
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening (co)polymerization of chiral seven-membered lactones mediated by achiral yttrium catalysts: insights into the catalyst stereocontrol by mass spectrometry†
Ali
Dhaini
a,
Jérôme
Ollivier
a,
Nicolas
Le Yondre
b,
Ali
Alaaeddine
c,
Sophie M.
Guillaume
 *a and
Jean-François
Carpentier
*a
*a and
Jean-François
Carpentier
*a
aUniv. Rennes, CNRS, Institut des Sciences Chimiques de Rennes, UMR 6226, F-35042 Rennes, France. E-mail: sophie.guillaume@univ-rennes.fr; jean-francois.carpentier@univ-rennes.fr
bUniv. Rennes, Centre Régional de Mesures Physiques de l'Ouest, UAR 2025 ScanMAT, F-35042 Rennes, France
cUniv. Libanaise, Campus Universitaire Rafic Hariri Hadath, Faculté des Sciences, Laboratoire de Chimie Médicinale et des Produits Naturels, Beirut, Lebanon
First published on 16th April 2024
Abstract
Ring-opening polymerization (ROP) of cyclic esters is a preferred approach for the preparation of various polyesters with controlled microstructures. In this work, the ROP of chiral seven-membered substituted-ε-caprolactones, namely 1-methyl-ε-caprolactone (CLMe) and 1-n-butyl-ε-caprolactone (CLnBu, aka ε-decalactone), was investigated to assess the potential stereoregularity of the resulting polylactones. The reactions mediated by yttrium complexes Y{ON(N)OR2} based on non-chiral diamino-bis(o,p-disubstituted-phenolate) ligands, associated with an exogeneous alcohol as a co-initiator, were effectively catalyzed – that is, with good control over molar mass values, narrow dispersity, and chain-end fidelity. However, the tacticity of the homopolymers obtained from racemic monomers rac-CLMe or rac-CLnBu could not be evidenced by NMR spectroscopy, as outlined in previous literature reports. Alternatively, the ring-opening copolymerization (ROCOP) of equimolar mixtures of (R)-CLnBu/(S)-CLMe enabled, indirectly, the assessment of the catalyst stereocontrol through the evaluation of the ultimate degree of alternation of the inserted units of each comonomer. While NMR spectroscopy again did not enable unambiguous evaluation of the copolymer topology/sequence, detailed MALDI-ToF and high-resolution ESI mass spectrometric analyses rewardingly revealed two major series of macromolecules, cyclic and linear ones. Both series of macromolecules showed randomly distributed units of both comonomers, thereby evidencing the absence of any significant stereocontrol from the yttrium catalyst over these large, seven-membered substituted ε-caprolactones. This latter lack of stereocontrol is assumed to arise from a too long range between adjacent chiral centers, preventing an effective chain-end stereocontrolled mechanism.
Introduction
The ring-opening polymerization (ROP) of cyclic esters is by now a well-established privileged synthetic method towards the synthesis of polyesters with controlled features and especially exhibiting tunable microstructures (in particular, tacticity).1 To this end, one of the most effective series of catalysts relies on rare earth metal complexes, such as those supported by achiral tripodal dianionic diamino- or amino-alkoxy-bisphenolate ligands {ON(X)OR2}2− (X = NMe2, OMe; R = o,p-substituents on phenolates).2 These achiral complexes, associated with an exogeneous alcohol as a co-initiator, have been extensively studied with a wide range of monomers, displaying high activities and high degrees of control (molar mass, dispersity, end-group fidelity, tacticity etc.).One of the ubiquitous illustrations of this chemistry is the stereoselective ROP of six-membered lactides.1,3 The ROP of racemic lactide (rac-LA) mediated by achiral yttrium Y{ON(N)OR2} complexes bearing sterically bulky substituents on the ancillary phenolate ligand (R = tBu, adamantyl, CMe2Ph, CMe2tBu, CPh3) returns highly heterotactic PLA (Pr up to 0.96‡) (Scheme 1). Also, the ROP of the four-membered β-butyrolactone (rac-BPLMe) mediated by the related Y{ON(X)OR2} yttrium catalysts proceeds with high activities and enables fine-tuning of the microstructure of the resulting poly(3-hydroxybutyrate)s (poly(BPLMe), aka PHB) from atactic to highly syndiotactic (Pr up to 0.96‡), depending on the nature of the ortho- (and to a much lesser extent the para-) R substituents on the tripodal ligand.4,5 Similarly, the ROP of four-membered 4-alkoxymethylene-substituted β-propiolactones, rac-BPLCH2OR (R = Me, All, Bn), offers syndiotactic poly(BPLCH2OR) (Pr up to 0.91‡) provided sterically crowded tert-butyl, cumyl or trityl substituents are installed on the yttrium complexes, or atactic poly(BPLCH2OR) with non-crowded complexes bearing methyl-substituted ligands. More uniquely, the corresponding achiral halogen-substituted yttrium catalysts Y{ON(X)OZ2} (Z = F, Cl, Br) give highly isotactic poly(BPLCH2OR)s (Pm up to 0.95‡), thanks to attractive non-covalent interactions (NCIs) between the ligand halogen and the acidic methylene hydrogens within the growing polymer chain (Scheme 1).6,7 Also, using a syndioselective catalyst, the ROP of equimolar mixtures of two different, enantiomerically pure 4-substituted-β-propiolactones, with opposite configurations, gives highly alternating polyhydroxyalkanoates (PHAs).8 For instance, the ROP of enantiopure allyl (S)-β-malolactonate and benzyl (R)-β-malolactonate (MLAR′; R′ = allyl, benzyl) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), using a Y{ON(X)OR2} yttrium catalyst bearing bulky R substituents, returns a nearly perfectly alternating copolymer, poly(MLAAllyl-alt-MLABenzyl) (Scheme 1).9 In another related approach to form microstructurally controlled PHAs, chiral salen-yttrium catalysts have been used by the group of Chen for the stereoselective ROP of alkyl-substituted eight-membered racemic cyclic diolides (rac-DLR).10 Hence, the ROP of rac-DLMe proceeds readily to offer perfectly isotactic poly(DLMe), that is, PHB (Pm up to 0.99‡) with a controlled and high molar mass (Mn up to 154000 g mol−1, ĐM = 1.01). Similarly, starting from unsymmetrical disubstituted eight-membered diolides rac-DLR,R′ (R ≠ R′) or by copolymerizing rac-DLMe with rac-DLR′ (R′ = Et, nBu), highly alternating isotactic poly(DLR,R′)s were produced (Pm up to 0.99‡). Access to syndiotactic PHAs (Pr up to 0.88‡) was also accomplished starting from meso-DLR (Scheme 1).10
:1), using a Y{ON(X)OR2} yttrium catalyst bearing bulky R substituents, returns a nearly perfectly alternating copolymer, poly(MLAAllyl-alt-MLABenzyl) (Scheme 1).9 In another related approach to form microstructurally controlled PHAs, chiral salen-yttrium catalysts have been used by the group of Chen for the stereoselective ROP of alkyl-substituted eight-membered racemic cyclic diolides (rac-DLR).10 Hence, the ROP of rac-DLMe proceeds readily to offer perfectly isotactic poly(DLMe), that is, PHB (Pm up to 0.99‡) with a controlled and high molar mass (Mn up to 154000 g mol−1, ĐM = 1.01). Similarly, starting from unsymmetrical disubstituted eight-membered diolides rac-DLR,R′ (R ≠ R′) or by copolymerizing rac-DLMe with rac-DLR′ (R′ = Et, nBu), highly alternating isotactic poly(DLR,R′)s were produced (Pm up to 0.99‡). Access to syndiotactic PHAs (Pr up to 0.88‡) was also accomplished starting from meso-DLR (Scheme 1).10
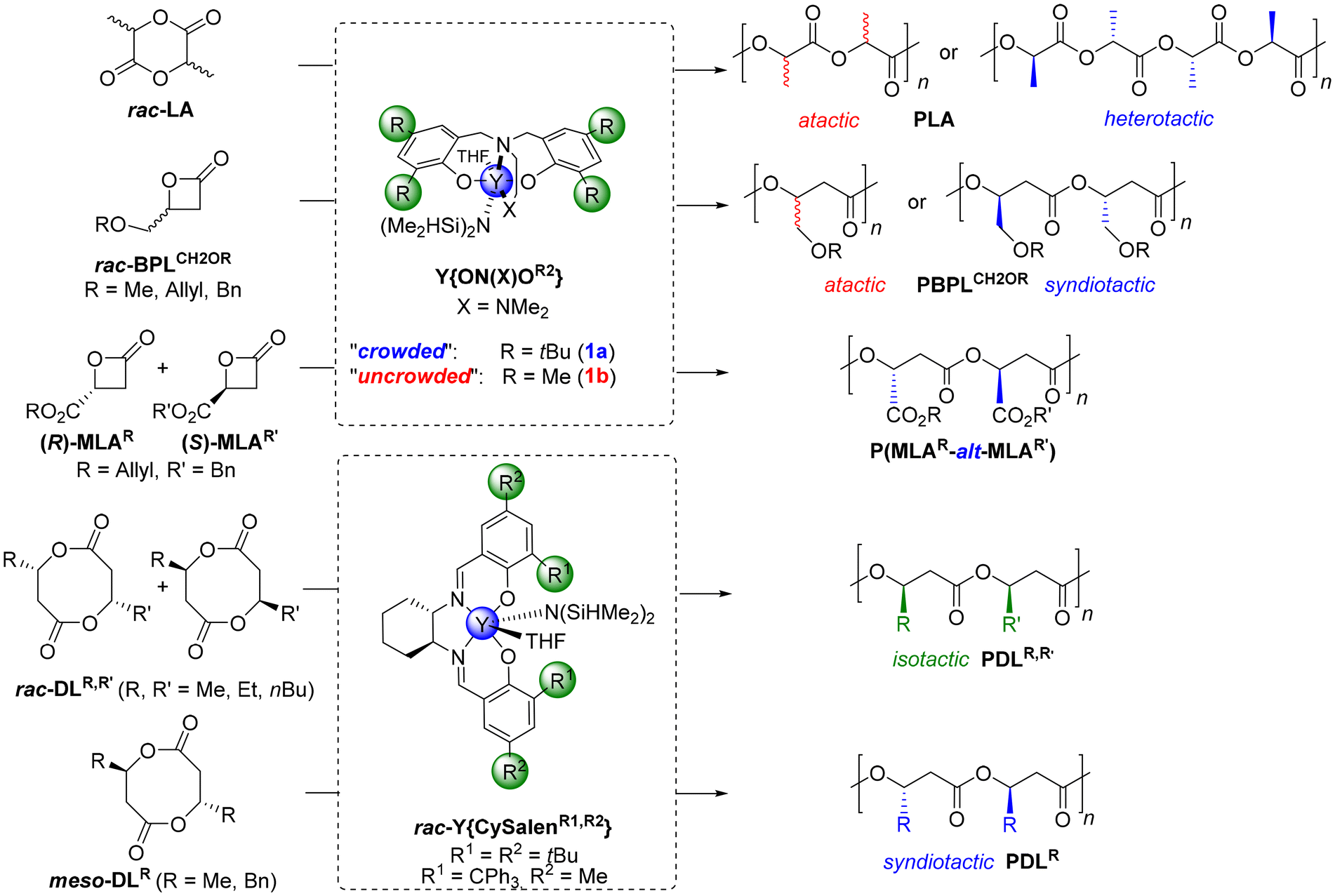 | ||
| Scheme 1 Examples of yttrium-mediated stereoselective ROP/ROCOP of rac-lactide, rac-4-substituted-β-propiolactones, β-malolactonates, or rac-diolides.3,6,9,10 | ||
On the other hand, poly(ε-caprolactone) (PCL) is another quite important polyester,11 first synthesized by Carothers’ group in the 1930s from the parent seven-membered lactone (CL).12 PCL is semi-crystalline (up to 70% depending on the weight-average molar mass, Mw) with a low melting point (59–60 °C); it is hydrophobic with good solubility, features exceptional blending compatibility, and shows good degradation abilities by microorganisms13 – a set of distinctive properties making it extensively valuable in different application fields. The ROP of CL and its alkyl-substituted derivatives such as 1-methyl-ε-caprolactone (CLMe), mediated either by metal-based or organo-catalysts, has been the method of choice to target PCL-type materials and related copolymers, with minimum backbiting and obtaining very high molar mass PCL (Mn up to 730000 g mol−1) with a narrow dispersity (Đ ∼ 1.1) (Scheme 2).14 For instance, γ-methyl-ε-caprolactone has been polymerized using an ω-hydroxy-poly(ethylene oxide) macroinitiator combined with triethylaluminium,15 or copolymerized with CL using tin(II) bis(2-ethylhexanoate).16 Recently, the ROP of a series of spirocyclic acetal-functionalized ε-caprolactone monomers with zinc- or yttrium-based catalysts has been shown to open the route towards readily depolymerizable (i.e., chemically recyclable) materials.17 The ring-opening copolymerization (ROCOP) of β-butyrolactone (BPLMe) and 1-n-butyl-ε-caprolactone (that is ε-decalactone, hereafter referred to as CLnBu) mediated by yttrium-salan-type complexes, forms di- and triblock copolymers (Mn up to 31600 g mol−1, Đ = 1.15–1.35).18 Also, CLMe has been homopolymerized using chiral phosphoric acids (CPAs), in kinetic resolution polymerizations, affording stereogradient PCLMe with a high chain-end fidelity and controlled molar mass (Mn up to 5000 g mol−1, Đ = 1.11–1.22).19 The microstructure of the synthesized PCLMe was determined using chiral HPLC to assess the enantiomeric excess (ee) values of the unreacted CLMe monomer at various conversion levels, revealing that (S)-CPA consumes (S)-CLMe faster while (R)-CPA polymerizes (R)-CLMe preferentially. Yet, despite the progress made in polymerizing substituted ε-caprolactones, determination of the microstructure of the resulting materials has remained a challenge. In fact, the tacticity of these polymers could not be successfully assessed from their 1H and 13C NMR spectra. It was suggested to arise from the chiral centers along the polymer backbone that are positioned too far apart to allow a clear differentiation by NMR analysis, and from an insufficient resolution of resonances even at 125 MHz.18,19
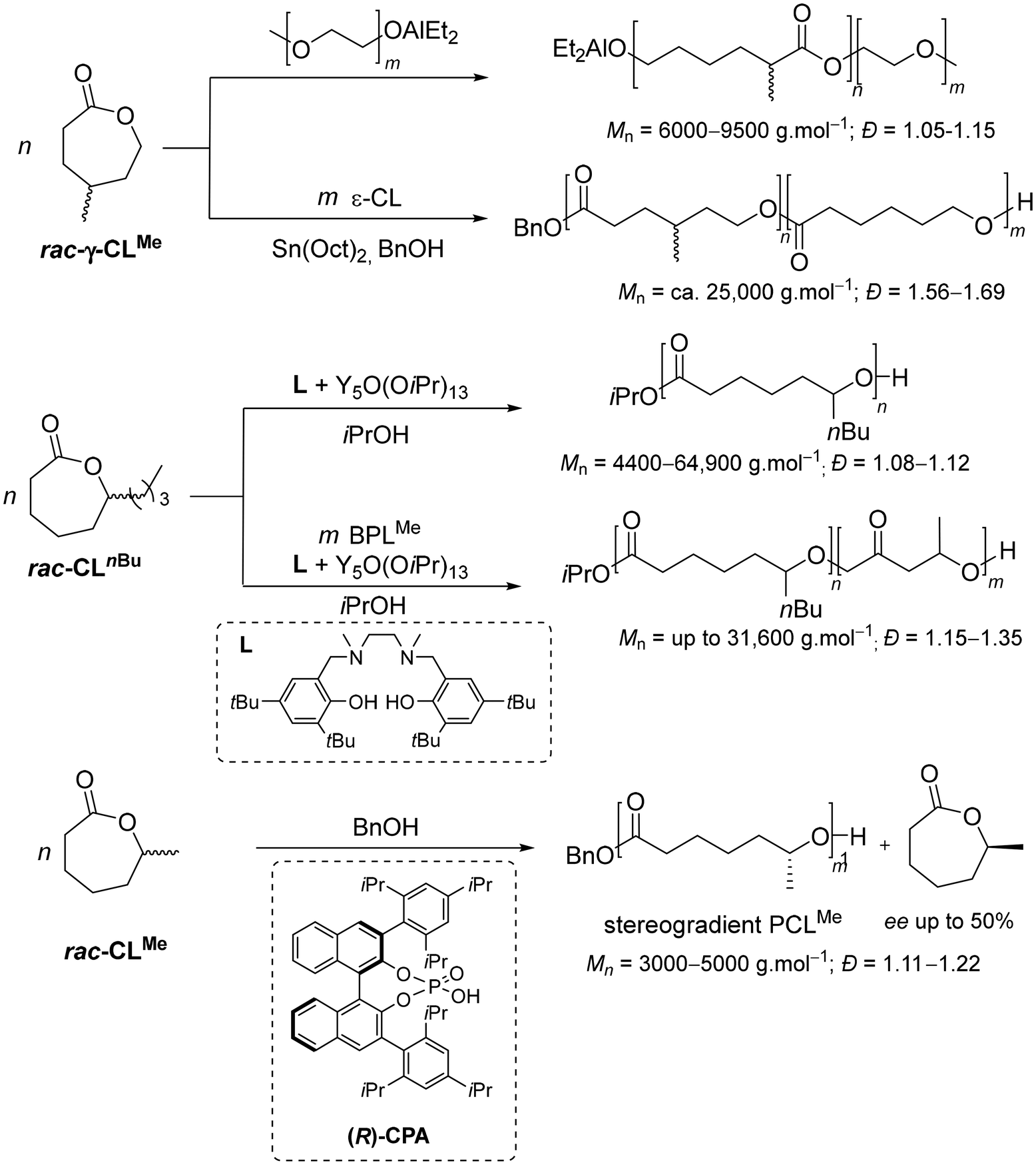 | ||
| Scheme 2 Examples of RO(CO)P of substituted-ε-caprolactones.15,16,18,19 | ||
In our study, we aimed at investigating the stereoselective ability of achiral yttrium catalysts of the type Y{ON(N)OR2} in the RO(CO)P of the seven-membered 1-substituted-ε-caprolactones, namely CLMe and CLnBu (Scheme 3). Conventionally, the tacticity of polyesters is determined through NMR spectroscopy, enabling us to infer the extent, and sometimes also to decipher the mechanism, of stereocontrol. However, as mentioned earlier and further documented in the present work, since NMR spectroscopy proved ineffective in assessing the tacticity in the PCLR homopolymers (R = Me, nBu) resulting from these large lactones, we implemented an alternative approach. This involved copolymerizing equimolar mixtures of the two enantiomerically pure, opposite-configuration 1-substituted-ε-caprolactones (S)-CLMe and (R)-CLnBu, to investigate whether or not there is alternation of the monomer units in the resulting copolymers. Such alternation would be a hallmark of syndiotactic control (vide infra).8,9 For this purpose, because NMR spectroscopy eventually turned out to be uninformative to assess the degree of alternation/randomness in the synthesized poly[(S)-CLMe/(R)-CLnBu] copolymers, extensive high resolution mass spectrometry (MS) analyses were conducted. With the aid of programmatic script, these MS studies enabled us to conclude on the stereoselective abilities of Y{ON(N)OR2} in the ROP of such 1-substituted-ε-caprolactones.
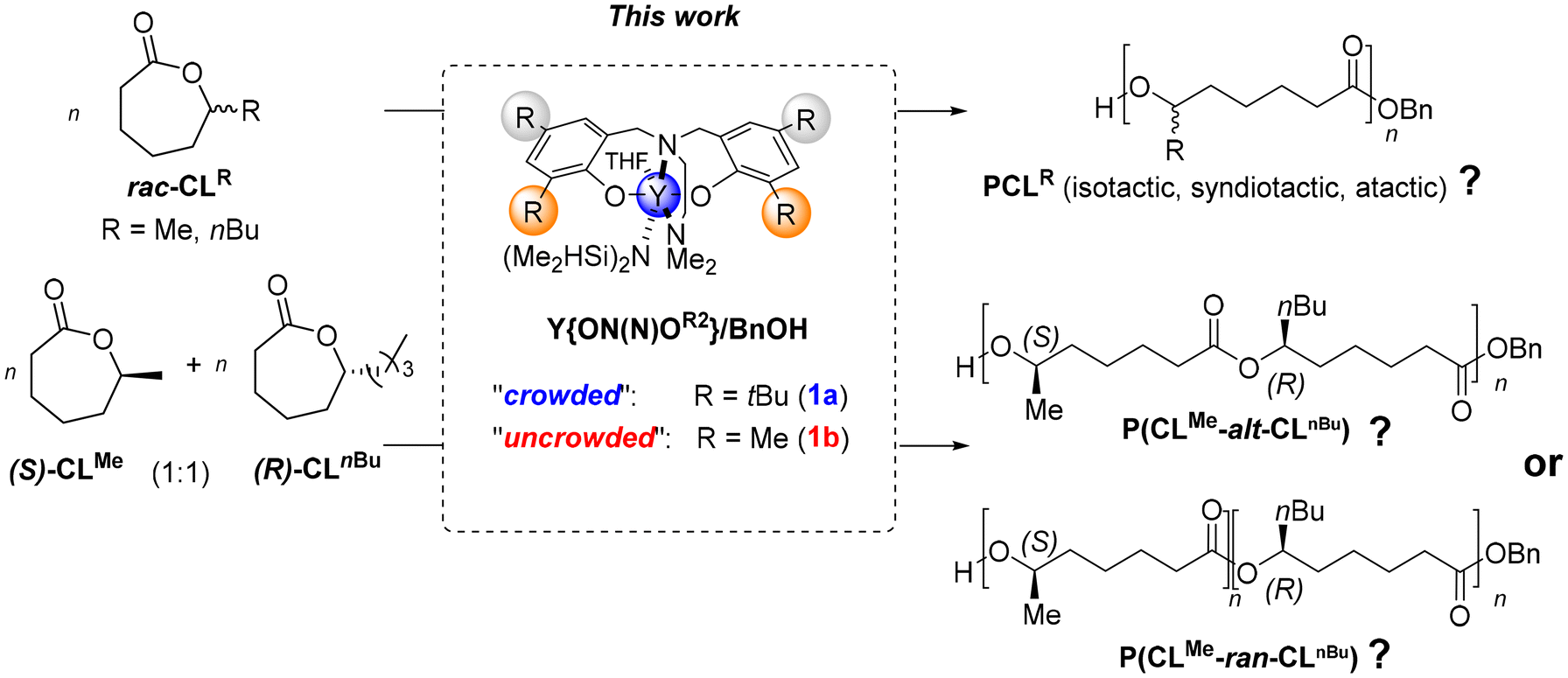 | ||
| Scheme 3 ROP of 1-substituted-ε-caprolactones (rac-CLR; R = Me, nBu) and ROCOP of equimolar mixtures of opposite-configuration, enantiopure monomers studied in the present work to assess the stereoselectivity ability of discrete yttrium complexes towards such seven-membered lactones. | ||
Experimental section
Materials and methods
All manipulations involving organometallic catalysts were performed under an inert atmosphere (argon, <5 ppm O2 and H2O) using standard Schlenk, vacuum line, and glovebox techniques. Toluene was freshly distilled from Na/benzophenone under argon and degassed thoroughly by freeze–thaw–vacuum cycles prior to use. Benzyl alcohol (BnOH) was distilled over Mg turnings under an argon atmosphere and kept over activated 3–4 Å molecular sieves. Proligands {ON(N)OtBu2}H2 and {ON(N)OMe2}H2 and precursor Y[N(SiHMe2)2]3(THF)2, used to prepare in situ yttrium amido complexes 1a–b, respectively, were synthesized according to the reported procedures.4b,20 Racemic ε-decalactone (rac-CLnBu) was purchased from Sigma Aldrich and dried over CaH2.Instrumentation and measurements
1H (500 and 400 MHz) and 13C{1H} (125 and 100 MHz) NMR spectra were recorded on Bruker Avance Ascend 400 spectrometers at 25 °C. 1H and 13C{1H} NMR spectra were referenced internally relative to SiMe4 (δ = 0 ppm) using the residual solvent resonances. Coupling constants are reported in Hz.The number-average molar mass (Mn,SEC) and dispersity (ĐM = Mw/Mn) values of the PCLR (R = Me, nBu) homopolymer and copolymer samples were determined by size-exclusion chromatography (SEC) in THF at 30 °C (flow rate = 0.8 mL min−1) on a Polymer Laboratories PL50 apparatus equipped with a refractometric detector and a UV detector at 254 nm, and a set of two ResiPore PLgel 3 μm MIXED-D 300 × 7.5 mm columns. The (co)polymer samples were dissolved in THF (5 mg mL−1). All elution curves were calibrated with polystyrene standards; the Mn,SEC values of the PCLR samples were uncorrected for the possible difference in the hydrodynamic radius vs. that of polystyrene. Representative SEC traces are given in the ESI as Fig. S26 and S27.†
The molar mass of PCLR samples was also determined by 1H NMR analysis in CDCl3 from the relative intensities of the signals of the PCLR repeating unit methine hydrogen (δ 4.84–4.90 ppm), –OCH((CH2)4CH(R)) and the benzyloxy chain-end (δ 5.09–5.11 ppm, –OCH2Ph). The accuracy of the Mn,NMR values thus determined is evaluated to be ±200 g mol−1. Monomer conversions were calculated from the 1H NMR spectra of the crude polymer samples in CDCl3 by using the integration (Int.) ratios [Int.RPCL/(Int.RPCL + Int.RCL)] of the methine hydrogens of PCLR (vide supra) and of the monomers (δ 4.32 ppm, CLMe; δ 4.17 ppm, CLnBu).
Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-ToF) high resolution (error <25 ppm) mass spectra were recorded using an ULTRAFLEX III TOF/TOF spectrometer (Bruker Daltonik Gmbh, Bremen, Germany) in positive ionization mode at CRMPO, ScanMat, Université de Rennes. Spectra were recorded using reflectron mode and an accelerating voltage of 25 kV. A mixture of a freshly prepared solution of the polymer in CH2Cl2 (HPLC grade, 10 mg mL−1) and DCTB (trans-2-(3-(4-tert-butylphenyl)-2methyl-2-propenylidene))-malononitrile, and an acetonitrile solution of the cationizing agent (CF3CO2Na or NaI, 10 mg mL−1) were prepared. The solutions were combined in a 1:1:1 v/v/v ratio of matrix-to-sample-to-cationizing agent. The resulting solution (0.25–0.5 μL) was deposited onto the sample target (entry 1: Prespotted AnchorChip PAC II 384/96 HCCA; entry 3: MTP 384 ground steel) and air- or vacuum-dried.
Electrospray Ionization (ESI) high resolution (error <3 ppm) mass spectra were recorded using an Orbitrap Q-Exactive spectrometer (Thermo Fisher Scientific, Waltham (MA), USA) in positive ionization mode at CRMPO, ScanMat, Université de Rennes. The sample was dissolved in MeOH before being analyzed by direct injection.
Differential scanning calorimetry (DSC) analyses were performed on a DSC 2500 TA instrument calibrated with indium using aluminum capsules (40 μL). The thermograms were recorded under a continuous flow of helium (25 mL min−1) according to the following cycles: −80 to +200 °C at 10 °C min−1; +200 to −80 °C at 10 °C min−1; −80 °C for 5 min; −80 to +200 °C at 10 °C min−1; +200 to −80 °C at 10 °C min−1. Representative DSC traces of homo- and copolymers are reported in the ESI as Fig. S28–S36.†
Program for automatic assignment of m/z peaks in HRMS spectra
A program was developed to process the high-resolution mass spectroscopy data of P(CLMe-co-CLnBu) copolymers, in order to establish the correspondence between the experimentally measured and theoretical m/z values, based on the measurement precision. The program supports as input Excel csv files representing the list of peaks (m/z and intensity), and aims to create: (a) as an in silico or real output, the table of calculated m/z values with the corresponding AxBy chemical formula (where A and B refer to the two comonomers), population type, and x and y values, and (b) as an output file, the table of measured m/z values and intensity of the MS peaks. The table of theoretical values (mono-isotopic exact masses) is generated from the different raw formulae (with different possible end-groups or as cyclic species) using the in-house software Polymers (see Fig. S15 in the ESI†), by considering some relevant MS parameters (ionization mode, polarity, type of adducts and charge state). The table of measured m/z values compiles the list of the peaks of the spectrum extracted using the Vendor software for each MS spectrum. The algorithm next determines the correspondence between the measured and the theoretical m/z values according to a measurement precision criterion expressed in ppm, as configured in the script; the measurement precision criterion can be set at 3 ppm, for the analysis of ESI HR-MS data, and 30 ppm for MALDI-ToF HR-MS data. When several matches are possible, only the closest one is kept. A table of results is automatically generated allowing the correspondence between the theoretical and measured m/z values to be recorded with their relative intensity and measurement precision. As output, the software also takes into account the theoretical isotope contributions and delivers a summation of intensities for the measured m/z values respecting the measurement precision criterion. Thus, one can interpret the spectrum according to the different compositions identified within each population considered and their corrected relative intensity.Separation of the enantiomers of rac-CLMe and rac-CLnBu by preparative chiral HPLC analyses
This was performed at Institut des Sciences Moléculaires de Marseille, Plateforme de chromatographie chirale (UMR 7313 CNRS-University of Aix-Marseille, France), using Chiralpak AD-H (250 × 10 mm) columns, an Agilent 1260 Infinity unit (pump G1311B, autosampler G1329B, DAD G1315D), with Igloo-Cil ovens, monitored by SRA Instruments Seleccol software (Version 1.2.3.0), Agilent OpenLAB CDS Chemstation LC and CE Drivers (A.02.08SP1) and Agilent OpenLAB Intelligent reporting (A.01.06.111). Chiroptical detection was performed using a Jasco OR-1590 polarimetric detector. n-Heptane and iPrOH, HPLC grade, were degassed and filtered on a 0.45 μm millipore membrane before use. Chiral HPLC traces for racemic and enantiopure CLMe and CLnBu are reported in the ESI as Fig. S3 and S4.†Determination of reactivity ratios following the Fineman–Ross method
Following reported procedures,21 the monomer reactivity ratios during the synthesis of the copolymers, r1 and r2, were calculated using the Fineman–Ross equation:| F/(f(f − 1)) = r1(F2/f) − r2 | (1) |
Synthesis of racemic 1-methyl-ε-caprolactone (rac-CLMe)
The synthesis of rac-CLMe was performed by the Baeyer–Villiger oxidation of 2-methylcyclohexanone, following a reported protocol10c that provides 1-methyl-ε-caprolactone as a major compound (95%; along 5% of 5-methyl-ε-caprolactone): 2-methylcyclohexanone (9.0 g, 79.8 mmol) was dissolved in CH2Cl2 (100 mL); m-CPBA (27.5 g, 159.7 mmol) was added and the solution was stirred in the dark for 48 h at 0 °C. A white suspension formed, which was filtered, and the filtrate was washed with a saturated aqueous NaHCO3 solution (3 × 100 mL) followed by a 5wt% Na2S2O3 solution (3 × 100 mL). The organic phase was dried over Na2SO4 and evaporated. The crude product was purified by chromatography using basic alumina and hexanes as eluents to give rac-CLMe as a colorless liquid (6.4 g, 62%). rac-CLMe was further dried over CaH2, vacuum-distilled (94 °C, 5 mmHg) and stored under argon. 1H NMR (400 MHz, CDCl3, 25 °C) (Fig. S1†): δ 1.20 (d, 3JH–H = 6.7, 3H, CH3CH), 1.43–1.53 (m, 3H, CHCH2CH2CH2), 1.72–1.82 (m, 3H, CHCH2CH2CH2), 2.44–2.51 (m, 2H, CH2C(O)O), 4.26–4.37 (m, 1H, CHCH3). 13C{1H} NMR (100 MHz, CDCl3, 25 °C) (Fig. S2†): δ 22.2 (CH3CH), 30.9 (CHCH2CH2CH2), 33.3 (CHCH2CH2CH2), 35.4 (CHCH2CH2CH2), 37.4 (CHCH2CH2CH2), 68.2 (CH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 176.1 (CO).
O), 176.1 (CO).
Typical procedure for the ROP of rac-CLR
In a typical experiment, in the glovebox, a Schlenk flask was charged with Y[N(SiHMe2)2]3(THF)2 (8.8 mg, 14 μmol) and {ON(N)OtBu2}H2 (7.4 mg, 14 μmol), and toluene (0.25 mL) was next added. To this solution, BnOH (107 μL of a 1% v/v solution in toluene, 1 equiv. vs. Y) was added under stirring at room temperature. After 5 min, a solution of rac-CLMe (150 mg, 0.84 mmol, 60 equiv. vs. Y) in toluene (0.5 mL) was added rapidly and the mixture was stirred at 20 °C for 1 h. The reaction was quenched by the addition of acetic acid (ca. 0.5 mL of a 1.6 mol L−1 solution in toluene). The mixture was concentrated to dryness under vacuum and the conversion was determined by the 1H NMR analysis of the residue in CDCl3. The crude polymer was then dissolved in CH2Cl2 (ca. 1 mL) and precipitated in cold pentane (ca. 5 mL), filtered and dried in a vacuum oven at 60 °C. The homopolymers were recovered as pale yellowish oils.Typical procedure for the ROCOP of (S)-CLMe with (R)-CLnBu
In a typical experiment, in the glovebox, a Schlenk flask was charged with Y[N(SiHMe2)2]3(THF)2 (3.7 mg, 5.8 μmol) and {ON(N)OtBu2}H2 (1a, 3.0 mg, 5.8 μmol), and toluene (0.25 mL) was next added. To this solution, BnOH (60 μL of a 1% v/v solution in toluene, 1 equiv. vs. Y) was added under stirring at room temperature. After 5 min, a solution of (S)-CLMe (>98% ee, 37.8 mg, 0.29 mmol, 50 equiv. vs. Y) and (R)-CLnBu (>98% ee, 50.0 mg, 0.29 mmol, 50 equiv. vs. Y) in toluene (0.25 mL) was added rapidly and the mixture was stirred at 20 °C for 3 h. The reaction was quenched by the addition of acetic acid (ca. 0.5 mL of a 1.6 mol L−1 solution in toluene). The mixture was concentrated to dryness under vacuum and the conversion was determined by the 1H NMR analysis of the residue in CDCl3. The crude polymer was then dissolved in CH2Cl2 (ca. 1 mL) and precipitated in cold pentane (ca. 5 mL), filtered and dried. P(CLMe-co-CLnBu) copolymers were recovered as pale yellowish oils. 1H NMR (400 MHz, CDCl3, 25 °C) (signals were labelled from a–a′ to h, as shown in Fig. 2) (see also Fig. S9 in the ESI†): δ 7.34 (broad m, 5H, C6H5CH2O, h), 5.11 (s, 2H, C6H5CH2O, g), 4.86 (2 overlapping m, 3JH–H = 6.4, 2H, CHCH3, e + e′), 2.28 (m, 4H, COCH2, a + a′), 1.61 (m, 6H, COCH2CH2CH2CH2 and OCOCH2CH2CH2CH2, d + d′), 1.50 (m, 4H, COCH2CH2 and OCOCH2CH2, b + b′), 1.30 (m, 8H, CH2CH2CHCH2CH2CH2CH3 and OCOCH2CH2CH2, c + c′), 1.19 (d, 3JH–H = 6.8, 3H, CH3, f′), 0.89 (t, 3JH–H = 7.1, 3H, CH3, f). 13C{1H} NMR (100 MHz, CDCl3, 25 °C) (signals were labelled from #1 to #12, as shown in Fig. 3) (see also Fig. S10 in the ESI†): δ 173.4 (CO, #3), 173.2 (CO, #3′), 129.2, 128.4, 124.9, 123.5 (4s, C6H5CH2O, #1), 74.0 (C(O)OCHCH2, #8), 70.7 (OCHCH2, #8′), 66.1 (C6H5CH2O, #2), 35.7 (COCH2, #4 and #4′), 34.6 (CH2CH2CHOC(O), #7, and CHCH2(CH2)CH3, #9), 33.9 (OCH(CH3)CH2, #7′), 27.6 (CHCH2CH2CH2CH3, #10), 25.1 (several signals, C(O)CH2CH2CH2CH2CH, #5, #6, and OC(O)CH2CH2CH2CH2CH, #5′, #6′), 21.7 (s, CH3CH2CH2, #11), 20.1 (CH3CH, #9′), 14.1 (CH3CH2CH2, #12).Results and discussion
Homopolymerization of rac-CLR
First, the homopolymerization of both rac-CLMe and rac-CLnBu was studied using the yttrium catalytic system Y{ON(N)OtBu2} (1a)/BnOH (Scheme 3), selected as a benchmark system considering its typical high productivity, activity and syndiotactic abilities toward a number of racemic 4- and 6-membered lactones (vide supra).2–7,9 The ROP reactions proceeded readily in toluene at room temperature to form the corresponding PCLMe and PCLnBu, respectively (Table S1†). The structures of the homopolymers were confirmed by 1H and 13C NMR spectroscopy (see the ESI, Fig. S3–S6†), indicating linear macromolecules capped with benzyloxycarbonyl and hydroxy end-groups. However, a close examination of the different signals in the 13C NMR spectra did not allow establishing the polymer tacticity as anticipated (Fig. 1). As mentioned earlier, this is in line with previous literature reports, which stated that no differentiated meso/racemo diad sequences are observable in the 13C NMR spectra of PCLRs recorded at a 100–125 MHz field.18,19 To further confirm this observation, two quite different benchmark ROP catalytic systems, namely {BDIDIPP}Zn(N(SiMe3)2)/BnOH (hereafter referred to as Zn{BDI}/BnOH)22 and potassium tert-butoxide, were used for comparison purposes (Table S1,† entries 2 and 3). Also, isotactic homopolymers (Pm > 0.98) were prepared starting from the enantiopure (R)-CLMe and (R)-CLnBu monomers, respectively (preliminarily obtained by preparative chiral HPLC; see the Experimental section), using the {BDIDIPP}Zn(N(SiMe3)2)/BnOH system (Table S1,† entry 4). This eventually ended up with a set of PCLMe and PCLnBu homopolymers, with obviously different tacticities, whose 13C NMR spectra yet were virtually identical and indistinguishable from those of polymers produced by Y{ON(N)OtBu2} (1a). This absence of differentiated, observable meso/racemo diad sequences in the 13C NMR spectra of PCLR-type polymers is likely due to the long-range separation from one chiral group to the next one, despite the short (two bonds) distance between the chiral groups and the carbonyl groups. Note that all polymers featured essentially the same thermal signature (as determined by DSC; see the ESI, Fig. S28–S36†).23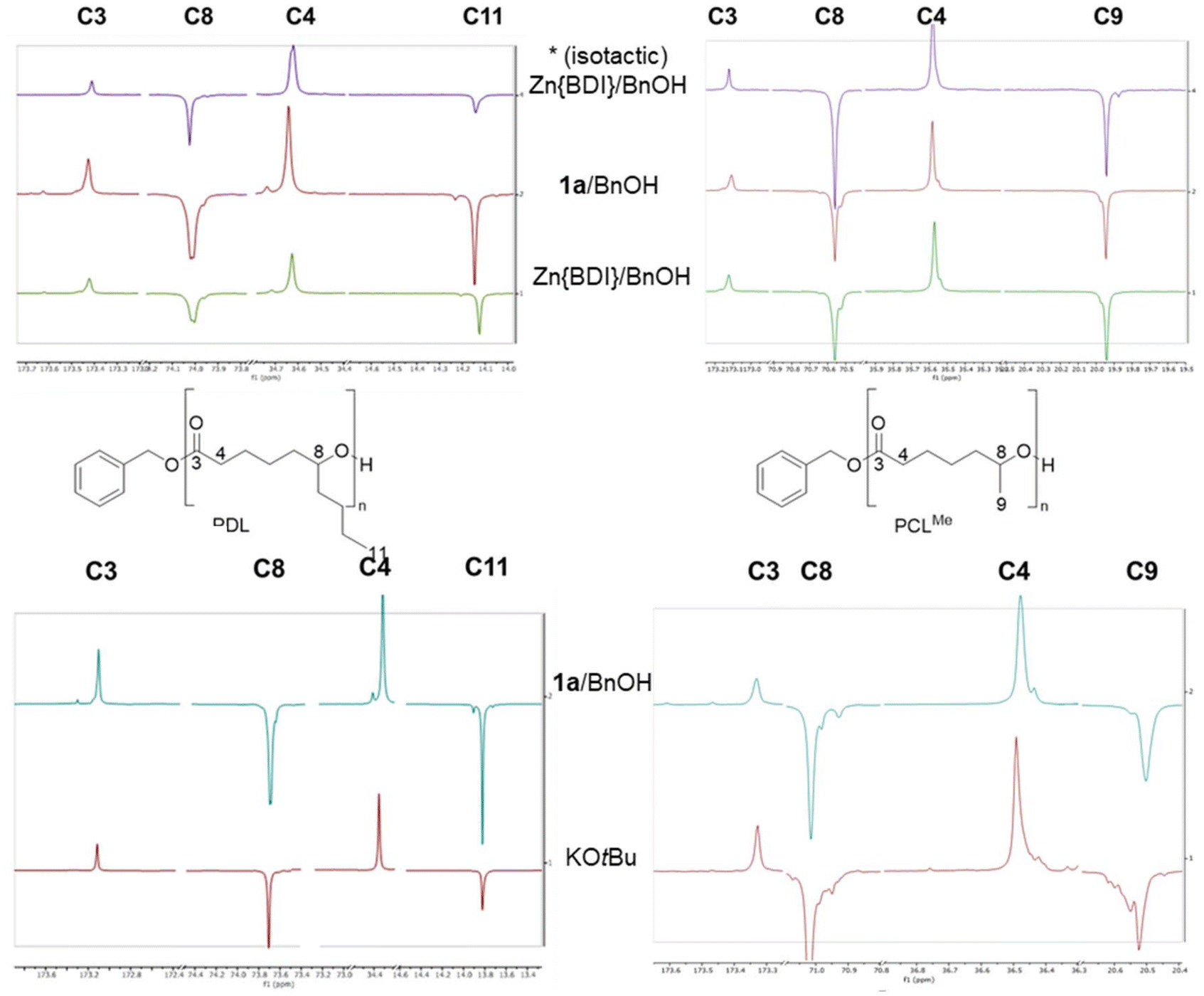 | ||
| Fig. 1 Zoomed regions of the 13C{1H} NMR spectra (100 MHz, CDCl3 (top) and (CD3)2CO (bottom), 25 °C) of PCLnBu (left) and PCLMe (right) homopolymers prepared by the ROP of rac-CLMe and rac-CLnBu, respectively, except for the spectra labeled (*), prepared from enantiopure (S)-CLMe and (R)-CLnBu, respectively, mediated by the Zn{BDIDIPP}/BnOH, Y{ON(N)OtBu2} (1a)/BnOH and KOtBu catalytic systems (Table S1,† entries 1–4). The low intensity signals observed are assigned to the terminal repeating units in these low molar mass polymers. | ||
Copolymerization of (S)-CLMe with (R)-CLnBu
An alternative way to assess the stereoselective ability of achiral Y{ON(X)OR2} catalysts towards racemic 1-substituted ε-caprolactones is through the investigation of the ROCOP of 1:1 mixtures of two chemically different, enantiopure CLR monomers with opposite configurations. Indeed, with such comonomer feed, (highly) syndioselective catalysts shall return (highly) alternating copolymers (as previously demonstrated with various chiral β-lactones and Y{ON(X)OR2}-type catalysts);8,9 on the other hand, (highly) isoselective catalysts shall provide (highly) blocky copolymers, while non-stereoselective catalysts shall give random copolymers. This is, of course, assuming that both monomers have essentially the same reactivity/polymerizability, that is, the reaction is under stereocontrol rather than being governed by kinetic considerations. This is anticipated in the present case because of the limited stereoelectronic difference in the R substituents (i.e., Me vs. nBu) of the two CLR monomers.24 Finally, it is expected that examining the comonomer sequence (alternation/randomness degree) in the resulting polycaprolactone copolymers would be easier than assessing tacticity in the corresponding homopolymers.
Hence, the ROCOP of 1:1 mixtures of (S)-CLMe and (R)-CLnBu was conducted with relatively low-to-moderate targeted molar mass (Mn = ca. 3000 g mol−1, DP = 20 up to Mn = ca. 11000 g mol−1, DP = 100), using either the 1a or 1b catalyst. The results are gathered in Table 1. The 1H and 13C NMR spectra of the resulting copolymers (Fig. 2 and 3, respectively; see also the ESI, Fig. S9 and S10,† and the Experimental section) featured the characteristic resonances for the repeating units. Taking into account the benzyloxy end-group introduced in the initiation step, the molar masses of the copolymers were calculated from the 1H NMR spectra (see the Experimental section for calculation details). The Mn,NMR values thus obtained were in good agreement with the Mn,theo values calculated from the monomer-to-initiator ratio and monomer conversion (Table 1). The Mn,SEC values determined in chloroform vs. polystyrene standards (values uncorrected for possible differences in hydrodynamic radii) followed almost the same trend. The dispersity of the copolymers obtained was rather narrow, with ĐM values ranging from 1.07 to 1.31. As typical of this chemistry,2–6,8 the longer the reaction time, the greater the extent of transesterification reactions (reshuffling and back-biting), resulting in broadening of the dispersity (compare entries 1 and 2 vs. 3) (note that reaction times were not necessarily optimized in this study); this transesterification was further corroborated by mass spectrometric studies (vide infra). The reactions mediated by 1b proceeded more slowly than those with 1a (compare entries 4 and 5 vs. 7). This is a behavior previously noted in the ROP of different lactones2 and accounted for by the likely aggregation of the sterically uncrowded 1b in contrast to the mononuclear 1a which is protected by ortho,para-tert-butyl substituents on the phenolate rings. Also, as mentioned earlier and anticipated considering the small increase in the substituent bulkiness, CLMe appears to be slightly more reactive than CLnBu (see entries 4, 5 and 7; for 1H NMR monitoring see Fig. S11 in the ESI†).
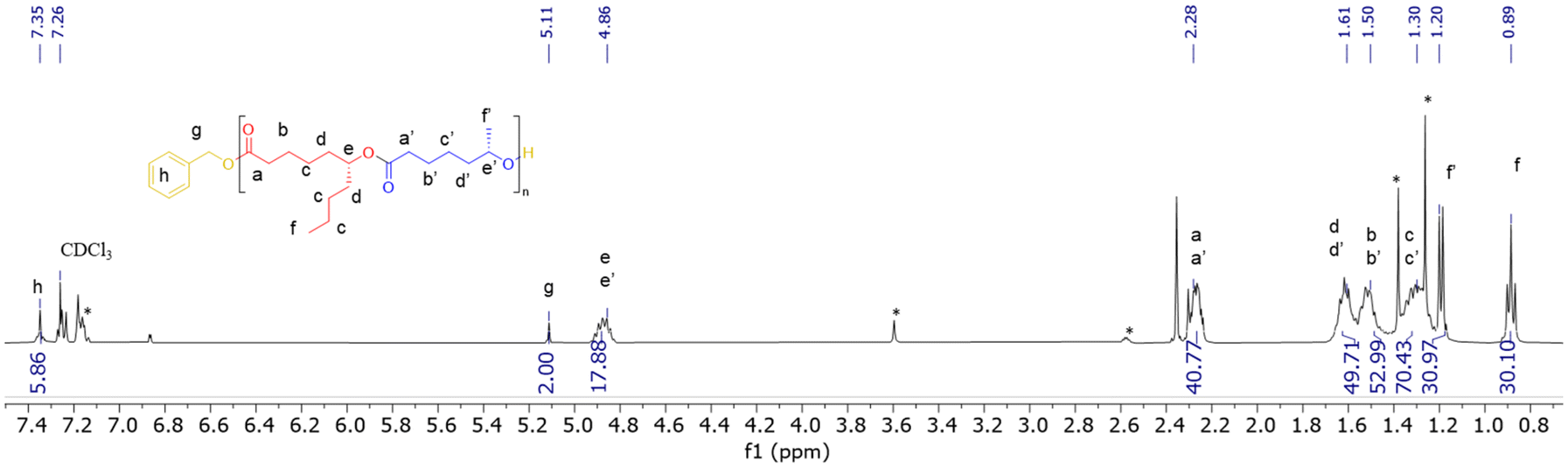 | ||
| Fig. 2 Representative 1H NMR spectrum (400 MHz, CDCl3, 25 °C) of a P(CLMe-co-CLnBu) copolymer prepared from the ROCOP of a 1:1 mixture of (S)-CLMe and (R)-CLnBu mediated by the 1a/BnOH (1:1) catalytic system (Table 1, entry 3). * stands for resonances of the residual solvent and/or catalyst. | ||
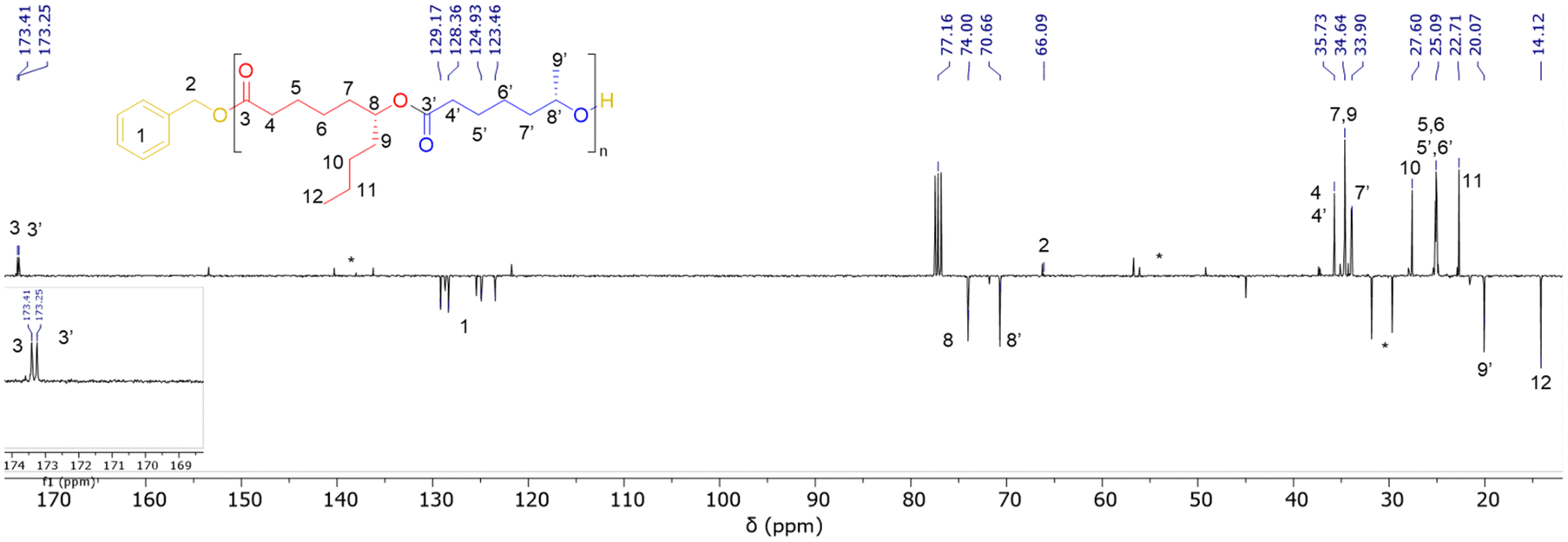 | ||
| Fig. 3 Representative JMOD 13C{1H} NMR spectrum (100 MHz, CDCl3, 25 °C) of a P(CLMe-co-CLnBu) copolymer prepared from the ROCOP of a 1:1 mixture of (S)-CLMe and (R)-CLnBu mediated by the 1a/BnOH (1:1) catalytic system (Table 1, entry 3). * stands for resonances of the residual solvent and/or catalyst. | ||
:1 mixtures of (S)-CLMe and (R)-CLnBu mediated by the 1a–b/BnOH catalytic systemsa
| Entry | Cat. | [(S)-CLMe]0/[(R)-CLnBu]0/[BnOH]0 | Reaction timeb (min) | [(S)-CLMe]0/[(R)-CLnBu]0 Conv.c (%) |
M
n,theod (g mol−1) |
M
n,NMRe (g mol−1) |
M
n,SECf (g mol−1) |
Đ
Mf |
T
gg (°C) |
|---|---|---|---|---|---|---|---|---|---|
|
a Reactions performed with [(S)-CLMe]0 = [(R)-CLnBu]0 = 1.0 M in toluene with [1a–b]/[BnOH]0 = 1:1.
b Reaction time was not optimized.
c Conversion of (S)-CLMe and (R)-CLnBu as determined by the 1H NMR analysis of the crude reaction mixture.
d Theoretical molar mass calculated according to Mn,theo = [CLMe]0/[1] × conv. (CLMe) × M(CLMe) + [CLnBu]0/[1] × conv. (CLnBu) × M(CLnBu) + M(BnOH), with M(CLMe) = 128 g mol−1, M(CLnBu) = 170 g mol−1 and M(BnOH) = 108 g mol−1.
e Molar mass as determined by the 1H NMR analysis of the isolated polymer, from the resonances of the terminal OBn group (refer to the Experimental section).
f Number-average molar mass (uncorrected) and dispersity (Mw/Mn) as determined by SEC analysis in THF at 30 °C vs. polystyrene standards.
g Glass transition temperature as determined by DSC; no melting transition was observed.
h Not determined.
i Reaction performed with (R)-CLMe and (S)-CLnBu.
j Reaction performed with rac-CLMe and rac-CLnBu.
k Reaction performed using iPrOH as a co-initiator rather than BnOH.
|
|||||||||
| 1 | 1a | 10:10:1 |
35 | 100:100 |
3100 | 3500 | 5500 | 1.17 | −45.7 |
| 2 | 1a | 10:10:1i |
40 | 100:100 |
3050 | 2900 | 4200 | 1.18 | ndh |
| 3 | 1a | 10:10:1 |
390 | 100:100 |
3000 | 3100 | 4850 | 1.31 | ndh |
| 4 | 1a | 50:50:1 |
180 | 100:86 |
10400 |
9100 | 13500 |
1.07 | ndh |
| 5 | 1a | 50:50:1j |
300 | 100:97 |
15200 |
10000 |
11500 |
1.11 | ndh |
| 6 | 1b | 10:10:1 |
15 h 30 | 100:100 |
3100 | 3000 | 4150 | 1.27 | −49.4 |
| 7 | 1b | 28:33:1k |
300 | 63:46 |
4900 | 8600 | 5700 | 1.09 | −59.8 |
Despite NMR spectroscopy confirming the formation of P(CLMe-co-CLnBu) copolymers, their detailed microstructure, in particular the alternation degree, could not be assessed using this technique. In fact, in the carbonyl region (C3,C3′) and in the methine region (C8,C8′) of all 13C NMR spectra, only two resonances were observed that arose from the two chemically different CLMe and CLnBu monomer units (Fig. 3 and S10†); if 13C NMR spectroscopy enables assessing alternation, more than two resonances would be observed, at least in the spectra of copolymers produced by catalyst 1b since this one is notoriously poorly syndioselective, if not completely non-stereoselective, due to its sterically small methyl substituents.9
Mass spectrometry (MS) is an alternative technique to NMR to study the sequence and microstructure of copolymers and, with proper data processing tools, more information concerning the composition, monomer sequence, polymer length, and reactivity ratios can be extracted.25–30 Hence, MS investigations were performed in order to better understand the microstructure of the P(CLMe-co-CLnBu) copolymers. Both MALDI-ToF and ESI mass spectra were recorded. One of the advantages of MALDI-ToF over ESI is that the range of m/z values of the returned mass spectra is greater; on the other hand, higher resolution can be often achieved with ESI.
In the MALDI-ToF mass spectra of our copolymers, the two major distributions observed featuring the most intense peaks refer to the linear α-BnO,ω-OH-[P(CLnBu)x-co-(CLMe)y] and cyclic [P(CLnBu)x-co-(CLMe)y] series (Fig. 4 and S13–S14†). Similar observations were made from the ESI HR-mass spectra (Fig. 5 and S16–S20†) from which both the aforementioned linear and cyclic populations could be unambiguously assigned.31 These MS data confirm the above NMR data regarding the formation of linear macromolecules with α-benzyloxy and ω-hydroxy end-groups. Noteworthily, cyclic populations cannot be assessed by NMR spectroscopy, since they have the same repeating units as linear chains, but no end-groups. The present observation of cyclic populations both in the MALDI-ToF and ESI mass spectra has two likely origins: (i) observation of cyclic macromolecules formed during the ROCOP process, arising from intramolecular back-biting/transesterification, as corroborated by the abovementioned broadening of the dispersities at longer reaction times, also in line being the MALDI-ToF mass spectrum of a copolymer prepared at short reaction time (35 min; Table 1, entry 1; Fig. 4a) features no cyclic population as compared to that of a copolymer exposed over a longer period to the catalytic system (390 min; Table 1, entry 3; Fig. 4b). (ii) Other possible reasons for the observation of a significant population of cyclic macromolecules by MALDI-ToF MS is likely the result of incidental over-expression of specific species during the crystallization process and/or the sample preparation for MS (in the acidic DCTB matrix, which promotes easy transesterification/cyclization).
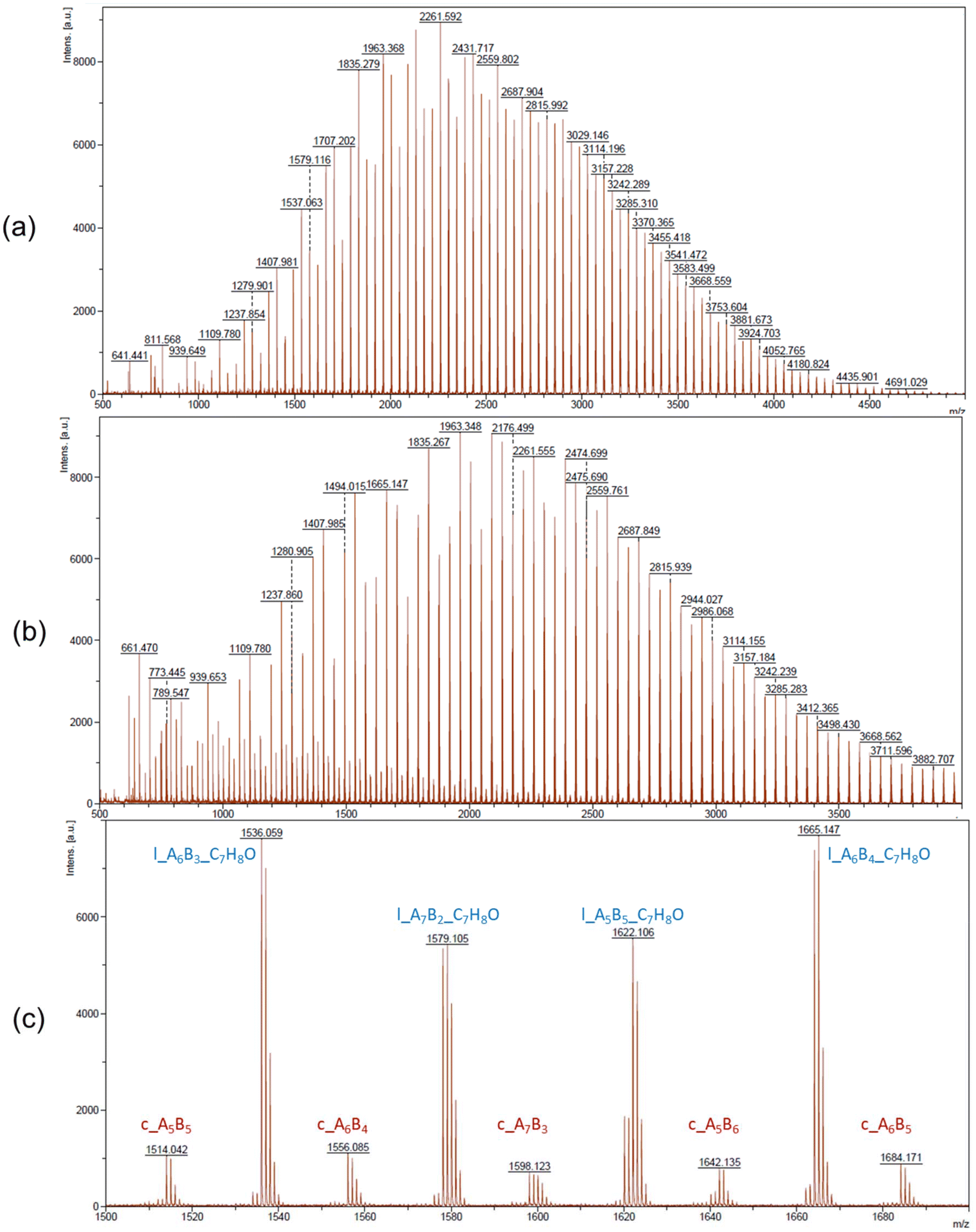 | ||
| Fig. 4 MALDI-ToF mass spectra (DCTB matrix, ionized by Na+) of P(CLnBu-co-CLMe) copolymers prepared by the ROCOP of a 1:1 mixture of (R)-CLnBu and (S)-CLMe with the 1a/BnOH catalytic system, over (a) 35 min and (b) 390 min, respectively (Table 1, entries 1 and 3, respectively) for m/z up to 4000, (c) zoomed region (m/z = 1500–1700) of the above MALDI-ToF mass spectrum (b) with corresponding assignments of the two major series observed. AxBy stands for x repeating units of A (CLnBu) and y repeating units of B (CLMe) in P(CLnBu-co-CLMe). | ||
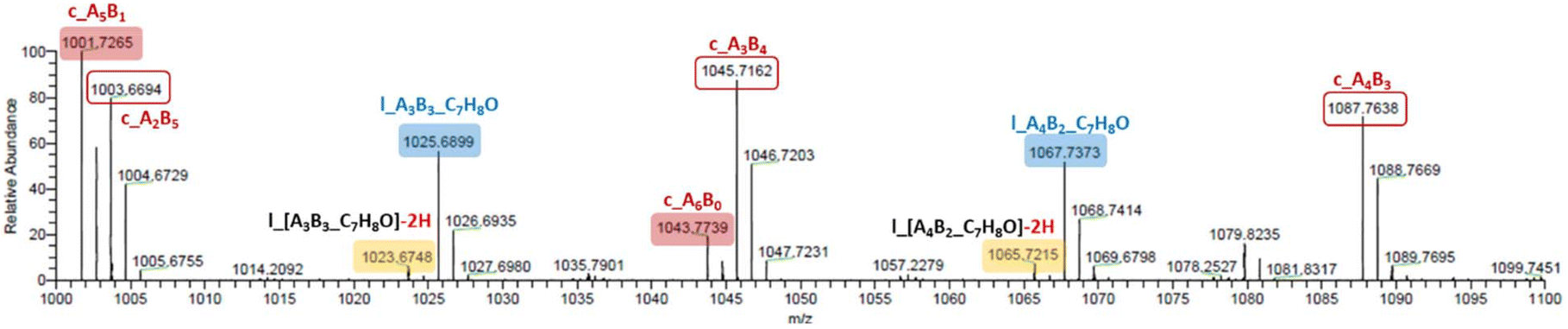 | ||
| Fig. 5 Details (m/z = 1000–1100) of the high resolution ESI mass spectrum of a P(CLnBu-co-CLMe) copolymer prepared by the ROCOP of a 1:1 mixture of (R)-CLnBu and (S)-CLMe with the 1a/BnOH catalytic system (Table 1, entry 3), showing resolved peaks for cyclic P[(CLnBu)5-co-(CLMe)] (m/zexp (all 12C) = 1001.7265, m/zcalcd = 1001.7263), cyclic P[(CLnBu)2-co-(CLMe)5] (m/zexp (all 12C) = 1003.6694, m/zcalcd = 1003.6692), linear P[(CLnBu)3-co-(CLMe)3]-C7H8O – 2H (m/zexp (all 12C) = 1023.6748, m/zcalcd = 1023.6743), linear P[(CLnBu)3-co-(CLMe)3]-C7H8O (m/zexp (all 12C) = 1025.6899, m/zcalcd = 1025.6900), cyclic P[(CLnBu)6-co-(CLMe)0] (m/zexp (all 12C) = 1043.7739, m/zcalcd = 1043.7733), cyclic P[(CLnBu)3-co-(CLMe)4] (m/zexp (all 12C) = 1045.7162, m/zcalcd = 1045.7162), linear P[(CLnBu)4-co-(CLMe)2]-C7H8O – 2H (m/zexp (all 12C) = 1065.7215, m/zcalcd = 1065.7213), linear P[(CLnBu)4-co-(CLMe)2]-C7H8O (m/zexp (all 12C) = 1067.7373, m/zcalcd = 1067.7369), cyclic P[(CLnBu)4-co-(CLMe)3] (m/zexp (all 12C) = 1087.7638, m/zcalcd = 1087.7631); the population at m/z = 1079.8325 could not be identified. | ||
For better depicting the distribution of different copolymer populations, assignments of the different series were established by the comparison of the experimental m/z values with the theoretical masses calculated for different copolymer compositions (i.e., by varying the number of CLnBu and CLMe units for both values of x and y, along with the two different families indicated) (Table S2†). Fig. 6 depicts the distribution of each population thus obtained in two different P(CLnBu-co-CLMe) copolymers.
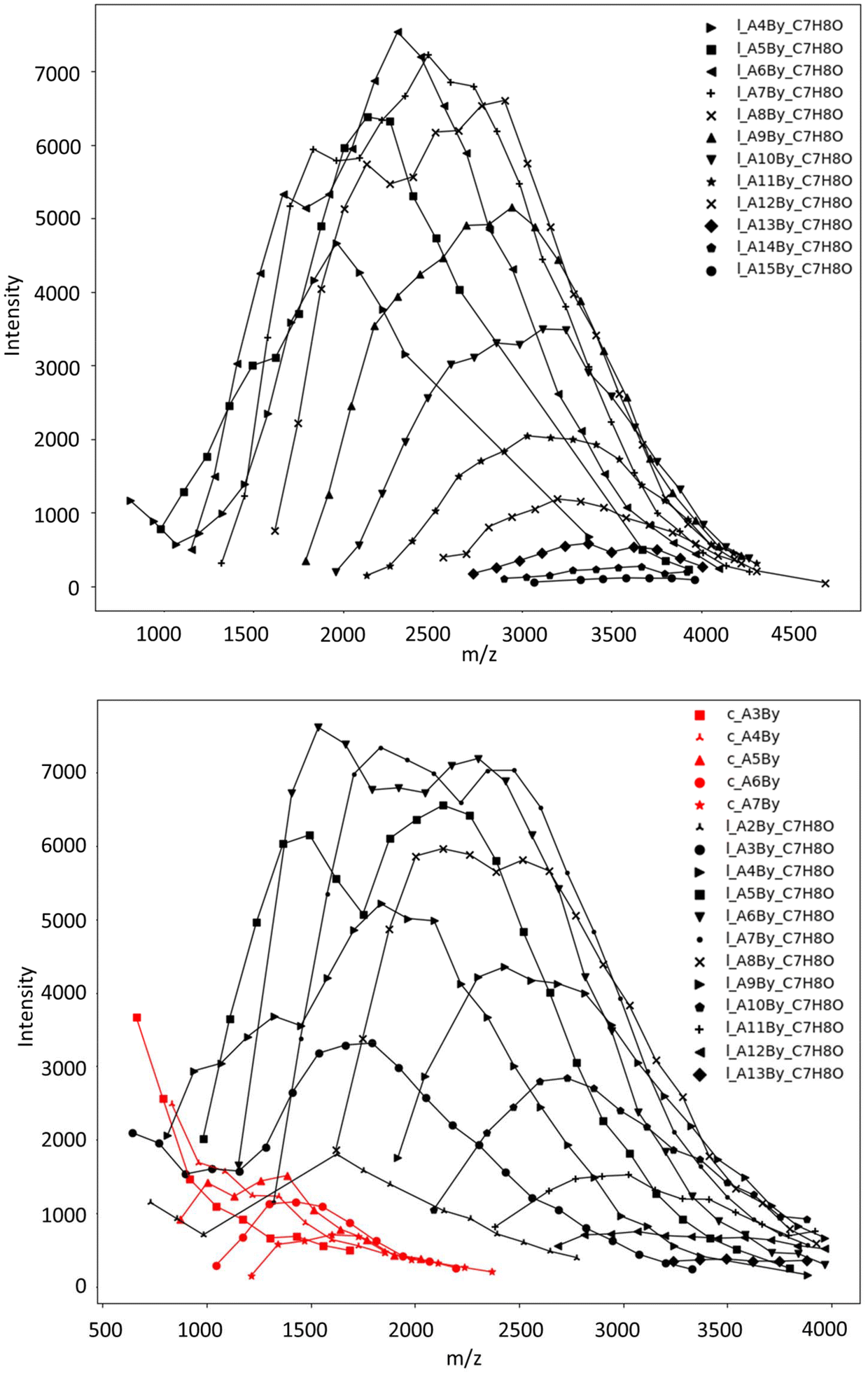 | ||
| Fig. 6 Distribution of individual polymer series experimentally observed in MALDI-ToF mass spectra (DCTB matrix, ionized by Na+, data from Fig. 4) of P(CLnBu-co-CLMe) copolymers prepared by the ROCOP of a 1:1 mixture of (R)-CLnBu and (S)-CLMe with the 1a/BnOH catalytic system, over (a) 35 min and (b) 390 min, respectively (Table 1, entries 1 and 3, respectively) for m/z up to 4000. One or two major series of macromolecules, i.e. linear α-BnO,ω-OH-P[(CLnBu)x-co-(CLMe)y] (black series) and cyclic P[(CLnBu)x-co-(CLMe)y] (red series) populations, are distinguished. AxBy stands for x repeating units of A (CLnBu) and y repeating units of B (CLMe). | ||
In our first attempts to determine the monomer sequence in the copolymers, MS/MS-ESI was applied on different selected polymer ions (Fig. S21†). However, the fragmentation spectra obtained showed different pathways for the same ion. In fact, the intensity values of the different peaks were affected by the overlap of signals within a narrow range, which makes this approach biased and eventually uninformative.
For this reason, the raw ESI and MALDI-ToF mass data were processed through a programmatic script Polymers (see the Experimental section). This enabled, for a given degree of polymerization (DP of P[(CLnBu)x-co-(CLMe)y] = x + y = constant), the identification of all possible ionized macromolecular chains with different values of x and y; these different possibilities were then adjusted in histograms displaying their relative intensities as a function of x. As illustrated in Fig. 7, the experimental data for both the linear (blue) and cyclic (red) macromolecule populations, as determined from the ESI and MALDI-TOF mass data, follow the same trend. They also match quite well the whole distribution for a random copolymer (plotted in green), computed following a binomial distribution law. Essentially the same trend was observed for different DPs up to 26. In other words, the ROCOP of equimolar mixtures of (R)-CLnBu and (S)-CLMe with the 1a/BnOH catalytic system leads to random-type copolymers; in case a highly alternating microstructure of the type P[(CLnBu-CLMe)n] had formed, the above experimental histograms would have been narrowly centered on the x ± 1 values.
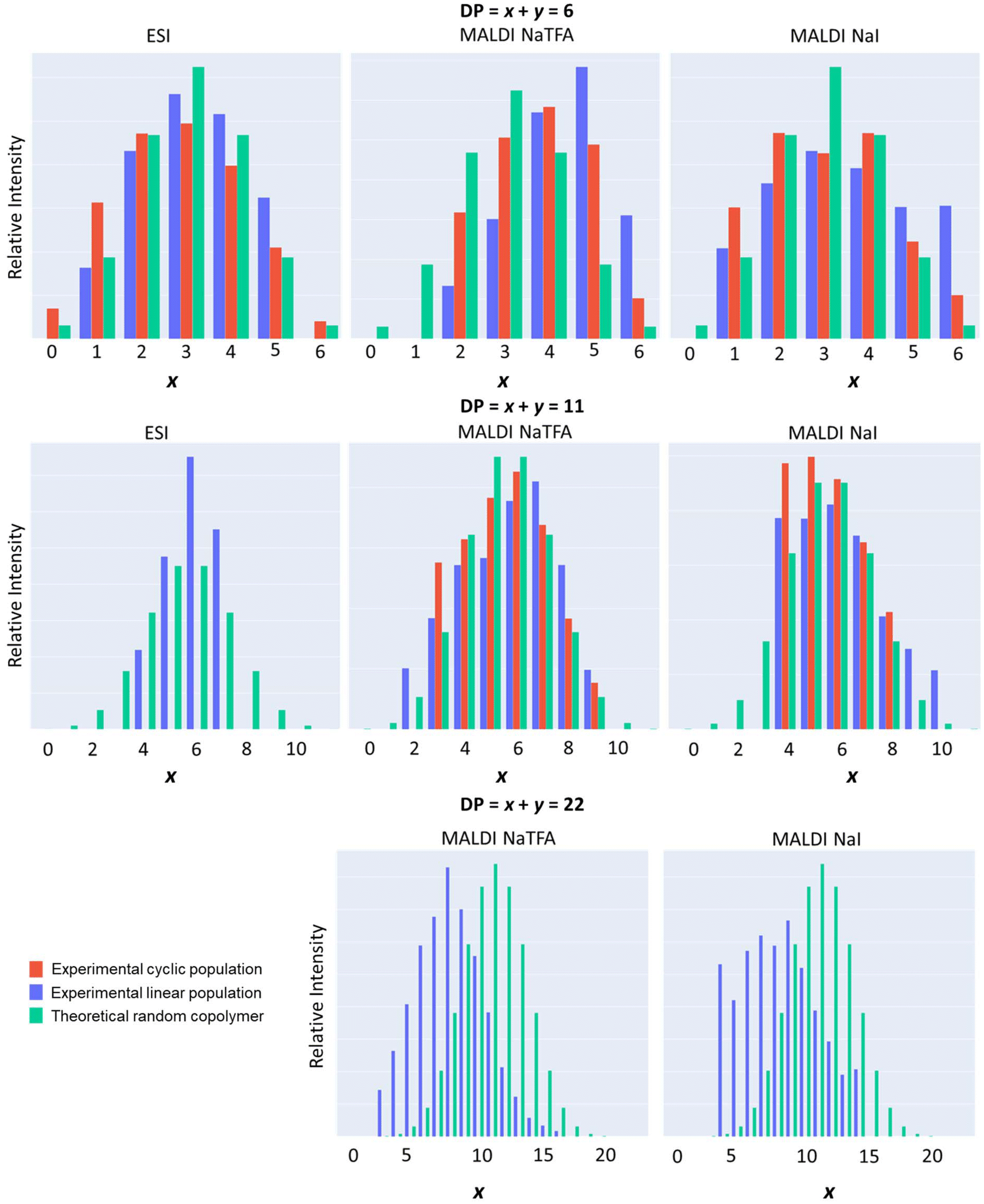 | ||
| Fig. 7 Histograms of experimental (blue: linear and red: cyclic) macromolecular ion distributions in a P[(CLnBu)x-co-(CLMe)y] copolymer (Table 1, entry 3) for selected DP = x + y = 6, 11, and 22 extracted from (left) ESI HRMS, (middle) MALDI-ToF MS using CF3CO2−Na+, and (right) MALDI-ToF MS using Na+I− as an ionizing source, and compared to theoretical (green) random copolymer distribution calculated from a binomial law. | ||
Fig. 7 also reveals interesting patterns in how the CLnBu and CLMe comonomers are arranged within the chains. Short chains (e.g. DP = 6) have a slightly higher proportion of CLnBu units compared to what is predicted theoretically. The distribution of the comonomers in medium-length chains (e.g. DP = 11) is closer to what is expected based on a binomial distribution for a random copolymer. Conversely, long chains (e.g. DP = 26) have a lower proportion of CLnBu units. Fig. 8 further exemplifies this trend by tracking the composition of the most intense monoisotopic m/z signal at different lengths (DPs). For chains with x + y = 13 units, the amounts of CLnBu and CLMe are roughly equal. For shorter chains (DP < 13), there is a higher proportion of CLnBu, while there is a higher content of CLMe for longer chains (DP > 13). These uneven distributions point to gradient-type macromolecules which are likely formed through preferential polymerization of the more reactive CLMe at early stages of the copolymerization reaction.
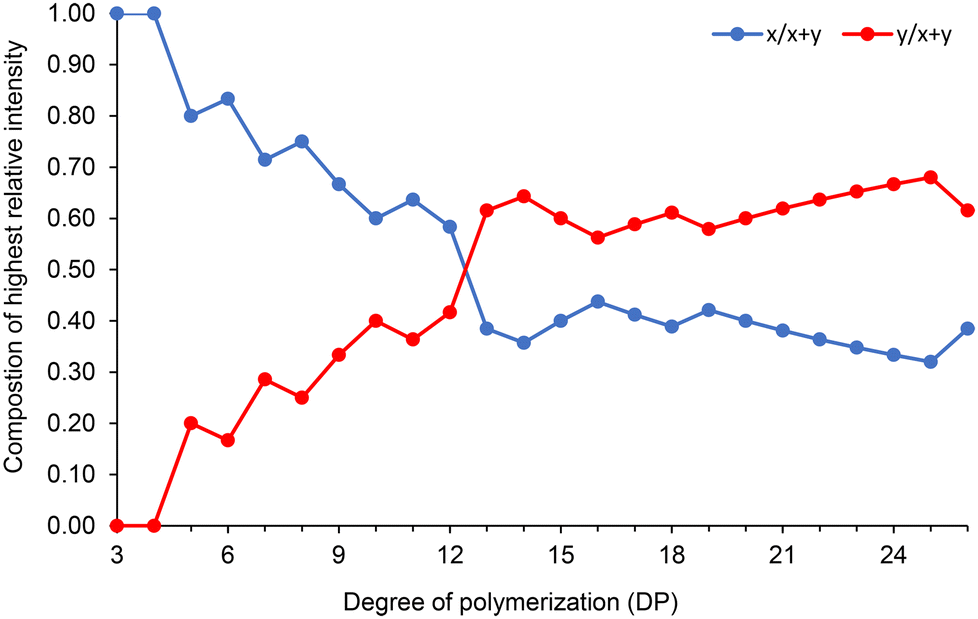 | ||
| Fig. 8 Variation of the composition (x, y) in P[(CLnBu)x-co-(CLMe)y] copolymers as a function of the degree of polymerization (DP). The composition was calculated from the most intense m/z signal for a defined DP, from MALDI-ToF MS of P(CLnBu-co-CLMe) copolymers prepared by the ROCOP of a 1:1 mixture of (R)-CLnBu and (S)-CLMe with the 1a/BnOH catalytic system, over 35 min (Table 1, entry 1). | ||
Böcker et al. developed a related method to determine similar parameters from a single MALDI-ToF MS analysis.27 Their software (COCONUT) plots the matrix of mass data in a 3D graph where the x- and y-axes show the amount of each monomer in the copolymer, with the peak intensity displayed on the z-axis. The shape of the thus obtained counter-plots reflects the microstructure of the copolymer: when the contour plot is centered on a diagonal straight line that crosses the origin, with a narrow distribution along this line, then an alternating copolymer is formed (the narrower the contour plot, the higher the alternation degree); however, if the distribution is broad, and passes through the origin, a random copolymer is obtained; on the other hand, if the contour plot deviates from the diagonal line, not going through the origin, then a gradient (composition drift) or a blocky structure is proposed.25 The COCONUT software allows also the determination of the reactivity ratios from monomer distribution contour-plots. We analyzed the MALDI-ToF MS data of our P(CLnBu)x-co-(CLMe)y copolymers using COCONUT, and examples of the resulting contour-plots are shown in Fig. 9. In all cases, the line drawn through the center of the broadly dispersed contour-plots is consistent with a gradient (i.e., random with a composition drift) structure, with no alternation. The offset of the contour plots toward the y-axis indicates the higher reactivity of CLMe compared to that of CLnBu. The COCONUT software also allowed calculating from the copolymer distribution the reactivity ratios of both co-monomers (rnBuCL = 0.10 and rMeCL = 1.94), which is in line with a random gradient structure and the aforementioned observations.
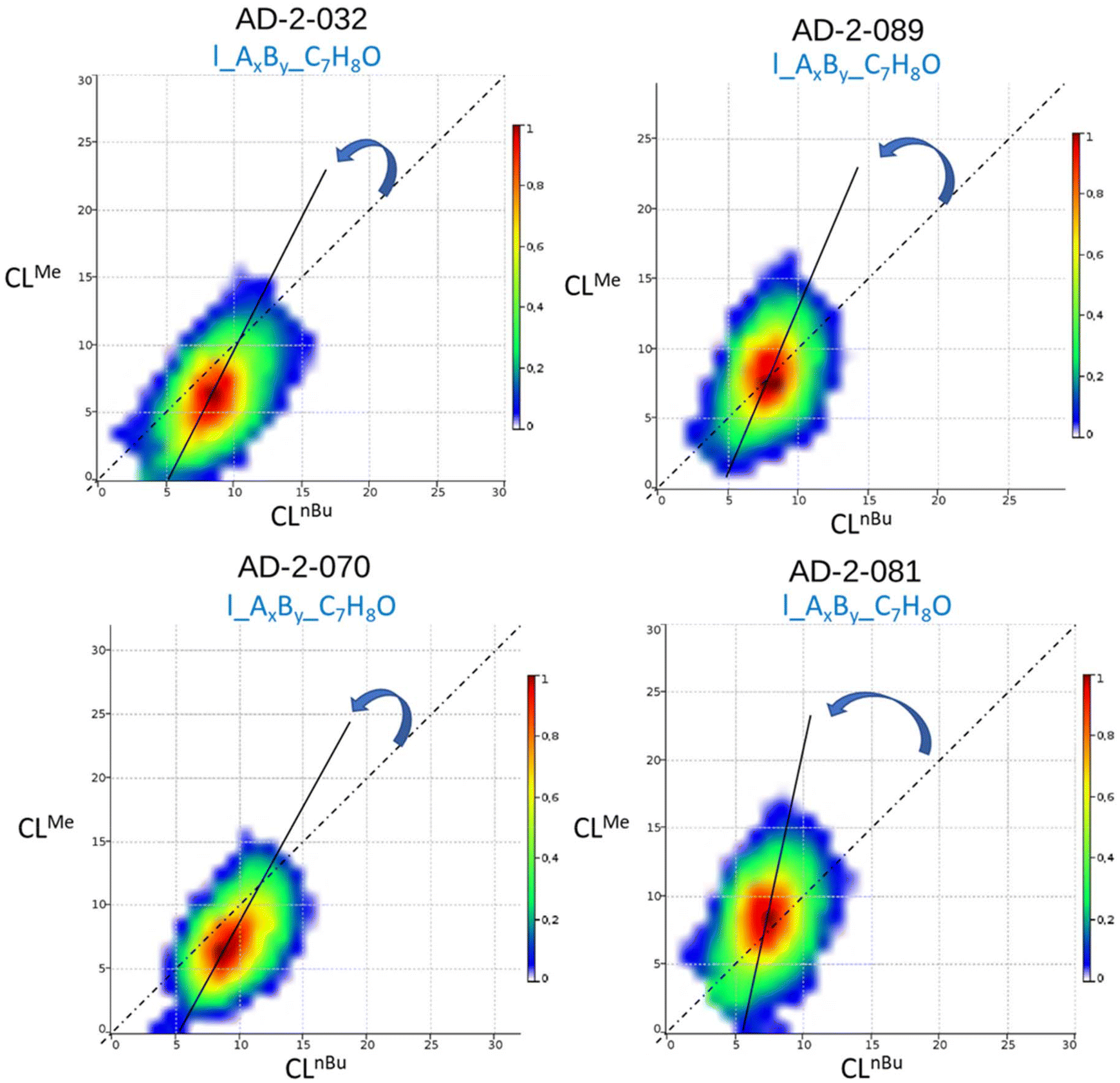 | ||
| Fig. 9 Examples of contour-plots generated using the COCONUT software24 from MALDI-ToF MS data of a P(CLnBu)x-co-(CLMe)y copolymer (Table 1, left: entry 3, right: entry 1). | ||
In order to correlate the reactivity ratios of the two monomers derived from MS analysis, further experiments based on the Fineman–Ross method were performed (refer to the Experimental section, Fig. S12†). The thus calculated reactivity ratios (rnBuCL = 0.19 and rMeCL = 1.02) are consistent with the above values determined from the MS data with COCONUT. They point out at the formation of random copolymers (with a composition drift), which eventually reflects the lack of stereocontrol of the yttrium achiral catalyst over seven-membered substituted-ε-caprolactones.
Conclusion
Yttrium complexes based on tetradentate non-chiral diamino-bis(o,p-disubstituted-phenolate) ligands effectively (co)polymerize 1-substituted-ε-caprolactones such as CLMe and CLnBu. 1H and 13C NMR spectroscopy confirmed the general structure of the (co)polymers formed and the good control over the molar masses, as also supported by SEC analyses. However, the NMR spectroscopic method remained insufficient for analyzing the (co)polymers’ microstructures and, in particular, for assessing the tacticity of the homopolymers. The ROCOP of equimolar mixtures of enantiopure comonomers with opposite configurations, (R)-CLnBu and (S)-CLMe, returned copolymers whose microstructure/comonomer sequence could be only, but effectively, deciphered by detailed MS analyses. The latter studies provided solid evidence for the formation of gradient copolymers. The non-alternating monomer sequence in these copolymers evidences that the yttrium catalysts used failed to differentiate between the two opposite-configuration monomers. In turn, this lack of alternation during copolymerization evidences the absence of significant syndioselectivity of these yttrium catalysts in the homopolymerization of rac-CLMe and rac-CLnBu. This contrasts drastically with the good stereoselectivities towards syndiotactic, or even isotactic, polyesters that these Y{ON(N)OR2} catalysts have shown in the ROP of a variety of racemic β-lactones, lactides and diolides. These observations made for the two catalysts and two monomers selected in the present study allow conclusions which, of course, should be extrapolated only with caution (as for all extrapolation in science). Despite the limited stereoelectronic difference in the R substituents (i.e., Me vs. nBu) of the two CLR monomers (corroborated by the small difference in their ROP reaction rates), a different outcome may be observed with even less differentiated monomers (e.g. R = Me vs. Et) or, obviously, different catalytic systems with better stereocontrol ability.Author contributions
Ali Dhaini: investigation, formal analysis, writing – original draft, review and editing. Jérôme Ollivier: mass spectrometry analysis, formal analysis, data curation, writing – original draft, review and editing. Nicolas Le Yondre: mass spectrometry analysis, MS software, and writing – original draft. Ali Alaaeddine: supervision. Sophie M Guillaume: writing – original draft, review & editing, supervision, formal analysis, and conceptualization. Jean-François Carpentier: writing – original draft, review and editing, supervision, formal analysis, and conceptualization.Data availability
The raw data required to reproduce these findings are available from the authors. The processed data required to reproduce these findings are available in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was financially supported in part by the University of Rennes (Ph.D. grant to A. D.) and the Lebanese University (Ph.D. grant to A. D.). We are grateful to CRMPO and UAR ScanMAT (CNRS, Univ. Rennes), especially to Philippe Jéhan and Elsa Caytan, for MS and NMR analyses, respectively. We thank Maxime Lanvin for the development of the MS data-processing script. We thank Dr Martin Engler-Lukajewski (Bright Giant Gmbh) for useful discussions regarding the exploitation of the COCONUT software.References
- For recent leading reviews on metal-catalyzed ROP of lactones and related derivatives, see:
(a) S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Beyond stereoselectivity, switchable catalysis: some of the last frontier challenges in ring-opening polymerization of cyclic esters, Chem. – Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed
; (b) X. Tang and E. Y.-X. Chen, Toward infinitely recyclable plastics derived from renewable cyclic esters, Chem, 2019, 5, 284–312 CrossRef CAS
- J.-F. Carpentier, Rare-earth complexes supported by tripodal tetradentate bis(phenolate) ligands: A privileged class of catalysts for ring-opening polymerization of cyclic esters, Organometallics, 2015, 34, 4175–4189 CrossRef CAS
-
(a) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Controlled ring-opening polymerization of lactide by group 3 metal complexes, Pure Appl. Chem., 2007, 79, 2013–2030 CrossRef CAS
-
(a) N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Syndiotactic-enriched poly(3-hydroxybutyrate)s via stereoselective ring-opening polymerization of racemic β-butyrolactone with discrete yttrium catalysts, Macromolecules, 2009, 42, 987–993 CrossRef CAS
- For recent examples of other highly efficient yttrium catalysts for stereoselective ROP of rac-BPLMe, see:
(a) H.-Y. Huang, W. Xiong, Y.-T. Huang, K. Li, Z. Cai and J.-B. Zhu, Spiro-Salen catalysts enable the chemical synthesis of stereoregular polyhydroxyalkanoates, Nat. Catal., 2023, 6, 720–728 CrossRef CAS
- R. Ligny, M. M. Hanninen, S. M. Guillaume and J.-F. Carpentier, Highly syndiotactic or isotactic polyhydroxyalkanoates by ligand-controlled yttrium-catalyzed
stereoselective ring-opening polymerization of functional racemic β-lactones, Angew. Chem., Int. Ed., 2017, 56, 10388–10393 CrossRef CAS PubMed
-
(a) R. M. Shakaroun, A. Dhaini, R. Ligny, A. Alaaeddine, S. M. Guillaume and J.-F. Carpentier, Stereo-electronic contributions in yttrium-mediated stereoselective ring-opening polymerization of functional racemic β-lactones: ROP of 4-alkoxymethylene-β-propiolactones with bulky exocyclic chains, Polym. Chem., 2023, 14, 720–727 RSC
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, Polymerization of enantiopure monomers using syndiospecific catalysts: A new approach to sequence control in polymer synthesis, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS PubMed
- C. G. Jaffredo, Y. Chapurina, S. M. Guillaume and J.-F. Carpentier, From syndiotactic homopolymers to chemically tunable alternating copolymers: Highly active yttrium complexes for stereoselective ring-opening polymerization of β-malolactonates, Angew. Chem., Int. Ed., 2014, 53, 2687–2691 CrossRef CAS PubMed
-
(a) X. Tang and E. Y.-X. Chen, Chemical synthesis of perfectly isotactic and high melting bacterial poly(3-hydroxybutyrate) from bio-sourced racemic cyclic diolide, Nat. Commun., 2018, 9, 2345–2356 CrossRef PubMed
-
(a) E. Archer, M. Torretti and S. Madbouly, Biodegradable polycaprolactone (PCL) based polymer and composites, Phys. Sci. Rev., 2023, 8, 4391–4414 Search PubMed
- F. J. van Natta, J. W. Hill and W. H. Carothers, Studies of Polymerization and Ring Formation. XXIII.1 ε-Caprolactone and its Polymers, J. Am. Chem. Soc., 1934, 56, 455–457 CrossRef CAS
-
(a)
Encyclopedia of polymer science and engineering, ed. F. Mark, N. Bikales, C. Overberger, G. Menges and J. Kroshwitz, John Wiley and Sons, New York, 1985, pp. 220–243 Search PubMed
-
(a) A. Watts, N. Kurkokawa and M. A. Hillmyer, Efficient polymerization of methyl-ε-caprolactone mixtures to access sustainable aliphatic polyesters, Biomacromolecules, 2017, 18, 1845–1854 CrossRef CAS PubMed
- P. Vangeyte and R. Jérôme, Amphiphilic block copolymers of high-molecular-weight poly(ethylene oxide) and either ε-caprolactone or γ-methyl-ε-caprolactone: Synthesis and characterization, J. Polym. Sci., Part A, 2004, 42, 1132–1142 CrossRef CAS
- C. Wang, Y. Xiao, A. Heise and M. Lang, Organometallic and enzymatic catalysis for ring opening copolymerization of ε-caprolactone and 4-methyl-ε-caprolactone, J. Polym. Sci., Part A, 2011, 49, 5293–5300 CrossRef CAS
- Y.-M. Tu, F.-L. Gong, Y.-C. Wu, Z. Cai and J.-B. Zhu, Insights into substitution strategy towards thermodynamic and property regulation of chemically recyclable polymers, Nat. Commun., 2023, 14, 3198 CrossRef CAS PubMed
- J. Kiriratnikom, C. Robert, V. Guerineau, V. Vendtiito and C. M. Thomas, Stereoselective ring-opening (co)polymerization of β-butyrolactone and ε-decalactone using an yttrium bis(phenolate) catalytic system, Front. Chem., 2019, 7, 301 CrossRef CAS PubMed
- C. Lv, G. Xu, R. Yang, L. Zhou and Q. Wang, Stereogradient polycaprolactones formed by asymmetric kinetic resolution polymerization of 6-methyl-ε-caprolactone, Polym. Chem., 2021, 12, 4856–4863 RSC
- M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Exploring electronic versus steric effects in stereoselective ring-opening polymerization of lactide and β-butyrolactone with amino-alkoxy-bis(phenolate)-yttrium complexes, Chem. – Eur. J., 2011, 17, 1872–1883 CrossRef CAS PubMed
- M. Fineman and S. D. Ross, Linear method for determining monomer reactivity ratios in copolymerization, J. Polym. Sci., 1950, 5, 259–265 CrossRef CAS
-
(a) L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, Single-site β-diiminate zinc catalysts for the ring-opening polymerization of β-butyrolactone and β-valerolactone to poly(3-hydroxyalkanoates), J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed
- DSC analyses of the prepared homo- and copolymers revealed no distinctive thermal properties (see Tables S1 and 1†): all polymers are essentially amorphous (no melting transition observed under our analytical conditions; Table S1†) and all have glass transition temperatures in the same range.
- As mentioned previously, the nearly perfect alternating copolymers obtained from 1:1 mixtures of (S)-MLABn and (R)-MLAAll using Y{ON(X)OR2} catalysts evidence limited stereoelectronic difference in the R substituents (i.e., benzyl vs. allyl).8 Also, using the same catalytic system, the ROCOP of 1:1 mixtures of (R)-BPLOMe and (S)-BPLOTBDMS (OTBDMS = OSitBuMe2) returned a copolymer with up to 67% alternation, despite the more pronounced steric difference between OMe and OTBDMS. Decreasing the stereoelectronic difference between R groups, as with mixtures of BPLOBn and BPLOMe, increased the degree of alternation to 89% (see: R. Ligny, S. M. Guillaume and J.-F. Carpentier, Yttrium-mediated ring-opening copolymerization of oppositely configurated 4-alkoxymethylene-β-propiolactones: Effective access to highly alternated isotactic functional PHAs, Chem. Eur. J., 2019, 25, 6412–6424 CrossRef CAS PubMed :1 mixtures of enantiopure, opposite-configuration BPLRs (R = Me and R = tBu), overcoming kinetic considerations where the reaction is under stereocontrol.7.
-
(a) S. Huijser, B. B. Staal, J. Huang, R. Duchateau and C. E. Koning, Chemical composition and topology of poly(lactide-co-glycolide) revealed by pushing MALDI-ToF MS to its limit, Angew. Chem., Int. Ed., 2006, 45, 4104–4108 CrossRef CAS PubMed
- R. X. Willemse, B. B. Staal, E. H. Donkers and A. M. van Herk, Copolymer fingerprints of polystyrene-block-polyisoprene by MALDI-ToF-MS, Macromolecules, 2004, 37, 5717–5723 CrossRef CAS
- M. S. Engler, S. Crotty, M. J. Barthel, C. Pietsch, K. Knop, U. S. Schubert and S. Böcker, COCONUT-An efficient tool for estimating copolymer compositions from mass spectra, Anal. Chem., 2015, 87, 5223–5231 CrossRef CAS PubMed
- J. Stouten, M. A. F. Delgove, N. De Vos, K. Matthyssen, G. G. P. Deroover and K. V. Bernaerts, Investigation of monomer reactivity, polymer microstructure and solubility in the copolymerization of 1,5-dioxepan-2-one with alkyl substituted lactones, Eur. Polym. J., 2023, 200, 112523 CrossRef CAS
-
B. Staal, Characterization of (co)polymers by MALDI-TOF-MS, Ph.D. Thesis, University of Technology Eindhoven, The Netherlands, 2005 Search PubMed
- O. Trhlíková, M. Janata, Z. Walterová, L. Kanizsová, E. Čadová and J. Horský, MALDI-ToF mass spectrometry detection of intramolecular composition gradient in copolymers, Talanta, 2019, 195, 215–220 CrossRef PubMed
- Besides the linear α-BnO,ω-OH-[P(CLnBu)x-co-(CL)y] and the cyclic [P(CLnBu)x-co-(CLMe)y] major series, another minor population was also observed in the ESI mass spectra – at e.g. m/z = 981.6271, 1023.6748, 1065.7215, that apparently corresponds to linear α-BnO,ω-OH-[P(CLnBu)x-co-(CL)y] macromolecules deprived of two hydrogens (see Fig. 5 and S14, S16, S18†). We assume that this population formed during the ESI MS ionization process (presumably by the loss of hydrogen atoms to form α,β-unsaturated ester within the main chain); no unsaturated groups were detected in the NMR spectra of the crude and reprecipitated copolymers.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00330f |
| ‡ Pr/m is the probability of racemo/meso enchainment between adjacent monomer units, as determined by 13C NMR spectroscopy, with Pr + Pm = 1; Pr = 1 for a perfectly syndiotactic polymer, Pr = Pm = 0.5 for an atactic one, and Pm = 1 for a perfectly isotactic polymer. |
| This journal is © The Royal Society of Chemistry 2024 |