).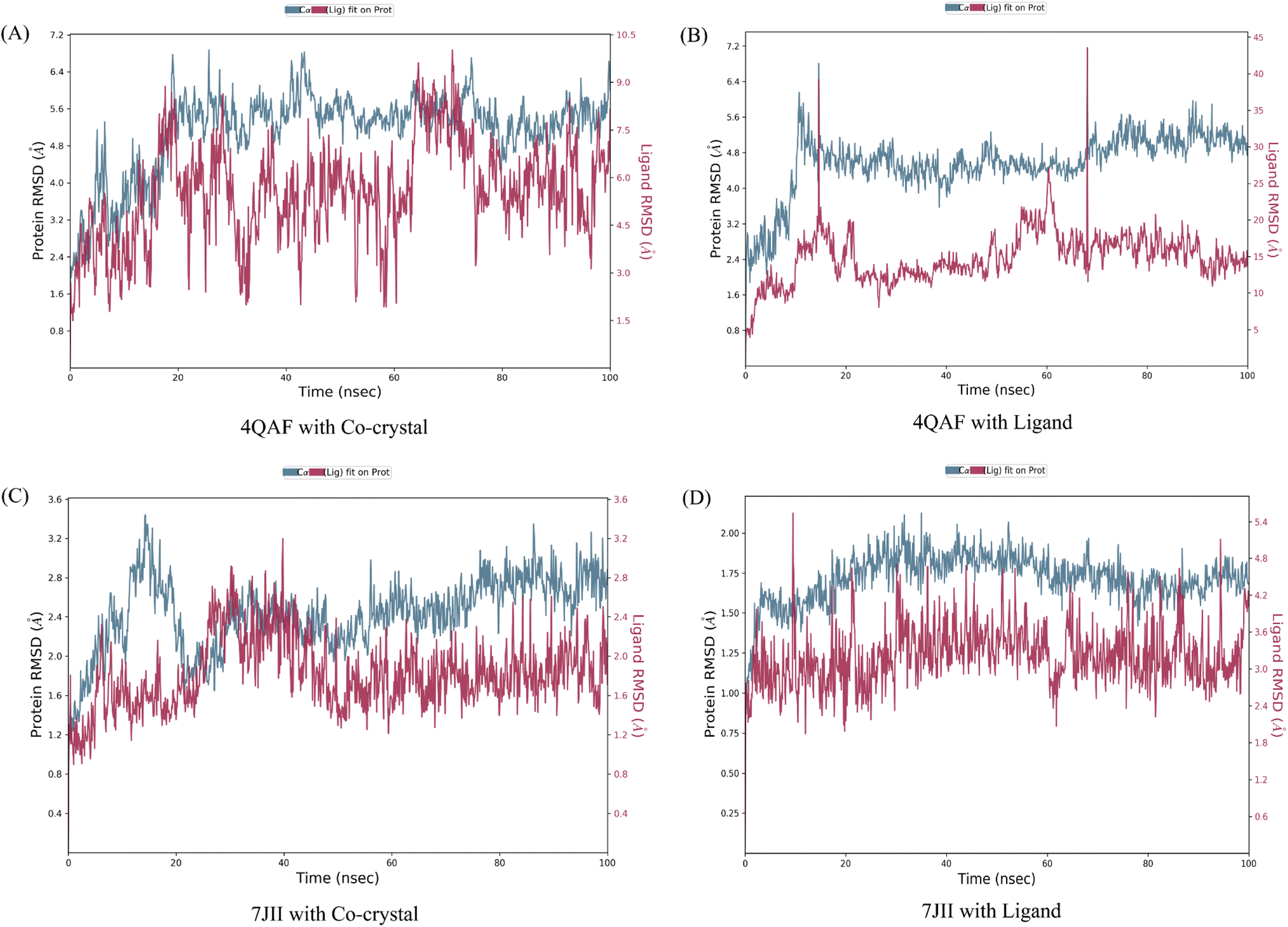 | ||
| Fig. 5 C-alpha root mean square deviation of (A) VEGF in complex with co-crystallized ligand (B) VEGF in complex with quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside (C) HRAS in complex with co-crystallized ligand (D) HRAS in complex with quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside. The root mean square deviations employed to assess the average alteration in the positioning of a chosen group of atoms within a specific frame when compared to a baseline frame. This value is computed across all frames present in the trajectory. | ||
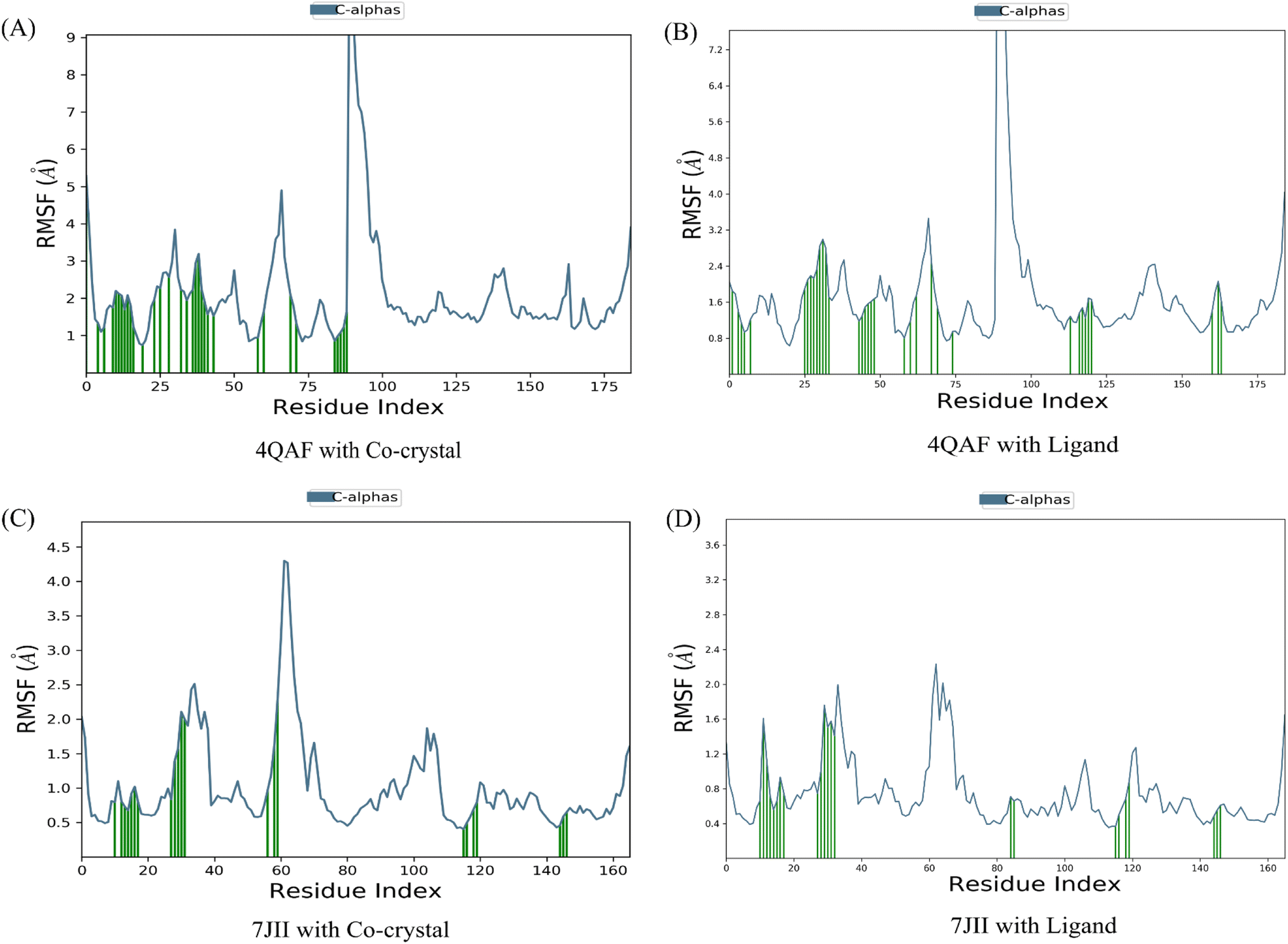 | ||
| Fig. 6 Root mean square fluctuation of C-α of amino acids of the target proteins in complex with Co-crystallized ligands and proteins in complex with quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside. The root mean square fluctuation is used to characterize local changes along the protein chain. | ||
The binding affinity of the ligand with the protein demonstrates similar RMSF values compared to the co-crystal structure. In the present study, simulation analysis elucidates the changes in the secondary structure elements (SSEs) during the interaction of the ligand–protein complex throughout the 100 ns of simulation time (Fig. S7†). The distribution of secondary structure elements (SSEs) in the protein structure, based on the residues index, was determined for both the co-crystal structure and the ligand–protein complex of VEGF. The α-helices and β-sheets exhibited distinct transitions into the loop regions of the secondary structure. In both the co-crystal structure and the ligand–protein complex, 150 residues of α-helices were consistently expressed throughout the simulation time. Similarly, the co-crystal structure and ligand–protein complex of HRAS demonstrated the percentage of SSEs for each residue of α-helices and β-sheets, exhibiting repetitive transitions throughout the 100 ns simulation time (Fig. S8†).
Intermolecular contacts of co-crystal structure and ligand–protein complex of VEGF, in the active site residues are presented in the histogram. The pattern of interaction represents hydrogen, hydrophobic, ionic bond, and water molecule interaction. Fig. 7A of the co-crystal structure shows the hydrogen bond interaction at amino acid positions Thr23, Asp25, Thr54, and Glu63. While the ligand–protein complex shows the hydrogen bond interaction at amino acid positions Val13, Ser14, Thr16, Tyr18, Pro38, Leu41, Thr43, Asn48, Ser69, Lys70, Thr71, Asp72, Lys87, Tyr97, Glu102, Pro40, Asp41, Glu44 respectively (Fig. 7B). The co-crystal structure of HRAS intermolecular contact of active site residues represents the hydrogen bond interaction at amino acid positions Gly13, Val14, Gly15, Lys16, Ser17, Ala18, Asp30, Glu31, Asn116, Lys117, Asp119, Ser145, Ala146, Lys147 respectively (Fig. 7C) whereas, ligand–protein complex of HRAS represents the hydrogen bond interaction at amino acid positions Gly12, Gly13, Gly15, Ser17, Ala28, Asp30, Glu31, Tyr32, Lys117, Asp119, Ala146, Lys147 respectively (Fig. 7D).
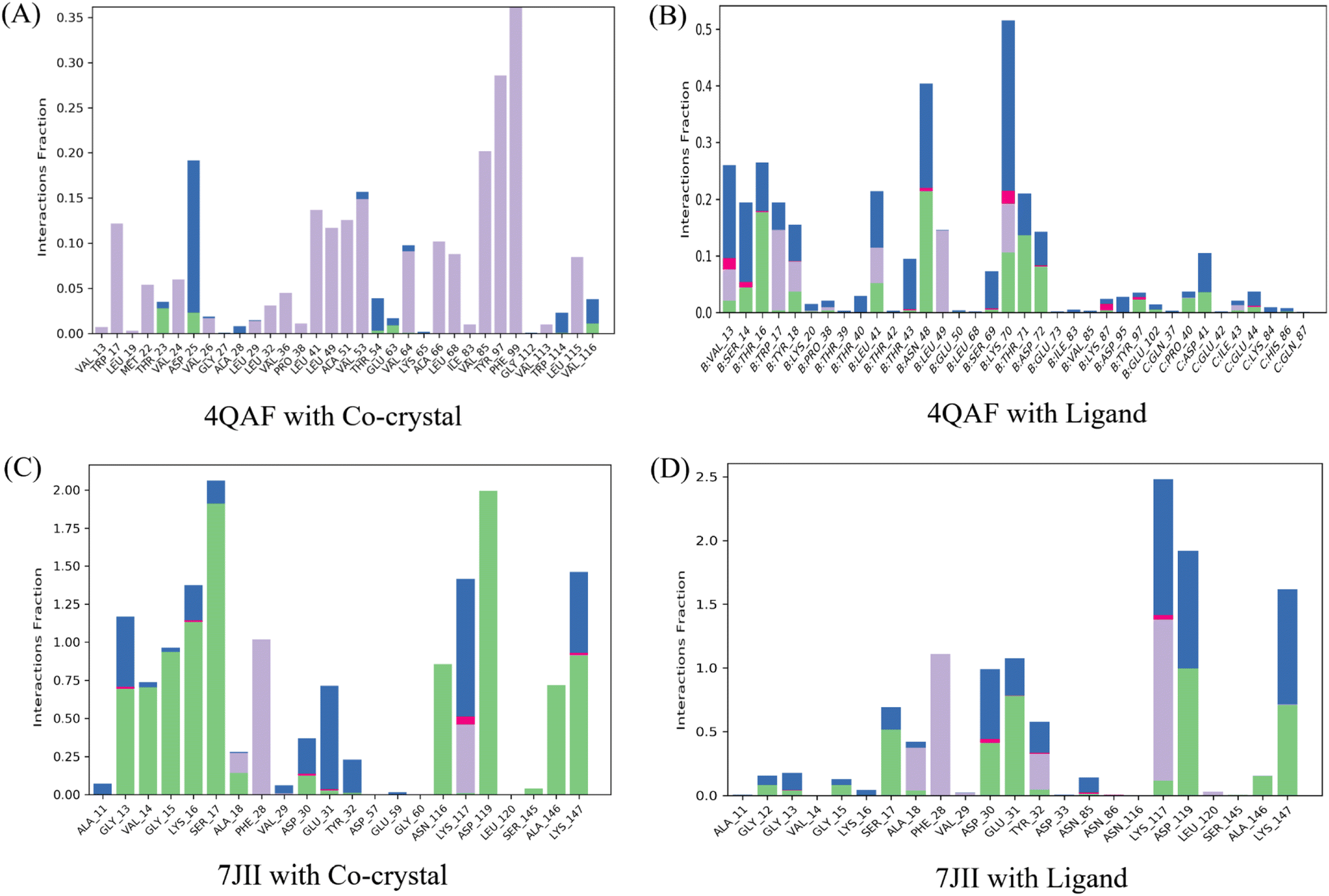 | ||
| Fig. 7 Histogram of protein-ligand contacts presenting hydrogen bonds, hydrophobic, ionic interactions, and water bridges during the 100 ns simulation. Hydrogen, hydrophobic, ionic interactions, and water bridges are represented in green, light purple, dark pink, and blue colors, respectively. The amino acids of the allosteric site involved in the interactions are noted on the respective x-axis. The stacked bar charts are scaled to represent the normalized duration of interactions during the trajectory. For instance, a value of 0.5 indicates that the specific interaction is maintained for approximately 50% of the simulation time. It is important to note that values greater than 1.0 are possible because some protein residues might form multiple contacts of the same subtype with the ligand. | ||
It was observed that during 100 ns simulation of co-crystal structure, minor interaction was determined including Trp17, Asp25, Val53, Val64, Ala66, Val85, Tyr97, Phe99, and Leu115 respectively. While, ligand protein complex of VEGF showed strong amino acid residue interaction via Val13, Ser14, Thr16, Trp17, Leu41, Asn48, Lys70. Similarly, co-crystal structure of HRAS represents strong intermolecular amino acid residues interactions by Gly13, Val14, Gly15, Lys16, Ser17, Ala18, Phe28, Asp30, Glu31, Tyr32, Asn116, Lys117, Asp119, Ala146, Lys147 respectively. Whereas, the interaction of protein HRAS with ligand also, showed strong interactions via Gly13, Gly15, Ser17, Ala18, Phe28, Asp30, Glu31, Lys117, Asp119, Ala146, Lys147 respectively. The result showed that both ligand–protein complexes have strong amino acid residue interactions throughout the simulation time (Fig. 8).
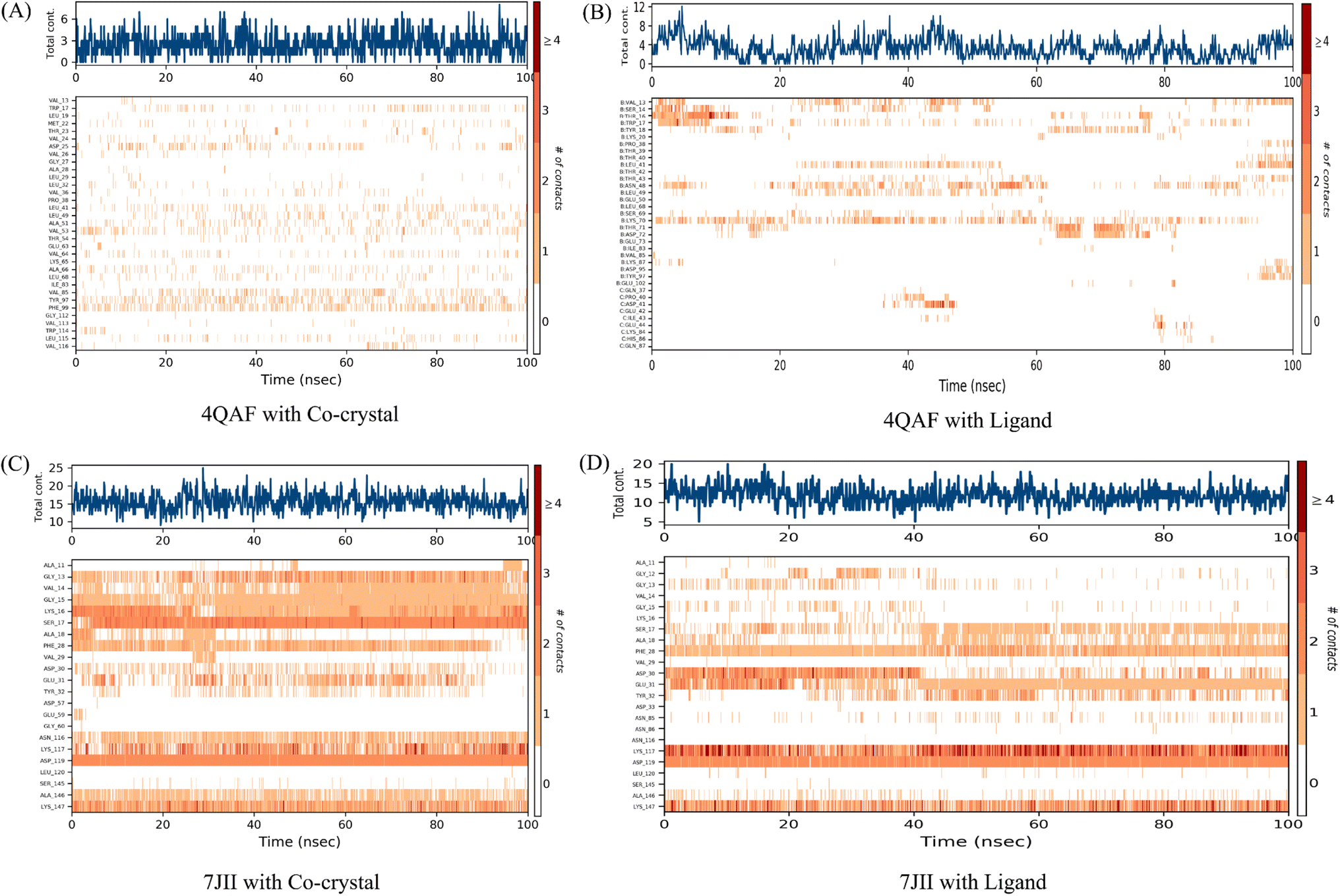 | ||
| Fig. 8 Number of contacts made by various residues of the allosteric site of the proteins in complex with co-crystallized ligands and quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside, respectively. The dark orange color indicates the maximum number of contacts, and the white colour indicates no contacts between the specific proteins' allosteric site residues and respective ligands over the 100 ns simulation time. On the right side, the scale shows the number of contacts. The increase in contacts is indicated by a change from white to dark orange colour. | ||
The rGYr value of both the co-crystal and ligand–protein complex of VEGF was stable between 5.4 to 6.6 Å and 4.4 to 4.6 Å. Similarly, the value of SASA at 0–100 ns simulation was steady between 80 to 120 Å2. Also, the PSA values were steady from the initial to 100 ns simulation time between 90 to 96 Å2 whereas SASA had fluctuation between 450-750 Å2 at 0 to 20 ns simulation time. After that, it was steady through simulation time. The PSA value was also steady throughout the simulation time ranging between 425 to 450 Å2. In addition there was minor RMSD fluctuation between 1 to 1.5 Å2 from the initial to 40 ns simulation time. After that, it was steady throughout the simulation time. Likewise, minor fluctuation was observed between 0.8 to 1.6 Å2 throughout 100 ns simulation time. The major fluctuation was observed at 40 ns simulation time for rGYr (4.2 to 4.6 Å2) and in PSA analysis, 465 to 495 Å2 fluctuation was observed at 100 ns simulation time whereas, the ligand–protein complex showed minor rGYr fluctuation in the 100 ns simulation time between 4.65 to 4.80 Å2. Initially, in 40 ns major fluctuation was observed between 425 to 450 Å2. After that, the fluctuation was steady throughout the simulation time. The result showed that ligand–protein complexes have good stability with minor fluctuation (Fig. S9†). After the simulation analysis, the co-crystal structure and ligand–protein complex interaction was visualized in 2D (Fig. S10†).
3.8. Molecular mechanics generalized Born surface area (MM-GBSA) calculations
The binding free energy and other contributing energies in the form of MM-GBSA were determined for the complexes of VEGF + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside and HRAS + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside by utilizing the MD simulation trajectory. The findings indicate that the primary factors influencing the stability of the simulated complexes are ΔGbindCoulomb, ΔGbindLipo, and ΔGbindvdW, while ΔGbindcovalent and ΔGbindSolvGB contribute to the instability of the corresponding complexes. Both VEGF + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside and HRAS + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside complexes exhibit higher binding free energies (Table S3†). The findings of this study support the substantial binding affinity of quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside towards VEGFA and HRAS proteins, highlighting its capacity to form stable complexes with these protein targets. The time series analysis of snapshots from the MD simulation trajectory of VEGF conformation reveals the movement of quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside from the exterior to the periphery between 0 ns and 100 ns. The terminal domain of VEGF undergoes a transformation from a diminishing helical turn to an open loop during this period. In HRAS, quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside initially appears straighter, but as the simulation progresses from 0 ns to 100 ns, the ligand demonstrates movement into a deeper arrangement within the binding pocket. Angular changes in the ligand's arrangement, along with relatively fewer changes in the protein loops, contribute to the rearrangement of the ligand (Fig. S11†).The MD simulation trajectories were analyzed to comprehend the dynamic cross-correlation33 among the domains of VEGF and HRAS chains that are linked with the quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside molecule. In particular, the amino acid residues that bind with quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside in the HRAS and VEGF proteins demonstrated a harmonized movement of residues (Fig. 9). The concept of free energy landscape (FEL) plays a pivotal role in elucidating the deterministic behavior of proteins as they transition to their lowest energy state.34 The energy state in question is often associated with high stability and optimal conformation, which is particularly true for VEGF in its quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside bound state. It is well recognized that proteins strive to achieve a global minimum, or the lowest free energy state, as part of their natural molecular mechanics. In the scenario presented, it has been observed that this global minimum is attained at a distance of approximately 3 Å with a radius of gyration (Rg) measured at 23.2 Å. While in the case of HRAS, the global minimum is attained at a distance ranging from 3.8 to 2.5 Å with a radius of gyration (Rg) measured at 26 Å. The free energy landscape thus serves as a critical indicator of the protein's folding pathway towards achieving this minimum energy state. The tendency of the protein to achieve its global energy minimum is significantly influenced by its interaction with quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside, which is clearly manifested in its bound state. Hence, in the context of this investigation, the FEL not only provides a landscape upon which the behavior of the protein can be studied and understood but also signifies the profound impact of quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside on facilitating the protein to reach its lowest free energy state. This corroborates the notion that FEL acts as a useful tool in deciphering protein folding mechanisms and the potential influence of external binding agents on this process (Fig. 9).
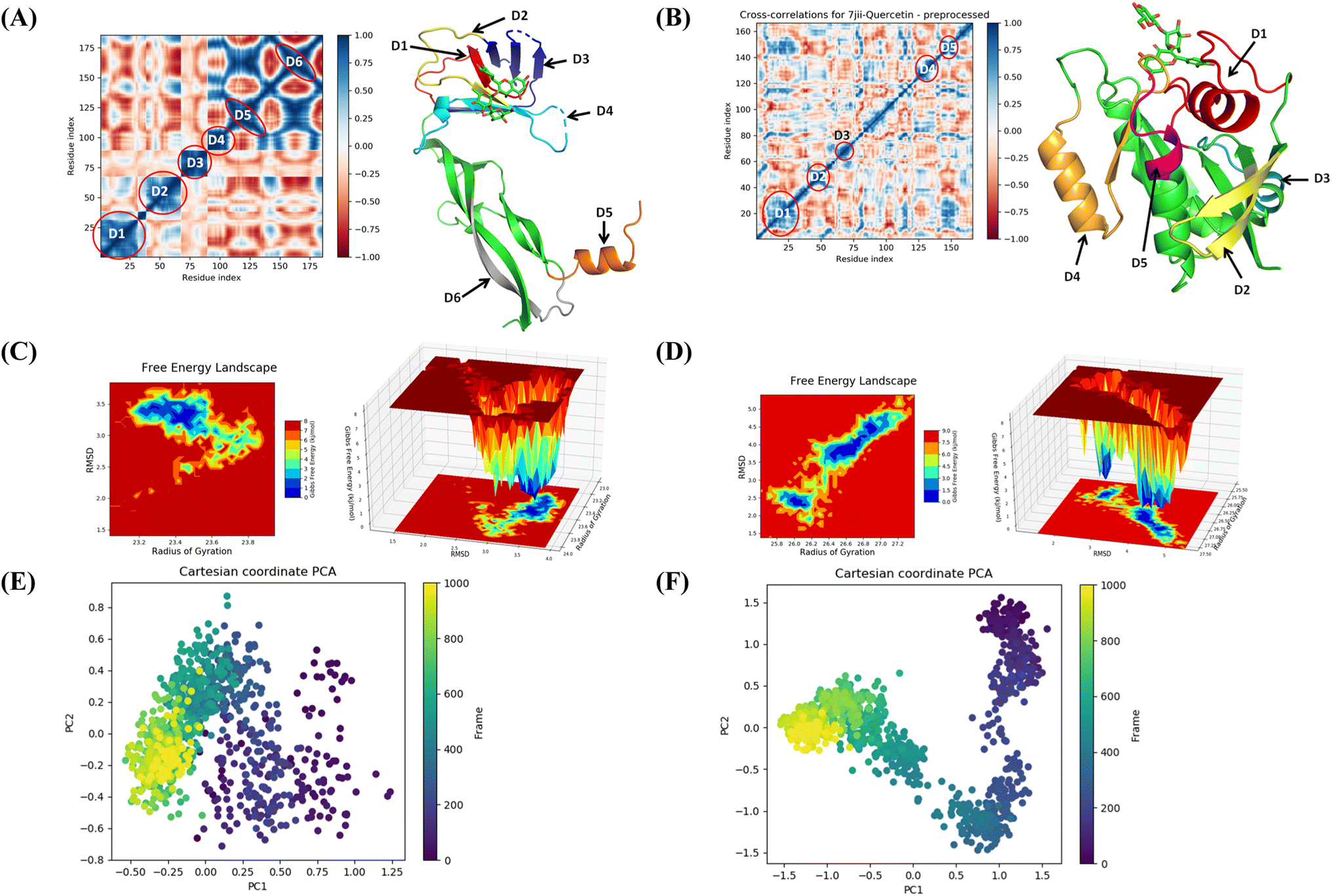 | ||
| Fig. 9 Dynamic cross correlation matrix (DCCM) of correlated amino acids conformed into secondary structural domains (colored) and non-correlated domains (green) of (A) 4QAF and (B) 7JII. 2D contour plot and 3D interpolation plot of the free energy landscape (FEL) of (C) 4QAF and (D) 7JII in the bound state. Principal component analysis (PCA) of (E) 4QAF + quercetin and (F) 7JII + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside in complex, displaying Eigen vector clusters of the simulation frames in PC1 and PC2. | ||
The use of principal component analysis (PCA) was employed to analyze the molecular dynamics (MD) simulation trajectories35 of VEGF + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside and HRAS + quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside. This approach provides an interpretation of the initially scattered trajectories that exhibit more flexibility, which can be attributed to the random nature of the protein structure resulting from non-correlated global motion. The covariance matrix was used to record the mobility of internal coordinates in three-dimensional space for a time period of 100 ns. The rational movement of every trajectory is analyzed through the utilization of orthogonal sets or Eigenvectors. The MD simulation trajectory of Cα atoms of VEGF bound to quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside displayed more unordered orientation in PC1 and PC2 modes and more toward negative correlation from initial 800 frames, but only the last 200 frames (from 800–1000) exhibited positive correlation motion while HRAS bound to quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside clustered into a more oriented manner, and clusters of the last 200 frames appeared to be a very ordered correlated motion. Hence, it can be inferred that the protein bound to quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside centering of the frames in a single cluster (shown by the color yellow) suggests the presence of periodic motion in the molecular dynamics trajectories, which is attributed to the stable conformational global motion (Fig. 9).
4. Conclusion
This study has successfully identified quercetin 3-O-β-D-glucopyranosyl-(1→2)-rhamnopyranoside as a promising inhibitor of vascular endothelial growth factor and HRas proto-oncogene for potential use in cervical cancer therapy. Employing a structure-based drug design approach, combined with docking, molecular dynamics simulations, and energy calculations, provided valuable insights into the behavior, stability, and energetic characteristics of the complexes formed between ligands and proteins. These findings hold the potential to significantly contribute to the development of novel compounds for combatting cervical cancer. However, it's important to note that the conclusions drawn in this study are based solely on computational findings, and further validation through experimental approaches is necessary.Ethical statement
This work doesn't include any animal or human studies.Conflicts of interest
All the authors of this manuscript declare that they do not have any conflicts of interest in any financial or non-financial means. All the authors of this manuscript have read and approved the final draft.References
- S. Zhang, H. Xu, L. Zhang and Y. Qiao, Chin. J. Cancer Res., 2020, 32, 720–728 CAS.
- M. Bezabih, F. Tessema, H. Sengi and A. Deribew, Ethiop. J. Health Sci., 2015, 25, 345–352 CrossRef PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- M. V. Sherer, N. V. Kotha, C. Williamson and J. Mayadev, Int. J. Gynecol. Cancer, 2022, 32, 281–287 CrossRef PubMed.
- A. Naeem, P. Hu, M. Yang, J. Zhang, Y. Liu, W. Zhu and Q. Zheng, Molecules, 2022, 27, 8367 CrossRef CAS PubMed.
- B. B. Shaik, N. K. Katari and S. B. Jonnalagadda, Chem. Biodiversity, 2022, 19, e202200535 CrossRef CAS PubMed.
- A. J. Akindele, I. F. Ibe and O. O. Adeyemi, Afr. J. Tradit. Complement. Altern. Med., 2012, 9, 25–35 CrossRef CAS PubMed.
- S. Singla, J. Pradhan, R. Thakur and S. Goyal, Phytomed. Plus, 2023, 3, 100469 CrossRef.
- H. Ji, K. Li, W. Xu, R. Li, S. Xie and X. Zhu, Front. Oncol., 2022, 11, 780387 CrossRef PubMed.
- H. S. Lee, I. H. Lee, K. Kang, S. I. Park, M. Jung, S. G. Yang, T. W. Kwon and D. Y. Lee, Nat. Prod. Commun., 2021, 16, 1–15 Search PubMed.
- X. Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2012, 7, 146–157 CrossRef PubMed.
- K. Bhattacharya, P. Khanal, V. S. Patil, P. S. R. Dwivedi, N. R. Chanu, R. K. Chaudhary and A. Chakraborty, J. Biomol. Struct. Dyn., 2023, 1–16 CrossRef PubMed.
- J. Ru, P. Li, J. Wang, W. Zhou, B. Li, C. Huang, P. Li, Z. Guo, W. Tao, Y. Yang, X. Xu, Y. Li, Y. Wang and L. Yang, J. Cheminf., 2014, 6, 1–6 Search PubMed.
- R. Xue, Z. Fang, M. Zhang, Z. Yi, C. Wen and T. Shi, Nucleic Acids Res., 2013, 41, 1089–1095 CrossRef PubMed.
- K. Bhattacharya, R. Bordoloi, N. R. Chanu and R. Kalita, J. Genet. Eng. Biotechnol., 2022, 20, 43 CrossRef PubMed.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, Nucleic Acids Res., 2018, 46, W257–W263 CrossRef CAS PubMed.
- J. Piñero, Á. Bravo, N. Queralt-Rosinach, A. Gutiérrez-Sacristán, J. Deu-Pons, E. Centeno, J. García-García, F. Sanz and L. I. Furlong, Nucleic Acids Res., 2017, 45, D833–D839 CrossRef PubMed.
- D. Gfeller, A. Grosdidier, M. Wirth, A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2014, 42, 32–38 CrossRef PubMed.
- D. S. Druzhilovskiy, A. V. Rudik, D. A. Filimonov, T. A. Gloriozova, A. A. Lagunin, A. V. Dmitriev, P. V. Pogodin, V. I. Dubovskaya, S. M. Ivanov, O. A. Tarasova, V. M. Bezhentsev, K. A. Murtazalieva, M. I. Semin, I. S. Maiorov, A. S. Gaur, G. N. Sastry and V. V. Poroikov, Russ. Chem. Bull., 2017, 66, 1832–1841 CrossRef CAS.
- Z. Wang, L. Liang, Z. Yin and J. Lin, J. Cheminf., 2016, 8, 1–10 Search PubMed.
- J. Pietsch, S. Riwaldt, J. Bauer, A. Sickmann, G. Weber, J. Grosse, M. Infanger, C. Eilles and D. Grimm, Int. J. Mol. Sci., 2013, 14, 1164–1178 CrossRef CAS PubMed.
- S. Paul, M. Andrew, O. Owen and S. Nitin, Genome Res., 1971, 13, 426 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- D. E. Shaw, P. Maragakis, K. Lindorff-Larsen, S. Piana, R. O. Dror, M. P. Eastwood, J. A. Bank, J. M. Jumper, J. K. Salmon, Y. Shan and W. Wriggers, Science, 2010, 330, 341–346 CrossRef CAS PubMed.
- K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, 2006, vol. 84 Search PubMed.
- E. Chow, C. A. Rendleman, K. J. Bowers, R. O. Dror, D. H. Hughes, J. Gullingsrud, F. D. Sacerdoti and D. E. Shaw, Simulation, 2008, 1–14 Search PubMed.
- D. Shivakumar, J. Williams, Y. Wu, W. Damm, J. Shelley and W. Sherman, J. Chem. Theory Comput., 2010, 6, 1509–1519 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- L. Piao, Z. Chen, Q. Li, R. Liu, W. Song, R. Kong and S. Chang, Int. J. Mol. Sci., 2019, 20, 224 CrossRef PubMed.
- G. Singh, D. Al-fahad, M. K. Al-zrkani, K. Chaudhuri, H. Soni, S. Tandon, C. Venkata, F. Azam and R. Patil, J. Biomol. Struct. Dyn., 2023, 1–18 Search PubMed.
- P. Khanal, V. S. Patil, B. M. Patil, K. Bhattacharya, A. K. Shrivastava, R. K. Chaudhary, L. Singh, P. S. Dwivedi, D. R. Harish and S. Roy, Comput. Biol. Chem., 2023, 107, 107957 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06297j |
| This journal is © The Royal Society of Chemistry 2024 |