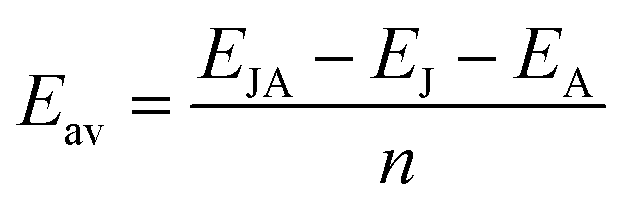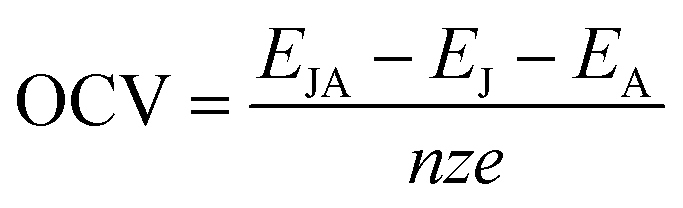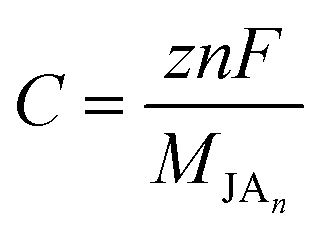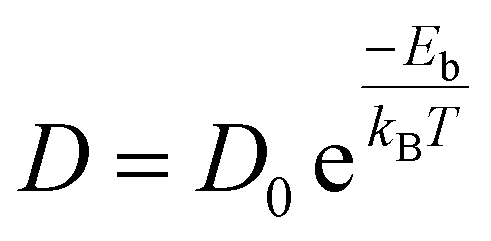DOI:
10.1039/D3RA04781D
(Paper)
RSC Adv., 2024,
14, 5771-5781
CrXY (X/Y = S, Se, Te) monolayers as efficient anode materials for Li and Na-ion batteries: a first-principles study†
Received
16th July 2023
, Accepted 25th January 2024
First published on 14th February 2024
Abstract
Over the last decade, two-dimensional (2D) materials have been of great interest in the energy storage field. Large-scale electrochemical energy storage is based on the intercalation of metal ions in layered materials having van der Waals gaps. In this work, by means of first-principles calculations, we explored the use of 2D Janus transition metal dichalcogenides (TMDs) CrSSe, CrSTe and CrSeTe as anode materials for lithium and sodium-ion batteries. To examine the electronic properties and electrochemical performance, density functional theory (DFT) calculation was used. Our research shows that lithium diffuses easily with short diffusion distances and prefers to bind effectively to the monolayer. These structures are metallic in their bare phases. The highest adsorption energy shown by CrSSe, CrSTe, and CrSeTe is −1.86 eV, −1.66 eV, −2.15 eV with a low diffusion barrier of 0.3 eV, 0.6 eV, and 0.1 eV for the Li atoms and 0.54 eV, 0.32 eV and 0.15 eV for the Na atoms, respectively. At different chemical stoichiometries, we discovered negligible average open-circuit voltages of 1.0 V, 0.52 V, 0.6 V for lithium and 0.1 V, 0.49 V, and 0.51 V for sodium atoms respectively. The storage capacities shown by CrSSe, CrSTe, and CrSeTe are 348 mA h g−1, 254 mA h g−1, 208 mA h g−1 for the Li atoms and 260 mA h g−1, 198 mA h g−1, 177 mA h g−1 for the Na atoms respectively.
Introduction
Over the past few decades, high-performance lithium-ion battery (LIB) storage materials have consistently been popular research areas due to their exceptional energy storage performance.1 Electronic gadgets, such as laptops and cell phones, are becoming more and more indispensable in today's world.2 This implies that there is a huge need for energy storage technology. Researchers have focused on enhancing design and finding new materials to enhance the performance of devices such as energy density, kinetics, and cycle life in order to meet this trend.3,4 Scientists are currently concentrating on two-dimensional (2D) materials, which are used in many different contexts due to their unique qualities with the characteristics of good conductivity and high specific surface area and a lot of different areas such as in electronics, catalysis, electrochemical energy storage, and particularly in rechargeable lithium-ion batteries.5–8 Since graphene was first exfoliated from graphite in 2004, numerous 2D materials, including phosphorene,9,10 arsenene11 transition metal dichalcogenides (TMDs),12–14 transition metal carbides15,16 and nitrides (MXenes)17 and other 2D nanomaterials, have been synthesized and studied for LIB electrode materials.
Transition metal dichalcogenides (TMDs) are a broad class of two-dimensional (2D) materials with high surface areas and exceptional electrical and chemical characteristics18,19 also TMDs have many excellent qualities, including an outstanding capacity for charge transfer,20 a high surface to volume ratio,21 an energy band gap that can be controlled by the number of layers,22 a potent interaction with light, and mechanical toughness.23,24 They have been proposed as suitable electrode materials for sodium-ion batteries (SIBs) as well as lithium-ion batteries (LIBs).25 For example, it has been proposed that 1T MoS2 monolayer is a suitable electrode material for sodium-ion batteries (SIBs) as well as lithium-ion batteries (LIBs) with a high specific capacity with an ultra-thin diffusion barrier.26,27 MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 has various crystalline phases (2H, 3R, 1T, etc.) with various atomic stacking arrangements and coordination modes. 3R-MoS2 has a rhombohedral unit cell with A–B–C layers (R3m), 2H-MoS2 adopts trigonal prismatic coordination in A–B sequence (P63/mmc space group), and 1T-MoS2 adopts octahedral coordination. The Janus monolayer TMXY is created when one of the two chalcogen atom (X) layers in monolayer TMX2 is completely replaced by another chalcogen atom (Y). A transition metal atom is bonded to three X atoms and three Y atoms in the Janus monolayer TMXY. There are four predominant ways to prepare T phase transition metal dichalcogenides: chemical vapour deposition (CVD),29 2H phase transformation,30 hydrothermal or solvothermal synthesis,31 and chemical exfoliation. Similarly, a new class of Janus transition-metal dichalcogenides monolayer CrSSe, CrSTe, and CrSeTe32,33 represented as CrXY (X, Y = S/Se/Te) has been reported which possesses T phase structure of transition-metal dichalcogenide. The Janus transition-metal dichalcogenides exhibit inherent ferromagnetism with high Curie temperatures34,35 and substantial spin polarisation36 with interesting spin-transport.37,38 But they have not been reported for their use in electrode material.
28 has various crystalline phases (2H, 3R, 1T, etc.) with various atomic stacking arrangements and coordination modes. 3R-MoS2 has a rhombohedral unit cell with A–B–C layers (R3m), 2H-MoS2 adopts trigonal prismatic coordination in A–B sequence (P63/mmc space group), and 1T-MoS2 adopts octahedral coordination. The Janus monolayer TMXY is created when one of the two chalcogen atom (X) layers in monolayer TMX2 is completely replaced by another chalcogen atom (Y). A transition metal atom is bonded to three X atoms and three Y atoms in the Janus monolayer TMXY. There are four predominant ways to prepare T phase transition metal dichalcogenides: chemical vapour deposition (CVD),29 2H phase transformation,30 hydrothermal or solvothermal synthesis,31 and chemical exfoliation. Similarly, a new class of Janus transition-metal dichalcogenides monolayer CrSSe, CrSTe, and CrSeTe32,33 represented as CrXY (X, Y = S/Se/Te) has been reported which possesses T phase structure of transition-metal dichalcogenide. The Janus transition-metal dichalcogenides exhibit inherent ferromagnetism with high Curie temperatures34,35 and substantial spin polarisation36 with interesting spin-transport.37,38 But they have not been reported for their use in electrode material.
In this study, we systematically investigated the main electrochemical properties, such as electronic structures, metal adsorption and diffusion properties, open-circuit voltage (OCV), and specific capacity, of Janus transition-metal dichalcogenide monolayer CrSSe, CrSTe, and CrSeTe as anode materials of metal-ion batteries (LIBs, SIBs) based on Density Functional Theory (DFT) calculations. We can employ this monolayer as a future anode for a metal ion battery due to its significant adsorption energy, good capacity, low OCV, and rapid diffusion characteristics.
Results and discussion
Computational methods
Utilizing the Quantum-ATK code,39,40 density functional theory (DFT)-based first-principles computations41 were used for this investigation. We utilize the linear combination of atomic orbital (LCAO) method as prescribed in Quantum ATK software.42 PseudoDojo pseudopotential with medium basis set and Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional43 with Fermi–Dirac occupation under the spin-polarized generalized gradient approximation (SGGA) method44 were utilized to optimize the structures. To carry out the optimization cutoff energy of 105 Hartree, a Monkhorst–Pack45 of 5 × 5 × 1 k-point grid has been employed to sample the Brillouin zone with the minimum force between the atom set as 0.01 eV A−1 and the maximum energy difference between the two phases is decreased to less than 10−5 eV. A Monkhorst–Pack of 7 × 7 × 1 k-point grid is used for the calculation of all the electronic structures.46 The effect of van der Waals (vdW) interaction is tackled by using the dispersion-corrected DFT method (DFT-D3 method).47 The effect of the periodic image is tackled by creating a significant vacuum by adopting a large lattice constant c = 23 Å in the z-direction. In order to investigate the potential consequences of on-site Coulomb interaction of localized electrons in the investigated transition metals, we used DFT + U (U = 2.5 eV) calculations.48,49
We have calculated the phonon band structure which has supported the structural integrity of this Janus monolayers.50 For a single metal atom on CrSSe, CrSTe, and CrSeTe monolayer, the adsorption energy (Ead) was determined in order to quantify the adsorption qualities between the metals and the bare structure. Which is given by51
where
EJA represents the total energy of the metal atom adsorbed Janus monolayer.
EJ represents the total energy of Janus monolayer in its pure form and
EA represents the total energies of a single metal atom.
The average adsorption energies in each layer (Eav) were computed as follows to determine the monolayer's maximum capacity for adsorption during the adsorption of metal atoms.52
| |
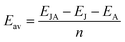 | (2) |
where
n represents the maximum adsorbed metal atom.
The climbing image nudged elastic band (CI-NEB),53,54 approach was used to determine the diffusion energy barriers and minimum energy paths (MEPs) of various metal ions on these monolayers.
The open circuit voltage (OCV)55 was also calculated using the following formula to indicate the performance of batteries:
| |
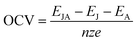 | (3) |
z is the electronic charge of the foreign metal ions (
z = 1 for Li/Na/K,
z = 2 for Mg,
z = 3 for Al), and
e represents charge of the electron.
The following formula could be used to calculate the corresponding specific capacity (C) of our monolayers:56
| |
 | (4) |
F is the Faraday constant (26
801 mA h mol
−1),
MJAn is the molar weights of the
n metal atom absorbed Janus monolayer.
Structural and electronic properties
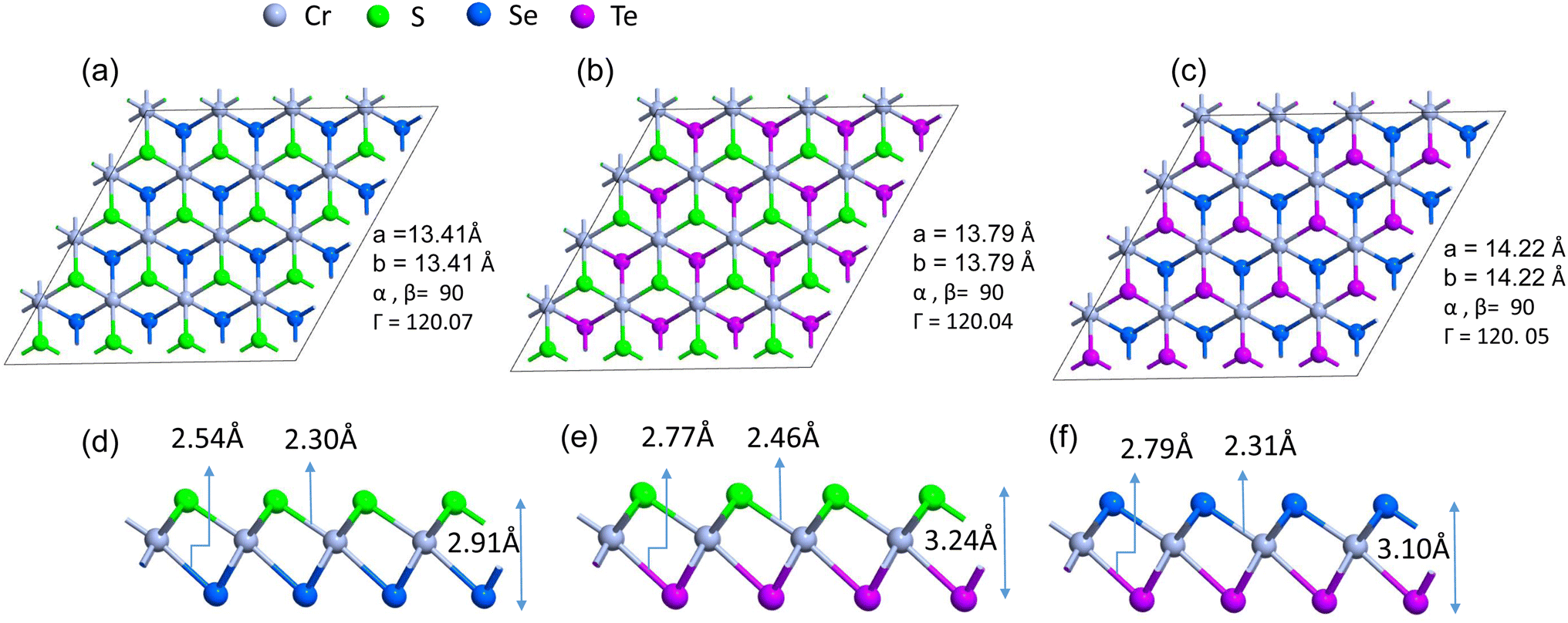
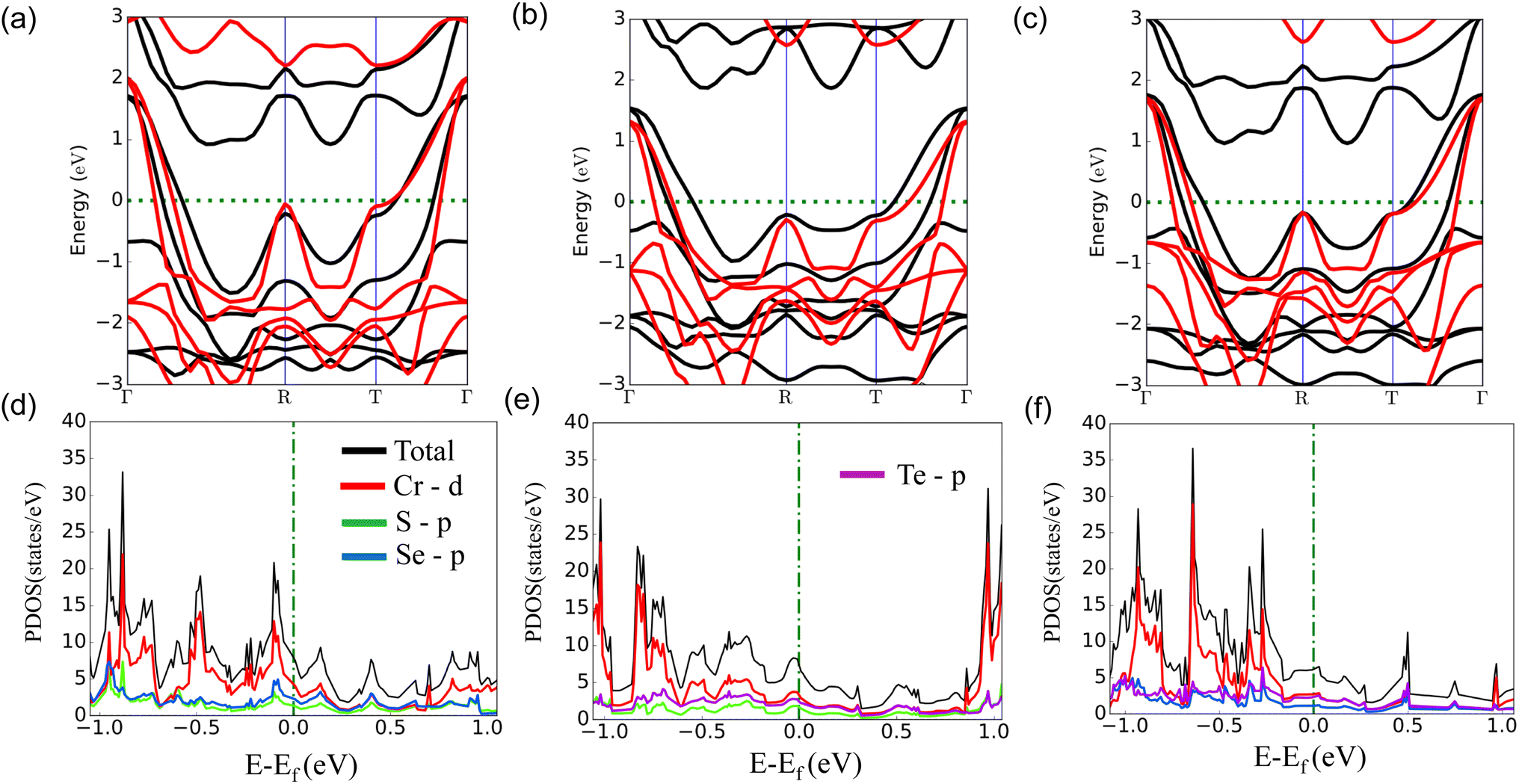
Firstly, we investigate the structures and electronic properties of three Janus transition-metal dichalcogenides monolayers CrSSe, CrSTe, and CrSeTe. The monolayer is a three-atom-thick layer in a sequence of X–TM–Y57 and has a D3d point-group symmetry, where TM is the transition metal atom Cr and X and Y are S, Se, and Te atoms based upon the configuration. Fig. 1 shows the optimized structure, bond length, and vertical distance between the top and bottom layers. Our calculated lattice parameters for CrSSe, CrSTe, and CrSeTe monolayer increase with the increasing atomic radius of chalcogen atoms. To demonstrate the dynamical stability of the Janus TMXY monolayer their phonon spectra were calculated and are displayed in Fig. 2. The entire Brillouin zone is devoid of negative frequency phonons. These findings demonstrate the dynamic stability of CrSSe, CrSTe, and CrSeTe monolayers and their potential to exist as free-standing 2D monolayers. It is crucial to look at the electrical characteristics of the Janus monolayer CrXY for any potential applications. In this work, we have taken 2 × 2 × 1 supercells of the Janus monolayers to study the electrical properties which contain 12 atoms (4 atoms of chromium, 4 chalcogen atoms S, Se and Te). To determine lattice constants we have performed geometrical optimization. The band structures and projected density of states (PDOS) of CrSSe, CrSTe, and CrSeTe monolayers are shown in Fig. 3. We have also calculated HSE band structure for all these Janus monolayers which is shown in Fig. S1 (ref. 58) in the ESI.† From the band structure, it can be clear that all these structures are metallic where the large electron density of states near the Fermi energy level signifies that there are many available channels. This contributes to the high electron conduction properties exhibited by 2D CrSSe, CrSTe and CrSeTe monolayers in their pure form representing good conductivity. From the spin-polarized calculation, we got both spin-up and spin-down bands crossing the Fermi level indicating metallic behavior for both spin parts (Fig. 3a–c). The PDOS shows that the states are primarily composed of the d orbital of the Cr atom and the p orbital of the X/Y (S, Se, or Te) atoms. From the PDOS we have clearly seen the significant hybridization between Cr 4d-orbital and p-orbitals of X/Y (S, Se, or Te) atoms.59 Finally, the structural stability and intrinsic metallic character of these monolayers are confirmed by our calculated results, indicating these deserve further investigation as anode materials.
 |
| | Fig. 1 (a)–(c) Top view of 4 × 4 × 1 supercells of the monolayers CrSSe, CrSTe, and CrSeTe with their lattice parameter and (d)–(f) are the side view of monolayers CrSSe, CrSTe, and CrSeTe with the respective bond lengths and vertical distance. | |
 |
| | Fig. 2 (a)–(c) Phonon band structure of CrSSe, CrSTe and CrSeTe monolayers. | |
 |
| | Fig. 3 (a)–(c) Band structure and (d)–(f) denotes PDOS of monolayers CrSSe, CrSTe, and CrSeTe respectively, black and red bands represent spin up and spin down contribution. | |
Adsorption properties of metal ions
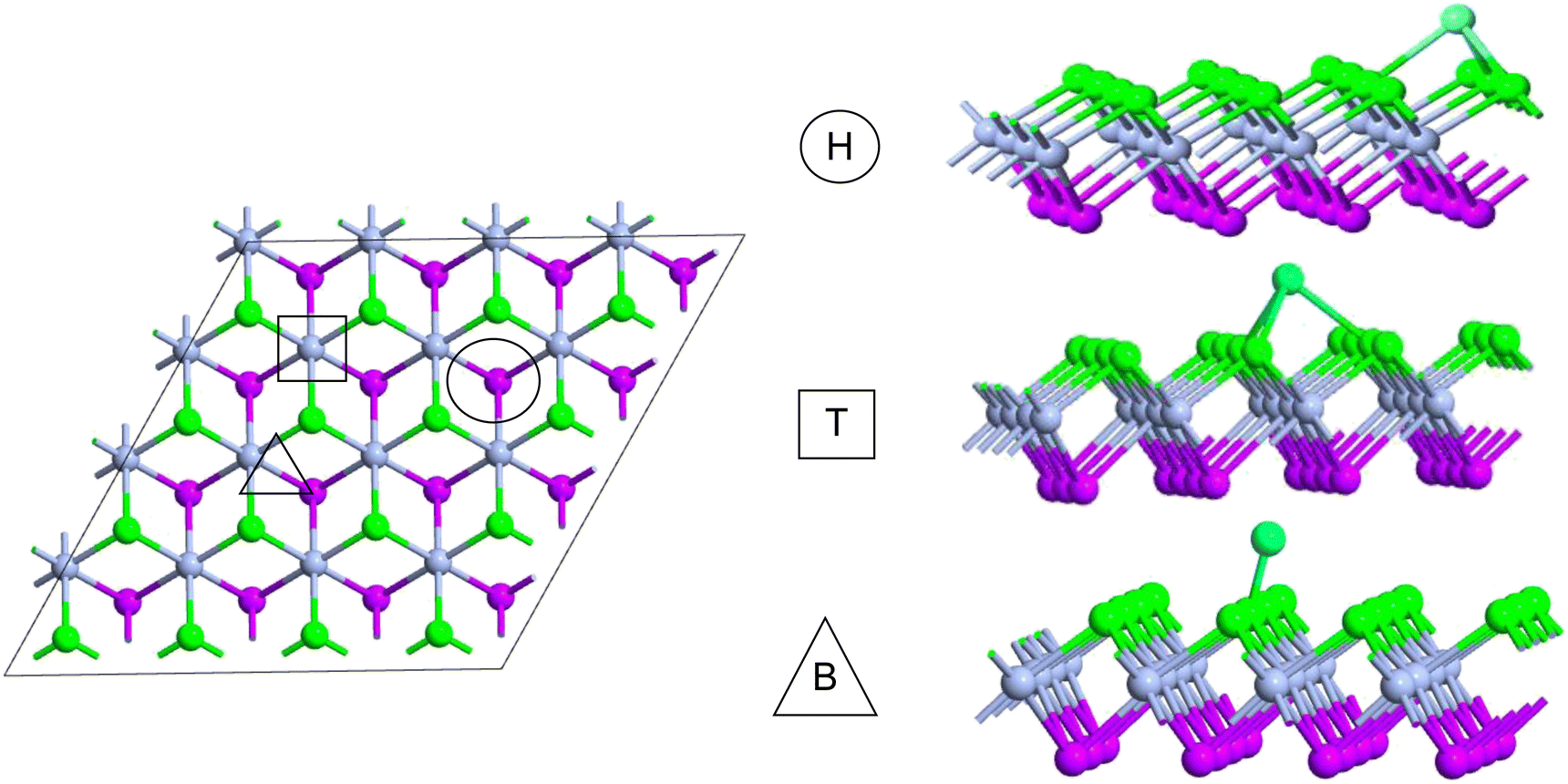
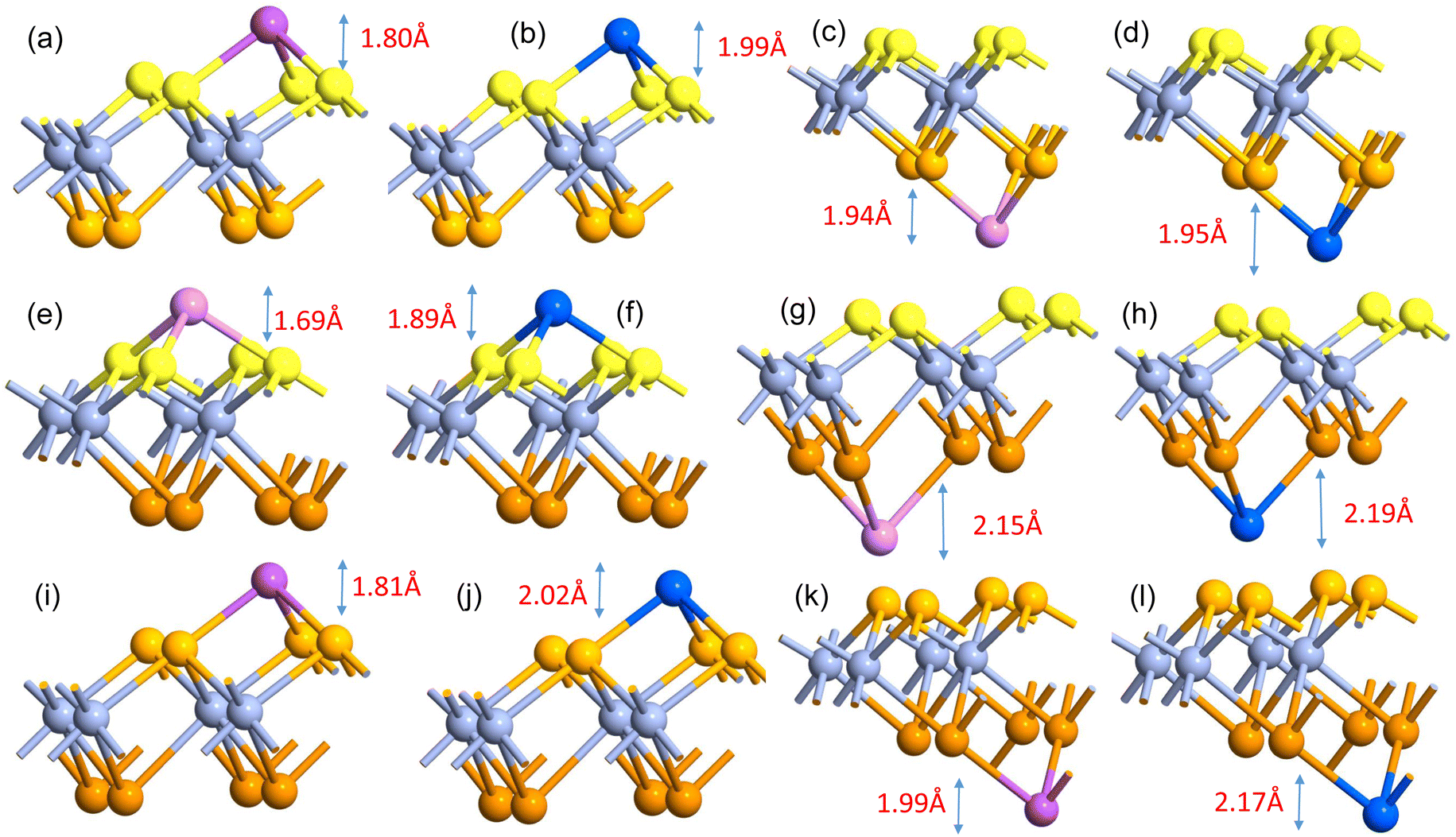
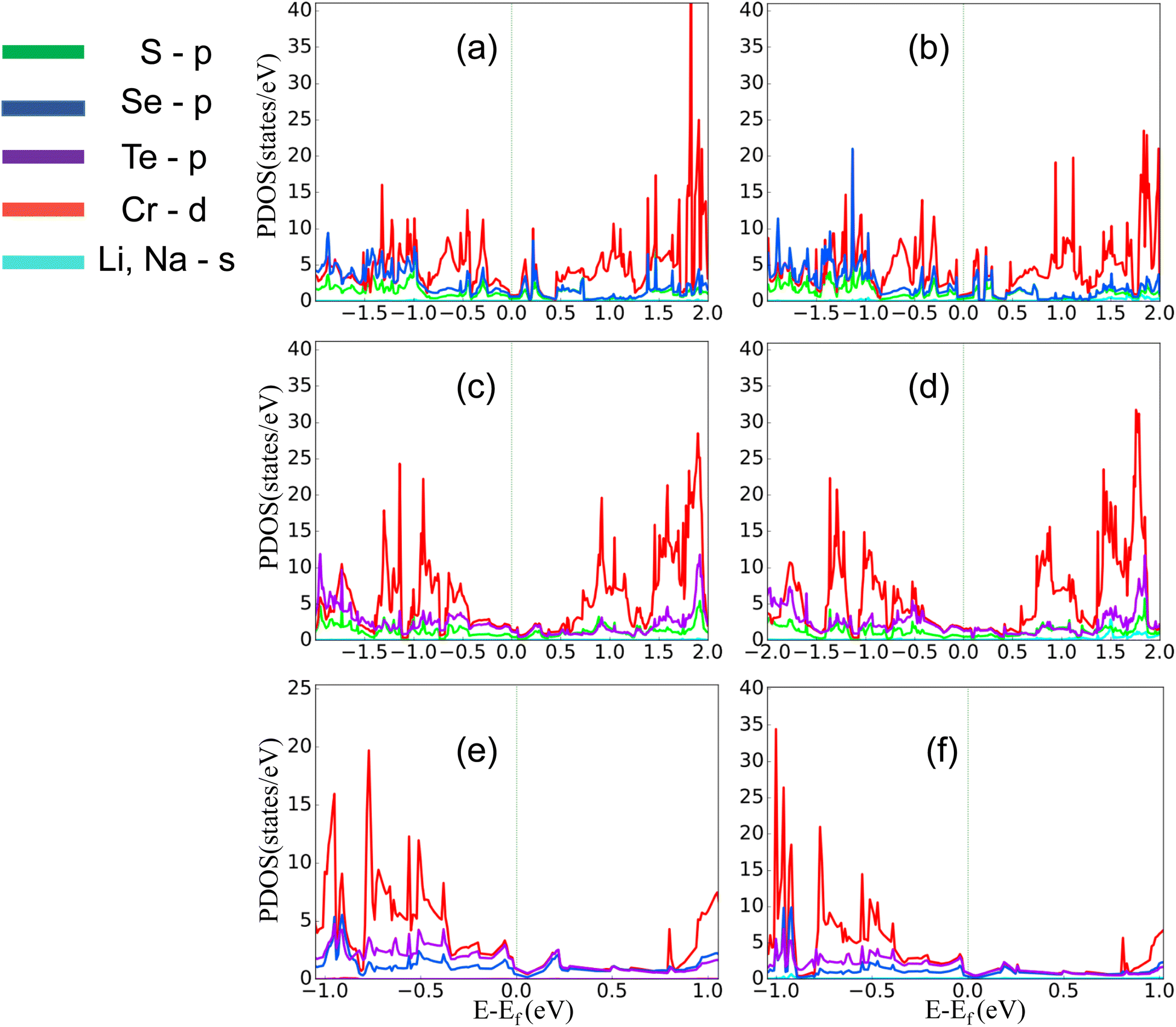
The comparatively high adsorption energy of the metal atom is a need as a potential contender for anode material in rechargeable lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs). We analyzed the adsorbed single metal atom on several potential sites. Considering the symmetry of the geometrical structure of monolayers, we have examined three different potential sites, namely the H site (three Cr–S/Se/Te bonds are created at the hollow location, where the metal is encased in the hollow of a six-membered ring), B site (the metal atom was positioned at the center of the Cr–S/Se/Te bond in the bridge site), T site (the metal is on top of the Cr atom in the top-TM site, and three Cr–S/Se/Te bonds have also been formed) as shown in Fig. 4. The adsorption energy of single alkali metal atoms (Li and Na) on H and T sites are mentioned in Table 1. The metal atoms at the T site and the H site have been seen to be completely optimized with no discernible movement but for metal atoms present on the B site moved to the T site after optimization indicating a meta-stable site of adsorption of metal atoms. For both H site and T site adsorption we have seen three bonds formed between the metal atom and surrounding S, Se, and Te atom (Fig. 4). The calculated adsorption energy is negative for site H and site T for both Li and Na atom adsorption indicating an exothermic reaction. From Table 1 we have concluded that the adsorption energy difference is small between H and T sites indicating both sites are preferable for adsorption. The alkali atoms show higher adsorption energy E(Top) at the top layers/side (S side for CrSSe and CrSTe and Se side for CrSeTe) than the adsorption at the bottom layer/side (Se side for CrSSe and Te side for CrSTe and CrSeTe monolayers respectively) E(Bot). To analyze the adsorption behavior of metal atoms with different functional we have calculated the adsorption energy with PBE and HSE functional as mentioned in Table S3.† This shows alkali atoms have higher interaction in the environment formed by the S atom followed by Se and Te atoms. In the case of CrSTe monolayers, the difference between the adsorption energy at top layer E(Top) and bottom layer E(Bot) is higher, both for Li and Na atom compared to CrSSe and CrSeTe. All the adsorption distances for Li and Na atoms are shown in Fig. 5. The higher adsorption distance is followed by lower adsorption energy due to lower interaction between monolayers and Li and Na atoms. The adsorption energies, and adsorption distance of Li and Na atoms for the Janus CrSSe, CrSTe, and CrSeTe monolayers are given in Table 2. From the projected density of states in Fig. 6, we concluded that a single metal atom contributes very little to the states in the states diagram, from the hybridization, which denotes the interaction of the metal atom and CrXY, where the d orbital of Cr atom and the s orbital of metal atom overlap. It is also clear that after metal atom adsorption the structure remains conducting. We also performed Barder charge analysis to confirm the charge transfer between the alkali atom and Janus monolayers at the H site, which is the most preferable adsorption site having the highest negative adsorption energy. Fig. 7 shows the isosurface plot of single Li and Na adsorption on CrSSe, CrSTe, and CrSeTe monolayer with the amount charge transfer (δQ) from Li and Na to Janus monolayer. We have taken an isosurface value of 0.002 eV Å−3. 0.57e fractional charge can be seen to be transferred to CrSSe monolayer followed by 0.38e and 0.31e to CrSTe and CrSeTe monolayer respectively by the Li atom where for Na 0.45e, 0.35e and 0.30e amount of charge is transferred to CrSSe, CrSTe and CrSeTe monolayer respectively. For a fully metal atoms adsorbed system, metal atoms contribute a very good amount to the density of states as cleared from Fig. 8.
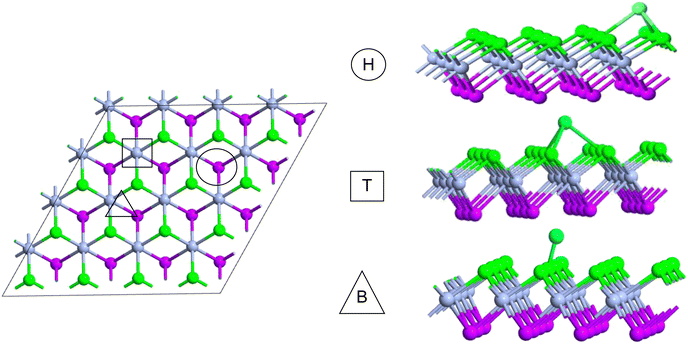 |
| | Fig. 4 Adsorption sites for metal atom (Li and Na) adsorption. | |
Table 1 Adsorption energy of metal atom at sites H and T
| Structure |
ELi–H (eV) |
ELi–T (eV) |
ENa–H (eV) |
ENa–T (eV) |
| CrSSe |
−1.86 |
−1.82 |
−0.77 |
−0.71 |
| CrSTe |
−1.66 |
−1.59 |
−1.2 |
−1.0 |
| CrSeTe |
−2.15 |
−2.10 |
−0.78 |
−0.71 |
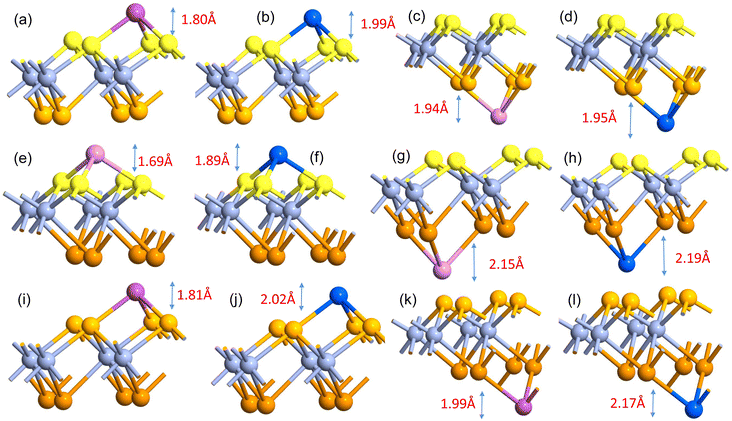 |
| | Fig. 5 (a) and (b) Schematic of adsorption of Li and Na on top side (S side) at H site and (c) and (d) adsorption of Li and Na on the bottom side at H site respectively on CrSSe monolayer; similarly (e), (f) and (i), (j) showing adsorption of Li and Na on top side (S or Se side) at H site on CrSTe and CrSeTe monolayers respectively; (g), (h) and (k), (l) showing adsorption of Li and Na on bottom side (Se or Te side) at H site on CrSTe and CrSeTe monolayers respectively. | |
Table 2 ELi-Top, ENa-Top = adsorption energy on top layer, ELi-Bot, ENa-Bot = adsorption energy on bottom layer, DLi-Top, DNa-Top = distance between metal atom and top layer, DLi-Bot, DNa-Bot = distance between metal atom and bottom layer, all adsorbed metal atoms are at H sites
| Structure |
ELi-Top (eV) |
ELi-Bot (eV) |
ENa-Top (eV) |
ENa-Bot (eV) |
DLi-Top (Å) |
DLi-Bot (Å) |
DNa-Top (Å) |
DNa-Bot (Å) |
| CrSSe |
−1.86 |
−1.81 |
−1.01 |
−0.77 |
1.80 |
1.94 |
1.99 |
1.95 |
| CrSTe |
−1.66 |
−0.71 |
−1.2 |
−0.8 |
1.69 |
2.15 |
1.89 |
2.19 |
| CrSeTe |
−2.15 |
−1.75 |
−1.56 |
−0.78 |
1.81 |
1.99 |
2.02 |
2.17 |
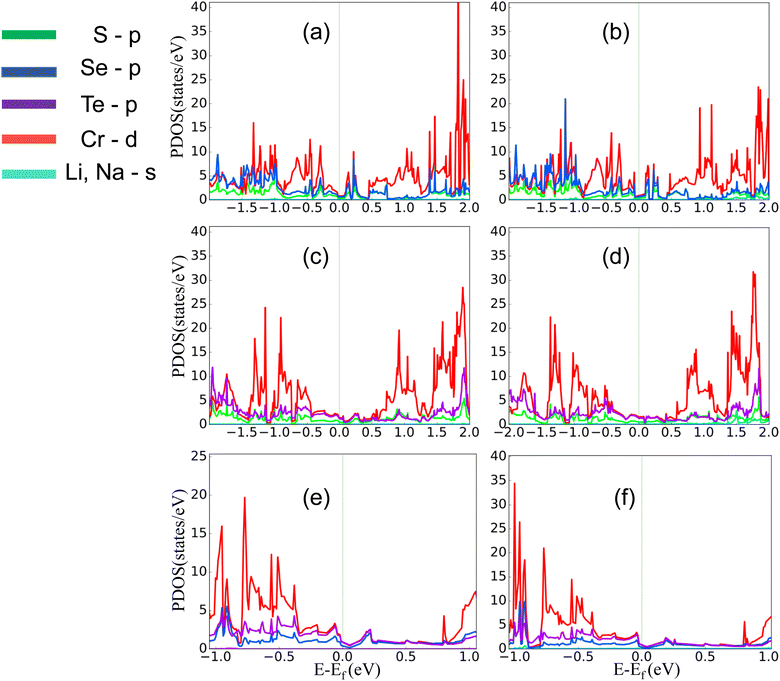 |
| | Fig. 6 (a) and (b) PDOS of single Li and Na atom adsorption on CrSSe monolayer on H site, similarly (c), (d) and (e), (f) represent PDOS of Li and Na atom of CrSTe, CrSeTe on H site respectively. | |
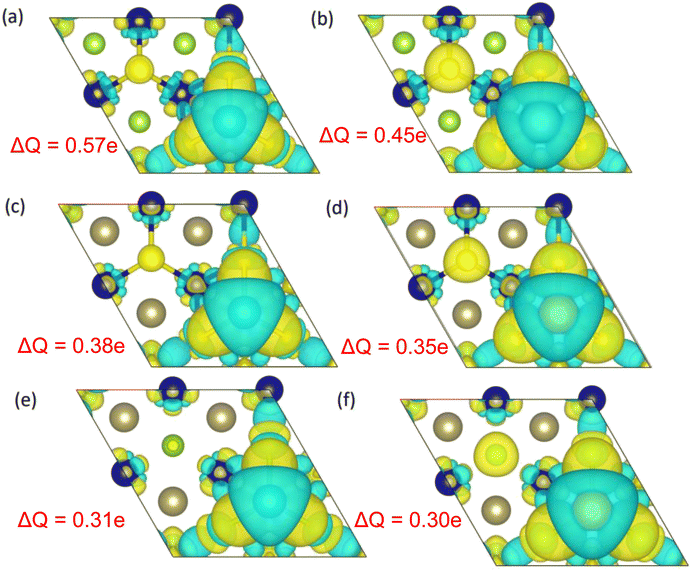 |
| | Fig. 7 (a) and (b) Represents the isosurface plot for Li and Na on the CrSSe monolayer at the H site with the charge transfer value (δQ) to the CrSSe monolayer on the top side, (c), (d) and (e), (f) are the isosurface plot for single Li and Na adsorbed structure at H site on CrSTe and CrSeTe monolayer with the charge transfer value of CrSTe and CrSeTe monolayers respectively. | |
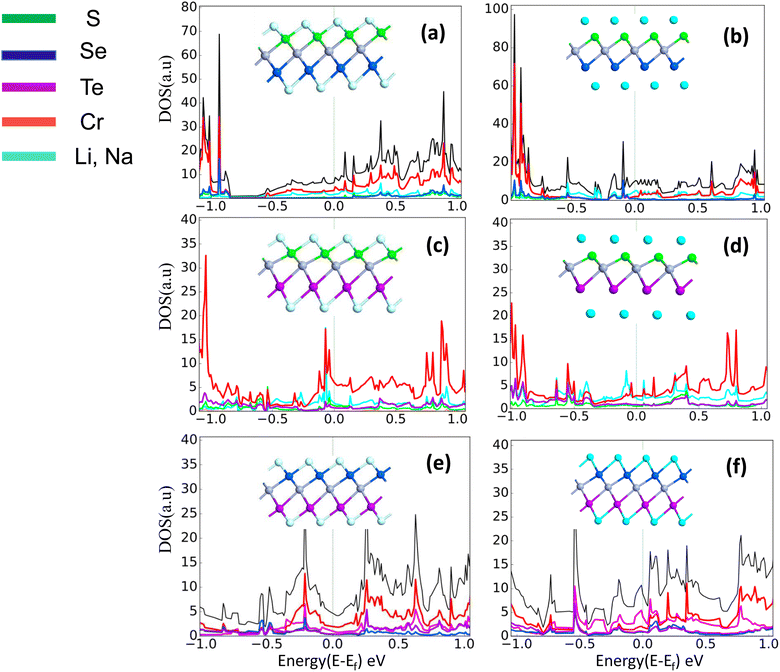 |
| | Fig. 8 (a) and (b) Show full adsorbed Li atom and Na atom structure with PDOS of CrSSe monolayers, (c), (d) and (e), (f) represents full adsorbed Li atom and Na atom system with PDOS of CrSTe and CrSeTe monolayers respectively. | |
The metal atom diffusion
By estimating diffusion paths and associated activation barriers, the kinetic properties of metal ions on Janus CrSSe, CrSeTe, and CrSTe monolayers were studied. Since the H site is most stable and has the lowest ground state energy, as was already indicated, we only take into account the diffusion between two neighboring H sites via the B site, which is a metastable site that serves as a diffusion channel shown in Fig. S4e.† The CI-NEB method measures the energy change when a metal atom moves between the relevant adsorption sites to assess the diffusion of a single Li and Na atom on monolayers. Even a small adjustment in barrier height has a huge impact on ion mobility. The Arrhenius equation60,61 is used to determine relative diffusion mobility. The following equation can be used to calculate the diffusion constant (D) of lithium and sodium ions.62| |
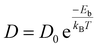 | (5) |
where T is the ambient temperature, Eb is the barrier energy, kB is the Boltzmann constant, and D0 is the temperature-independent pre-exponential constant and we took the D0 value same for all path.
The calculated lowest possible diffusion barrier for each monolayer for both Li and Na ions is shown in Fig. S3 and S4.† For the CrSSe monolayer, the lowest diffusion barrier shown by Li is 0.55 eV, and 0.35 eV for Na atom. However, Li and Na atoms faced a higher diffusion barrier on the top side (above the layers formed by the S atoms) than the bottom side (below the layers formed by Se or Te atoms) which can be confirmed from Fig. S3.† Similar trends also have been found in CrSTe and CrSeTe monolayers where the barrier faced above the space formed by the S atom is larger in CrSTe followed by space formed by the Se atom in CrSeTe monolayer (Fig. S4a–d†).58 The schematic of the diffusion path is shown in Fig. S4e.†
Theoretical specific capacity and voltage profile
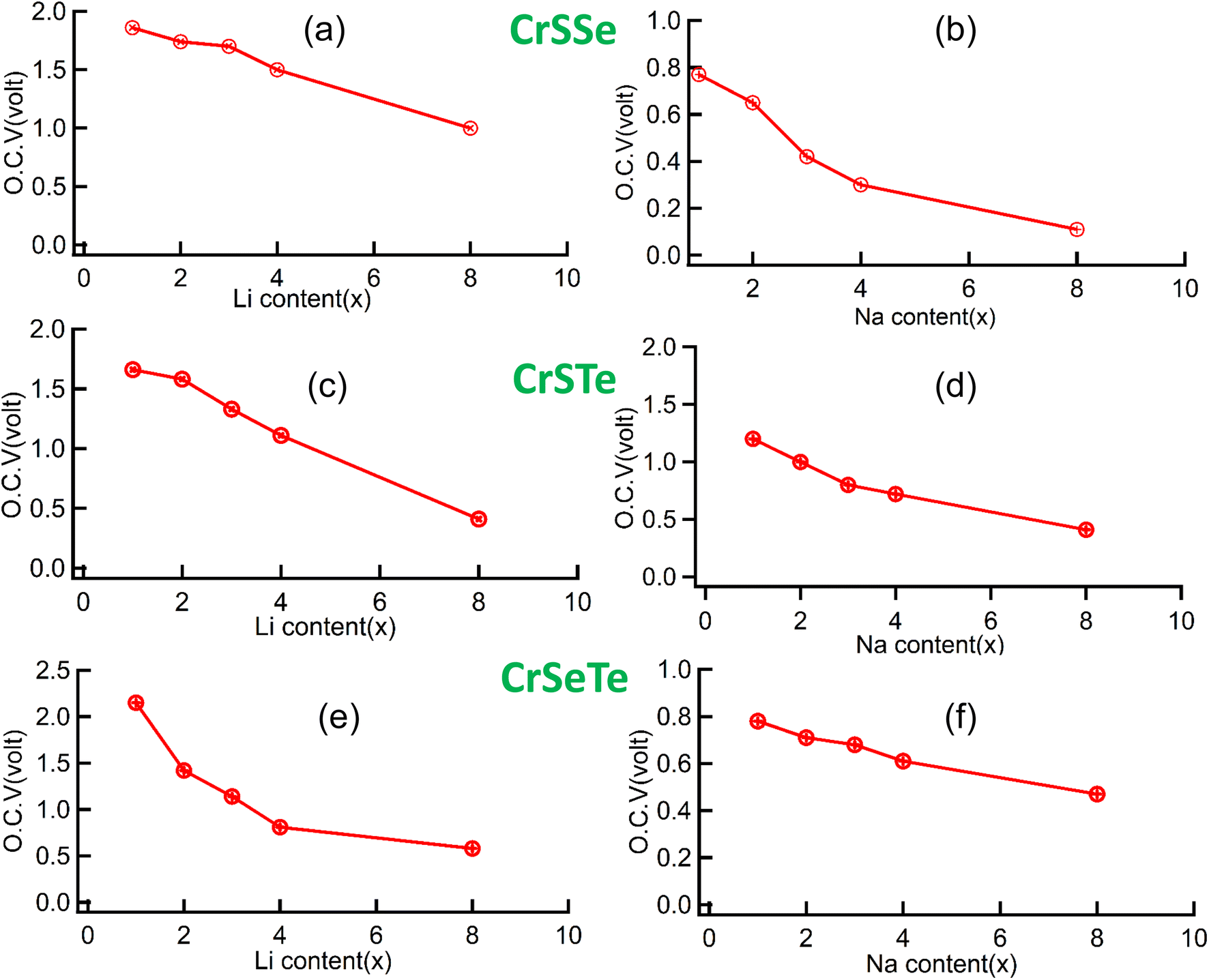
Theoretical specific capacity and open-circuit voltage (OCV) are two crucial measurements of a rechargeable battery's performance. By analyzing the properties while gradually increasing metal atoms, the two properties are evaluated. We used the 2 × 2 × 1 supercell of the monolayers as the substrate, which allowed us to make changes in the charging process to the adsorption of metals on both sides of the monolayer until it achieves its full capacity. In our study, each monolayer accepts 8 metal atoms giving rise to their respective highest specific capacity. The interaction between the adsorbed layers caused the absolute value of the average adsorption energy to steadily decrease as the amount of Li is increased.
By using eqn (2) we have calculated the average adsorption energy for each monolayer and then using the average adsorption energy we have calculated the open circuit voltage using eqn (3). The OCV for CrSSe is found to be 1.0 V for Li, 0.15 V for Na, similarly, the calculated OCV for CrSTe and CrSeTe are 0.53 V, 0.49 V and 0.6 V, and 0.51 V for Li and Na atom respectively as shown in Fig. 9. Li dendrite growth can be efficiently stopped by potentials between 0.1 V and 1.5 V, which also ensures the battery's comparatively high output voltage. So all our studied structures perform excellently as an anode material. We have calculated the specific capacity of each structure by using eqn (4). The calculated specific capacity are 348 mA h g−1, 254 mA h g−1, 208 mA h g−1 for Li atom and 260 mA h g−1, 198 mA h g−1, 177 mA h g−1 for Na atom for CrSSe, CrSeTe, and CrSTe monolayers respectively.
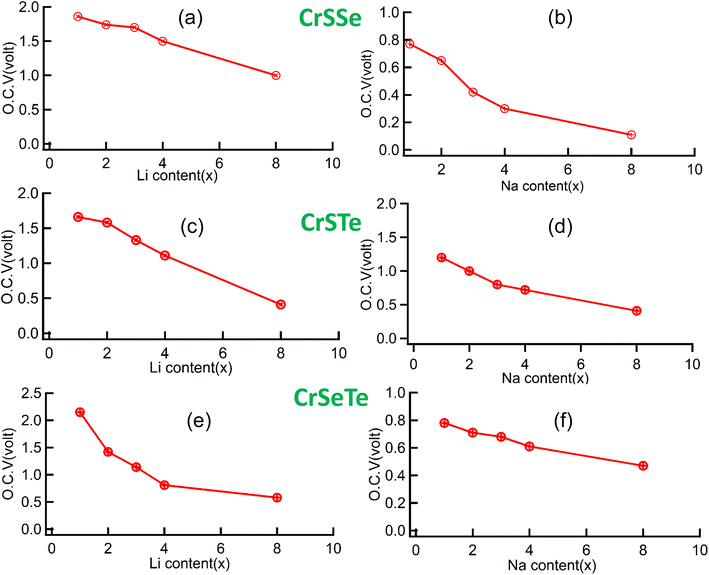 |
| | Fig. 9 (a) and (b) OCV vs. metal atom content of CrSSe, (c), (d) and (e), (f) represent OCV vs. metal atom content of CrSTe and CrSeTe monolayer respectively. | |
Conclusion
In the current study, the structural, electrical, and electrochemical properties of Janus CrSSe, CrSeTe, and CrSTe monolayers have been examined for prospective usage as anode material in lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs) using a first-principles based DFT technique. The dynamical stability of the structures is confirmed from phonon band structures. After metal atom adsorption, the material continues to be metallic with rising metal atom concentrations, ensuring strong electronic conductivity and serving as an anode material. A low diffusion barrier of 0.3 eV, 0.6 eV, and 0.1 eV for the Li atom and 0.54 eV, 0.32 eV, and 0.15 eV for Na atom were found in CrSSe, CrSeTe, and CrSTe monolayers respectively. The calculated low OCV of all the structures allows the efficient use as anode materials. Furthermore, the calculated highest theoretical specific storage capacity values correspond to the CrSSe monolayer, both for Li and Na atoms. These values are 348 mA h g−1 and 260 mA h g−1 respectively.
Conflicts of interest
There are no conflicts to declare.
References
- T. Bashir, S. A. Ismail, Y. Song, R. M. Irfan, S. Yang, S. Zhou, J. Zhao and L. Gao, A review of the energy storage aspects of chemical elements for lithium-ion based batteries, Energy Mater., 2021, 1, 100019 CrossRef CAS.
- S. Ray, Single molecular transistor as a superior gas sensor, J. Appl. Phys., 2015, 118, 034303 CrossRef.
- A.-I. Stan, M. Świerczyński, D.-I. Stroe, R. Teodorescu and S. J. Andreasen, Lithium ion battery chemistries from renewable energy storage to automotive and back-up power applications—an overview, in 2014 International Conference on Optimization of Electrical and Electronic Equipment (OPTIM), 2014, pp. 713–720 Search PubMed.
- S. Sahoo, P. Kumari and S. Jyoti Ray, Promising cathode material MnO2/CoO2 heterostructure for the Li and Na ion battery: a computational study, J. Appl. Phys., 2023, 134, 104302 CrossRef CAS.
- K.-S. Chen, I. Balla, N. S. Luu and M. C. Hersam, Emerging opportunities for two-dimensional materials in lithium-ion batteries, ACS Energy Lett., 2017, 2, 2026–2034 CrossRef CAS.
- L. Peng, Y. Zhu, D. Chen, R. S. Ruoff and G. Yu, Two-dimensional materials for beyond-lithium-ion batteries, Adv. Energy Mater., 2016, 6, 1600025 CrossRef.
- C. Ye, D. Chao, J. Shan, H. Li, K. Davey and S.-Z. Qiao, Unveiling the advances of 2D materials for Li/Na-S batteries experimentally and theoretically, Matter, 2020, 2, 323–344 CrossRef CAS.
- B. Li, H. Xu, Y. Ma and S. Yang, Harnessing the unique properties of 2D materials for advanced lithium–sulfur batteries, Nanoscale Horiz., 2019, 4, 77–98 RSC.
- S. Zhao, W. Kang and J. Xue, The potential application of phosphorene as an anode material in Li-ion batteries, J. Mater. Chem. A, 2014, 2, 19046–19052 RSC.
- M. M. K. Dad, R. P. Shahri and S. Ebrahimi, Ab-initio study of nanoporous phosphorene as anode material in rechargeable Li/Na ion batteries, Appl. Surf. Sci., 2021, 564, 150155 CrossRef.
- H. Benzidi, M. Lakhal, M. Garara, M. Abdellaoui, A. Benyoussef and O. Mounkachi, et al., Arsenene monolayer as an outstanding anode material for (Li/Na/Mg)-ion batteries: density functional theory, Phys. Chem. Chem. Phys., 2019, 21, 19951–19962 RSC.
- T. Zhao, H. Shu, Z. Shen, H. Hu, J. Wang and X. Chen, Electrochemical lithiation mechanism of two-dimensional transition-metal dichalcogenide anode materials: intercalation versus conversion reactions, J. Phys. Chem. C, 2019, 123, 2139–2146 CrossRef CAS.
- R. Sahoo, M. Singh and T. N. Rao, A review on the current progress and challenges of 2D layered transition metal dichalcogenides as Li/Na-ion battery anodes, ChemElectroChem, 2021, 8, 2358–2396 CrossRef CAS.
- E. Yang, H. Ji and Y. Jung, Two-dimensional transition metal dichalcogenide monolayers as promising sodium ion battery anodes, J. Phys. Chem. C, 2015, 119, 26374–26380 CrossRef CAS.
- X. Chen, Z. Kong, N. Li, X. Zhao and C. Sun, Proposing the prospects of Ti3CN transition metal carbides (MXenes) as anodes of Li-ion batteries: a DFT study, Phys. Chem. Chem. Phys., 2016, 18, 32937–32943 RSC.
- E. Yang, H. Ji, J. Kim, H. Kim and Y. Jung, Exploring the possibilities of two-dimensional transition metal carbides as anode materials for sodium batteries, Phys. Chem. Chem. Phys., 2015, 17, 5000–5005 RSC.
- X. Zhao, P. Wang, E. Lv, C. Wu, K. Ma, Z. Gao, I. D. Gates and W. Yang, Screening MXenes for novel anode material of lithium-ion batteries with high capacity and stability: a DFT calculation, Appl. Surf. Sci., 2021, 569, 151050 CrossRef CAS.
- R. de Alencar Rocha, W. F. da Cunha and L. A. Ribeiro, Electronic structure properties of transition metal dichalcogenide nanotubes: a DFT benchmark, J. Mol. Model., 2019, 25, 1–7 CrossRef PubMed.
- S. A. Yamusa, A. Shaari, N. A. Alsaif, I. M. Alsalamah, I. Isah and N. Rekik, Elucidating the structural, electronic, elastic, and optical properties of bulk and monolayer MoS2 transition-metal dichalcogenides: a DFT approach, ACS Omega, 2022, 7, 45719–45731 CrossRef CAS PubMed.
- S. Kar, P. Kumari, M. V. Kamalakar and S. Ray, Twist-assisted optoelectronic phase control in two-dimensional (2D) Janus heterostructures, Sci. Rep., 2023, 13, 13696 CrossRef CAS PubMed.
- S. Sachin, P. Kumari, N. Gupta, S. Rani, S. Kar and S. J. Ray, Van der Waals twistronics in a MoS2/WS2 heterostructure, Computational Condensed Matter, 2023, 35, e00797 CrossRef.
- S. Sachin, S. Rani, P. Kumari, S. Kar and S. J. Ray, Twist-engineered tunability in vertical MoS2/MoSe2 heterostructure, Appl. Phys. A: Mater. Sci. Process., 2023, 129, 46 CrossRef CAS.
- S. Tajik, Z. Dourandish, F. G. Nejad, H. Beitollahi, P. M. Jahani and A. Di Bartolomeo, Transition metal dichalcogenides: synthesis and use in the development of electrochemical sensors and biosensors, Biosens. Bioelectron., 2022, 114674 CrossRef CAS PubMed.
- A. Daus, S. Vaziri, V. Chen, Ç. Köroğlu, R. W. Grady, C. S. Bailey, H. R. Lee, K. Schauble, K. Brenner and E. Pop, High-performance flexible nanoscale transistors based on transition metal dichalcogenides, Nat. Electron., 2021, 4, 495–501 CrossRef CAS.
- H. Tributsch, Layer-Type Transition Metal Dichalcogenides—A New Class of Electrodes for Electrochemical Solar Cells, Berichte der Bunsengesellschaft für physikalische Chemie, 1977, 81, 361–369 CrossRef CAS.
- X. He, R. Wang, H. Yin, Y. Zhang, W. Chen and S. Huang, 1T-MoS2 monolayer as a promising anode material for (Li/Na/Mg)-ion batteries, Appl. Surf. Sci., 2022, 584, 152537 CrossRef CAS.
- M. Song, Q. Shi, D. Kan, S. Wei, F. Xu, W. Huo and K. Chen, The low symmetry 1T-MoS2 enabling the lithium directional diffusion through ferroelastic domain switching, Appl. Surf. Sci., 2023, 612, 155761 CrossRef CAS.
- H. Shu, F. Li, C. Hu, P. Liang, D. Cao and X. Chen, The capacity fading mechanism and improvement of cycling stability in MoS2-based anode materials for lithium-ion batteries, Nanoscale, 2016, 8, 2918–2926 RSC.
- R. Kappera, D. Voiry, S. E. Yalcin, W. Jen, M. Acerce, S. Torrel, B. Branch, S. Lei, W. Chen and S. Najmaei, et al., Metallic 1T phase source/drain electrodes for field effect transistors from chemical vapor deposited MoS2, APL Mater., 2014, 2, 092516 CrossRef.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Recent development of metallic (1T) phase of molybdenum disulfide for energy conversion and storage, Adv. Energy Mater., 2018, 8, 1703482 CrossRef.
- J. Strachan, A. F. Masters and T. Maschmeyer, Critical review: hydrothermal synthesis of 1T-MoS2–an important route to a promising material, J. Mater. Chem. A, 2021, 9, 9451–9461 RSC.
- M.-S. Chiou, P.-Y. Ho and H.-Y. Li, Adsorption of anionic dyes in acid solutions using chemically cross-linked chitosan beads, Dyes Pigm., 2004, 60, 69–84 CrossRef CAS.
- S.-Y. Yang, D.-R. Shi, T. Wang, X.-Y. Yue, L. Zheng, Q.-H. Zhang, L. Gu, X.-Q. Yang, Z. Shadike and H. Li, et al., High-rate cathode CrSSe based on anion reactions
for lithium-ion batteries, J. Mater. Chem. A, 2020, 8, 25739–25745 RSC.
- S. Kar, S. Rani and S. Ray, Stimuli assisted electronic, magnetic and optical phase control in CrOBr monolayer, Phys. E, 2022, 143, 115332 CrossRef CAS.
- A. Nair, S. Rani, M. V. Kamalakar and S. J. Ray, Bi-stimuli assisted engineering and control of magnetic phase in monolayer CrOCl, Phys. Chem. Chem. Phys., 2020, 22, 12806–12813 RSC.
- P. Kumari, T. Mukherjee, S. Kar and S. Ray, VClBr2: a new two-dimensional (2D) ferromagnetic semiconductor, J. Appl. Phys., 2023, 133, 183901 CrossRef CAS.
- S. Rani, A. Nair, M. V. Kamalakar and S. J. Ray, Spin-selective response tunability in two-dimensional nanomagnet, J. Phys.: Condens. Matter, 2020, 32, 415301 CrossRef CAS PubMed.
- P. Kumari, S. Rani, S. Kar, M. V. Kamalakar and S. Ray, Strain-controlled spin transport in a two-dimensional (2D) nanomagnet, Sci. Rep., 2023, 13, 16599 CrossRef CAS PubMed.
- M. Brandbyge, J.-L. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Density-functional method for nonequilibrium electron transport, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef.
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorff, J. Schneider, T. Gunst, B. Verstichel, D. Stradi, P. A. Khomyakov and U. G. Vej-Hansen, et al., QuantumATK: an integrated platform of electronic and atomic-scale modelling tools, J. Phys.: Condens. Matter, 2019, 32, 015901 CrossRef PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136, B864 CrossRef.
- J. Soler, E. Artacho, J. Gale, A. Garcıa, J. Junquera, P. Ordejón and D. Sánchez-Portal, Density-functional method for very large system with LCAO basis sets, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- D. Wang, Y. Gao, Y. Liu, D. Jin, Y. Gogotsi, X. Meng, F. Du, G. Chen and Y. Wei, First-principles calculations of Ti2N and Ti2NT2 (T = O, F, OH) monolayers as potential anode materials for lithium-ion batteries and beyond, J. Phys. Chem. C, 2017, 121, 13025–13034 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. U. Rahman, Strain Induces Ferromagnetism in a Janus Transition Metal Dichalcogenides: CrSTe-1H Monolayer, J. Electron. Mater., 2023, 52, 1036–1049 CrossRef CAS.
- J. L. Kaufman and A. Van der Ven, First-principles investigation of phase stability in layered NaxCrO2, Phys. Rev. Mater., 2022, 6, 115401 CrossRef CAS.
- O. I. Malyi, K. V. Sopiha and C. Persson, Energy, phonon, and dynamic stability criteria of two-dimensional materials, ACS Appl. Mater. Interfaces, 2019, 11, 24876–24884 CrossRef CAS PubMed.
- X. Lv, W. Wei, Q. Sun, B. Huang and Y. Dai, A first-principles study of NbSe2 monolayer as anode materials for rechargeable lithium-ion and sodium-ion batteries, J. Phys. D: Appl. Phys., 2017, 50, 235501 CrossRef.
- M. Wan, S. Zhao, Z. Zhang and N. Zhou, Two-Dimensional BeB2 and MgB2 as High Capacity Dirac Anodes for Li-Ion Batteries: A DFT Study, J. Phys. Chem. C, 2022, 126, 9642–9651 CrossRef CAS.
- B. Ge, B. Chen and L. Li, Ternary transition metal chalcogenides Ti2PX2 (X = S, Se, Te) anodes for high performance metal-ion batteries: a DFT study, Appl. Surf. Sci., 2021, 550, 149177 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- P. Panigrahi, S. B. Mishra, T. Hussain, B. Nanda and R. Ahuja, Density functional theory studies of Si2BN nanosheets as anode materials for magnesium-ion batteries, ACS Appl. Nano Mater., 2020, 3, 9055–9063 CrossRef CAS.
- X. Lv, F. Li, J. Gong, J. Gu, S. Lin and Z. Chen, Metallic FeSe monolayer as an anode material for Li and non-Li ion batteries: a DFT study, Phys. Chem. Chem. Phys., 2020, 22, 8902–8912 RSC.
- H. Ma, W. Zhao, S. Yuan, H. Ren, H. Zhu, H. Ma, F. Ding and W. Guo, Reconstructed edges of T phase transition metal dichalcogenides, Mater. Today Phys., 2021, 19, 100411 CrossRef CAS.
- S. Sahoo, P. Kumari and S. J. Ray, Supporting information of CrXY (X/Y = S, Se, Te) Monolayers as Efficient Anode Material for Li and Na-Ion Batteries: A First-Principles Study, 2024.
- S. Liu and Z. Liu, Hybridization induced metallic and magnetic edge states in noble transition-metal-dichalcogenides of PtX2 (X = S, Se) nanoribbons, Phys. Chem. Chem. Phys., 2018, 20, 21441–21446 RSC.
- Z. Wang, A. P. Ratvik, T. Grande and S. M. Selbach, Diffusion of alkali metals in the first stage graphite intercalation compounds by vdW-DFT calculations, RSC Adv., 2015, 5, 15985–15992 RSC.
- U. Germgård, The Arrhenius Equation is Still a Useful Tool in Chemical Engineering, Nord. Pulp Pap. Res. J., 2017, 32, 21–24 CrossRef.
- Q. Gao, X.-J. Ye and C.-S. Liu, Monolayer α-beryllene as an anode material for magnesium ion batteries with high capacity and low diffusion energy barrier, Phys. Chem. Chem. Phys., 2023, 25, 6519–6526 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Puja Kumari and
Soumya Jyoti Ray
,
Puja Kumari and
Soumya Jyoti Ray