
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in transition metal based catalysts and mechanism analysis for alcohol electrooxidation reactions
Yuguo
Zhao
ab,
Emma M.
Björk
 b,
Yong
Yan
*a,
Peter
Schaaf
c and
Dong
Wang
*c
b,
Yong
Yan
*a,
Peter
Schaaf
c and
Dong
Wang
*c
aCenter of Excellence for Environmental Safety and Biological Effects, Beijing Key Laboratory for Green Catalysis and Separation, Department of Chemistry, Beijing University of Technology, 100124 Beijing, China. E-mail: yong.yan@bjut.edu.cn
bNanostructured Materials, Department of Physics, Chemistry and Biology (IFM), Linköping University, SE 58183 Linköping, Sweden
cChair Materials for Electrical Engineering and Electronics, Institute of Materials Science and Engineering and Institute of Micro and Nanotechnologies MacroNano, TU Ilmenau, Gustav-Kirchhoff-Str. 5, 98693 Ilmenau, Germany. E-mail: dong.wang@tu-ilmenau.de
First published on 1st April 2024
Abstract
In order to address energy and environmental challenges effectively, there is a need to promote renewable energy-driven electrochemical conversion technologies, particularly electrosynthesis. Electrosynthesis has the potential to convert abundant molecules into valuable chemicals and fuels. However, the widespread adoption of electrosynthesis is often hindered by the slow oxygen evolution reaction (OER). To overcome this limitation, we can employ the more efficient alcohol electrooxidation reaction (AOR), utilizing renewable biomass-derived alcohols as an alternative to OER for producing high-value chemicals. Consequently, the development of efficient AOR catalysts, in conjunction with cathodic reduction reactions (hydrogen evolution, oxygen, and nitrogen electroreduction, etc.), is crucial for sustainable and environmentally-friendly advancements. A thorough understanding of AOR mechanisms is essential for catalyst design and can be achieved through the utilization of in situ characterization techniques and density functional theory (DFT) calculations. This review summarizes recent progress in AOR catalysts, with a particular focus on the electrooxidation of monohydric alcohols, polyols, and associated studies on reaction mechanisms. Additionally, the review identifies key factors impeding AOR development and provides insights into future prospects.
1. Introduction
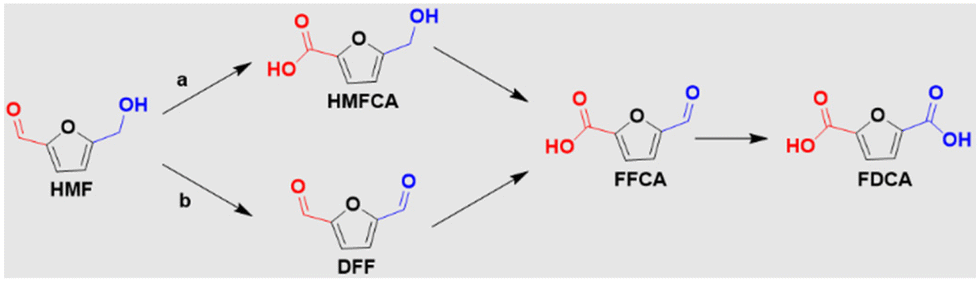
As the global energy and environmental crisis escalates, the advancement of electrochemical conversion technologies, powered by renewable energy sources like solar and wind, has become increasingly crucial.1–3 Electrosynthesis is recognized as an effective strategy to mitigate this crisis, as it enables the transformation of Earth-abundant molecules into valuable chemical products and transportable fuels.4,5 Often, this process involves coupling numerous cathodic electrochemical reduction reactions, such as the hydrogen evolution,6,7 oxygen,8,9 nitrogen,10,11 or carbon dioxide electroreduction,12,13 with the oxygen evolution reaction (OER). However, the slow kinetics and thermodynamics of OER at the anode present significant challenges for large-scale application of these processes. Furthermore, the limited value of the oxygen (O2) product exacerbates this issue.14 As a result, extensive research has been conducted to explore organic electrooxidation reactions (OORs) as more efficient alternatives to OER.15,16 These reactions not only exhibit lower overpotentials compared to OER but also yield high-value products at the anode.One prominent reaction within OORs is the alcohol electrooxidation reaction (AOR). This reaction involves the oxidation of biomass-derived alcohols, which are renewable and abundant. These alcohols are widely utilized in the manufacturing of commodity chemicals, such as formic acid, 2,5-furandicarboxylic acid, and dihydroxyacetone, among others.17,18 However, converting biomass-derived alcohols to value-added chemical products and fuels in traditional industries often requires harsh conditions, such as high temperatures and pressures, leading to the depletion of fossil energy resources and environmental pollution.19 Given the rising consumption of fossil energy and the negative impacts of environmental pollution, developing efficient AOR systems coupled with cathodic electroreduction is imperative for promoting sustainable global development. The success of this approach largely depends on the design and fabrication of highly efficient and selective catalysts for AOR. Thus, a timely and comprehensive review of this subject is vital for a deeper understanding of AOR and its potential implications.
This review aims to provide a detailed summary of recent advancements in transition metal-based catalysts. It primarily focuses on transition metals and their compounds, which have shown a high potential in the field of AOR. This potential arises from their well-regulated d orbitals and abundant presence on Earth.20 The review specifically summarizes the electrooxidation processes of monohydric alcohols and polyols in the context of AOR. Additionally, characteristic comparisons between representative AOR and OER are provided in Table 1. The applications of the products from these reactions, along with their respective reaction pathways, are also discussed. Understanding the reaction mechanisms of AOR is crucial for designing efficient catalysts, necessitating investigations into these mechanisms through advanced in situ characterization techniques and density functional theory (DFT) calculations. Therefore, this review emphasizes mechanism studies utilizing in situ characterization and DFT calculations, aiding comprehension of the relationship between catalyst structure and catalytic performance. Finally, the review concludes with an analysis of the challenges hindering AOR development and insights into future prospects in this field, hoping to guide efficient catalyst design.
| Reactions | Theoretical oxidation potential | Disadvantages/Challenges |
|---|---|---|
| HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid. | ||
| Water → Oxygen | 1.23 V (ref. 21) | Low energy conversion efficiency |
| 4 OH− → 2 H2O + O2 + 4 e− | O2 is less valuable | |
| Methanol → HCOOH | 0.103 V (ref. 21) | HCOOH is easily overoxidized to CO2 |
| CH3OH + 4 OH− → HCOOH + 3 H2O + 4 e− | ||
| Benzyl alcohol → Benzoic acid | 0.48 V (ref. 22) | The product separation process is complex |
| Ph-CH2OH + 4 OH− → Ph-COOH + 3 H2O + 4 e− | ||
| HMF → FDCA | 0.30 V (ref. 23) | HMF is unstable in the electrolyte |
| HMF + 6 OH− → FDCA + 4 H2O + 6 e− | ||
| Glycerol → HCOOH | 0.69 V (ref. 24) | The selectivity of aldehydes or ketones is low |
| C3H8O3 + 8 OH− → 3 HCOOH + 5H2O + 8 e− | ||
2. Alcohol electrooxidation reaction
The development of AOR systems has gained significant attention worldwide in recent years. Beyond the previously mentioned advantages, AOR also offers benefits such as safety, increased current density, and potential for fine chemical production. AOR is safer than OER in traditional water-splitting systems, as it minimizes O2 production, reducing risks associated with O2 and hydrogen (H2) mixing. This aspect mitigates safety concerns and potential hazards. AOR's ability to achieve higher current densities at the same voltage conditions, due to reduced overpotential, enhances the energy utilization efficiency of the system. Additionally, AOR is highly significant in the production of fine chemicals, laying the foundation for future development of high-efficiency electrocatalytic synthesis of fine chemicals.25–29 These advantages make AOR an attractive and promising electrochemical reaction system with the potential to contribute to clean and efficient chemical synthesis.This section offers an overview of representative catalysts for AOR, covering both monohydric alcohols and polyols. It investigates the reaction pathways associated with these catalysts and compiles a summary of recently reported catalyst performances for AOR in Table 2.
| Catalyst | Electrolyte | Main product | J (mA cm−2)/E (V vs. RHE) | Conversion (%) | FE (%) | Ref. |
|---|---|---|---|---|---|---|
| BAc: benzoic acid; HMF: 5-hydroxymethylfurfural; FDCA: 2,5-furandicarboxylic acid; DFF: 2,5-diformylfuran; EG: ethylene glycol; GA: glycolic acid; NA: not available. | ||||||
| Fe-NF-500 | 1 M KOH/1 M methanol | Formate | 10/1.328 | NA | >98 | 34 |
| h-NiSe/CNTs | 1 M KOH/1 M methanol | Formate | 10/1.37 | NA | >95 | 33 |
| NiB-400 | 1 M KOH/1 M methanol | Formate | 10/1.465 | NA | ∼100 | 14 |
| Ni3C particles/C | 1 M KOH/1 M methanol | Formate | 10/1.43 | ∼100 | NA | 48 |
| Ni0.75Fe0.25Se2 | 1 M KOH/1 M methanol | Formate | 53.5/1.5 | NA | 99 | 49 |
| β-Co0.1Ni0.9(OH)2/NF | 1 M KOH/0.05 M ethanol | Acetate | 100/1.35 | 100 | 95 | 50 |
| CoNi-PHNs | 1 M KOH/1 M ethanol | Acetate | 57/1.5 | NA | 99 | 51 |
| Ni3S2 NWs/C | 1 M KOH/1 M ethanol | Acetate | 107/1.5 | NA | NA | 52 |
| NiOx/CNx | 1 M KOH/1 M ethanol | Acetate | 10/1.354 | NA | NA | 53 |
| Cu-doped NiOOH | 1 M KOH/1 M ethanol | Acetate | 227/1.72 | NA | >98 | 54 |
| Ni0.75Co0.25Se2 | 1 M KOH/1 M ethanol | Acetate | 81.6/1.55 | NA | 82.2 | 55 |
| NPs | ||||||
| A–Ni–Co–H/NF | 1 M KOH/0.1 M BA | BAc | 100/1.35 | 99.6 | 93.5 | 22 |
| N–Mo–Ni/NF | 1 M KOH/0.1 M BA | BAc | 100/1.338 | 99.1 | 98.7 | 56 |
| h-Ni(OH)2 | 1 M KOH/0.04 M BA | BAc | 40/>1.4 | 99.99 | 98.62 | 57 |
| NiO/Ni3S2 | 1 M KOH/0.2 M BA | BAc | 50/1.391 | 99.11 | 94 | 58 |
| Ni–OH/NF | 1 M KOH/0.1 M BA | BAc | 100/1.33 | 84.5 | 99 | 59 |
| hp-Ni | 1 M KOH/0.01 M BA | BAc | 10/1.35 | NA | 98 | 60 |
| NiBx–Py | 1 M KOH/0.01 M HMF | FDCA | NA | 99 | 92.5 | 61 |
| NiCoP | 1 M KOH/0.3 M HMF | FDCA | 10/1.26 | 98.7 | 95.8 | 62 |
| NiFe LDH | 1 M KOH/0.01 M HMF | FDCA | 10/1.32 | 99 | 99.4 | 63 |
| NiSx/Ni2P | 1 M KOH/0.01 M HMF | FDCA | 10/1.34 | 100 | 95.1 | 64 |
| NiBx | 1 M KOH/0.01 M HMF | FDCA | 10/1.33 | 99.8 | 99.5 | 65 |
| NiSe@NiOx | 1 M KOH/0.01 M HMF | FDCA | 50/1.35 | 100 | 99 | 66 |
| NiCo2O4 | 1 M KOH/0.005 M HMF | FDCA | 10/1.47 | 99.6 | 87.5 | 67 |
| MoO2–FeP@C | 1 M KOH/0.01 M HMF | FDCA | 10/1.359 | 99.4 | 97.8 | 68 |
| Ru1–NiO | 1 M PBS/0.05 M HMF | DFF | 10/1.283 | 72.4 | 43.3 | 69 |
| Rh–SA/NiFe | 1 M PBS/0.05 M HMF | FDCA | 50/1.27 | 98 | 98.5 | 70 |
| NMLDH | ||||||
| Ni-VN/NF | 1 M KOH/0.01 M HMF | FDCA | 10/1.352 | >99 | 98.8 | 71 |
| CoNiP-NIE | 1 M KOH/0.01 M HMF | FDCA | 20/1.29 | NA | 87.2 | 72 |
| CoNi0.25P | 1 M KOH/0.3 M EG | Formate | 10/1.24 | 100 | 82.5 | 73 |
| Pd–Ni(OH)2/NF | 1 M KOH/1 M EG | GA | 100/0.69 | 93.2 | 86 | 74 |
| Branched NiSe2/C | 1 M KOH/1 M EG | Formate | 103.6/1.6 | NA | ∼80 | 75 |
| Ni–Mo–N/NC | 1 M KOH/0.1 M glycerol | Formate | 50/1.25 | 86 | 96.7 | 76 |
| E-NiV LDH | 1 M KOH/0.1 M glycerol | Formate | 10/1.23 | 98 | 94 | 77 |
| CoMoO4 | 1 M KOH/0.1 M Glycerol | Formate | 10/1.239 | NA | 90 | 78 |
| Bi–Co3O4 | 1 M KOH/0.1 M glycerol | Formate | 10/1.223 | NA | 97.05 ± 2.55 | 79 |
| Mn–CoSe2/CFC | 1 M KOH/0.1 M glycerol | Formate | 10/1.27 | NA | ∼95 | 80 |
| Ni–Mo–N/CFC | 1 M KOH/0.1 M glycerol | Formate | 10/1.3 | NA | ∼97 | 24 |
| MnO2 | 0.005 M H2SO4/0.2 M glycerol | Formic acid | 10/1.36 | NA | 53 | 81 |
| NiVRu-LDHs NAs/NF | 1 M KOH/0.1 M glycerol | Formate | 10/1.24 | NA | 97 | 82 |
| NiFeOx–NF | 1 M KOH/0.5 M glucose | Glucaric acid | NA | 98.3 | 87 | 83 |
| Co@NPC800 | 1 M KOH/0.1 M glucose | Lactate | 10/1.46 | NA | NA | 84 |
| Cu(OH)2 | 1 M KOH/0.1 M glucose | Gluconic acid | NA | NA | 98.7 | 85 |
2.1 Monohydric alcohols
Among the catalysts explored for MOR, those based on earth-abundant transition metals, especially in alkaline media, have received notable attention. A study by Li et al. introduced the Ni3B/Ni catalyst, termed NiB-400, synthesized by reacting sodium borohydride (NaBH4) with nickel nitrate, followed by annealing.14 NiB-400's crystal structure, shown in Fig. 1a, features numerous interfaces. Regarding MOR performance, NiB-400 demonstrated efficient catalytic activity in 1 M KOH electrolyte, achieving a current density of 175 mA cm−2 at a relatively low overpotential of 1.61 V vs. RHE (Fig. 1b). Additionally, the catalyst exhibited nearly 100% faradaic efficiency (FE) over a broad potential range (1.41–1.71 V vs. RHE), as depicted in Fig. 1c. To better understand NiB-400's efficient MOR performance, additional DFT calculations was conducted. The potential-determining steps for MOR was identified as the *CH3O → *CH2O + H+ + e− reaction on Ni, Ni3B, and Ni3B/Ni surfaces. Notably, the Ni3B(001)/Ni(111) heterostructure displayed the lowest Gibbs energy barrier for MOR, suggesting optimal activity on this surface. Additionally, the partial density of states (PDOS) was calculated to analyze the adsorption behavior of the *CH2O intermediate on the Ni3B(001)/Ni(111) heterostructure, as well as on Ni3B and Ni structures. Fig. 1e illustrates the d-band center values of Ni3B/Ni heterostructure, Ni3B, and Ni as −1.22, −1.26, and −1.34 eV, respectively. Importantly, the d-band center of the Ni3B/Ni heterostructure is closest to the Fermi energy level, indicating its superior adsorption capacity for *CH2O. This effectively promotes the binding of the key intermediate and reduces the associated energy barrier, thus, enhancing catalytic activity. The synthesis of NiB-400 and subsequent DFT studies offer profound insights into the exceptional MOR performance of the nickel Ni3B/Ni catalyst.
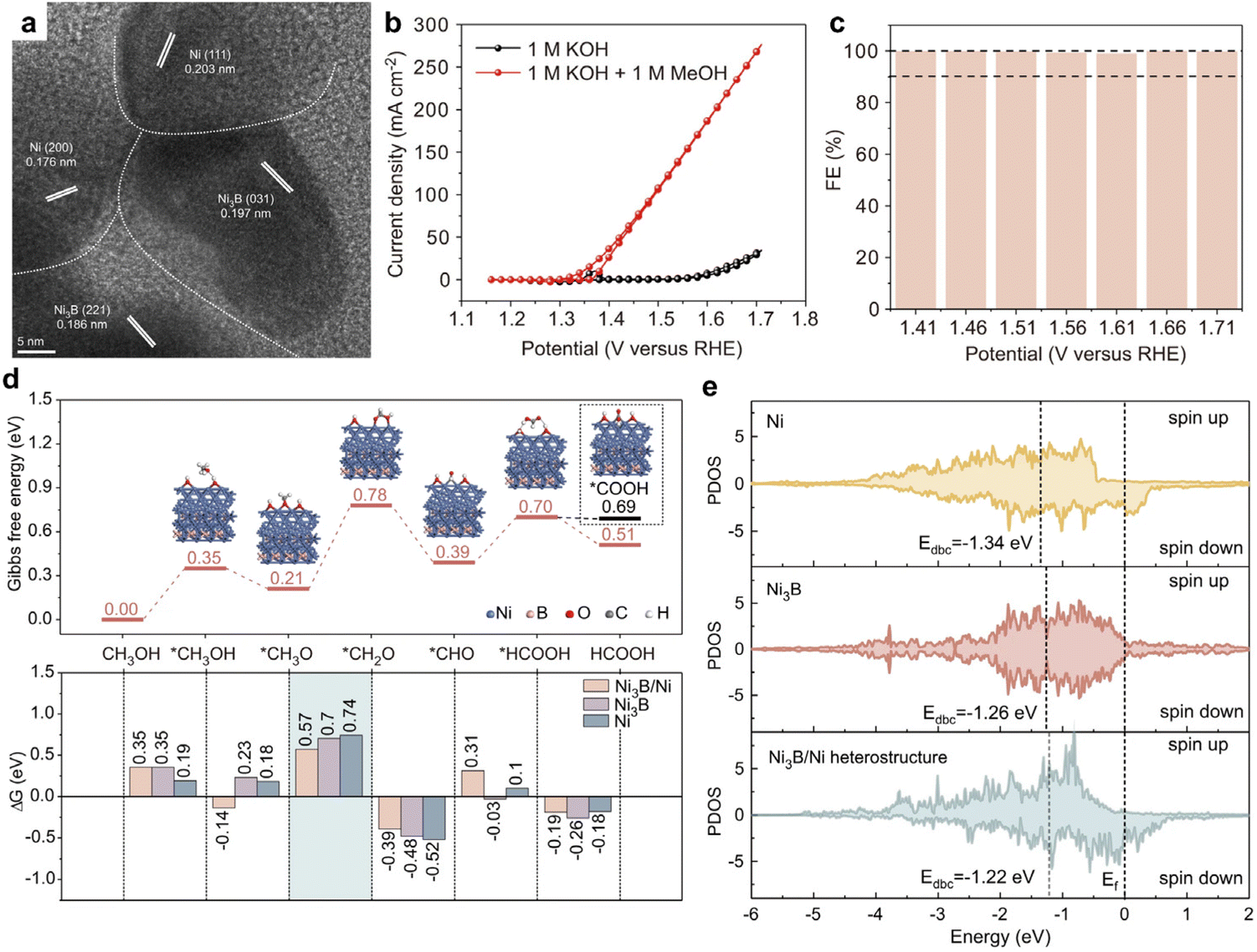 | ||
| Fig. 1 (a) TEM image of NiB-400. (b) CV curves of NiB-400 for MOR and OER. (c) The faradaic efficiency of formate was obtained by MOR at different potentials for 1 h. (d) Gibbs free energy diagram of MOR occurring on Ni3B(001)/Ni(111) heterostructure. (e) Partial density of states (PDOS) of Ni active sites for different structures. Reproduced with permission from ref. 14. Copyright 2022 Springer Nature. | ||
Similar enhancements in MOR performance have been observed in various catalysts with heterostructure configurations, including Fe2O3/NiO,34 NMS/CC,35 h-NiSe/CNTs,33 FeNi2S4/NiS-NG-2,36 NiFe-LDH/NiFe-HAB,37 NiCoP/MoS2,38 and 3D Ni/NiO/RG-400.39 These studies show that the heterogeneous structure of these catalysts significantly improves their electronic configuration, enhancing adsorption capacity for key intermediates and reducing associated energy barriers.
Core–shell catalysts like CuS@CuO/CF,40 Cu/NiCu NWs-220,41 Au@PdRu RNs,42 and Mo2C/MWCNT43 have also exhibited effcient MOR performance. They modulate electronic structure through lattice strain modification at the heterointerface, influencing interaction with intermediates as explained by d-band theory.44,45 This tunability of electronic structure in core–shell catalysts represents a promising direction for optimizing the catalytic activity in MOR.
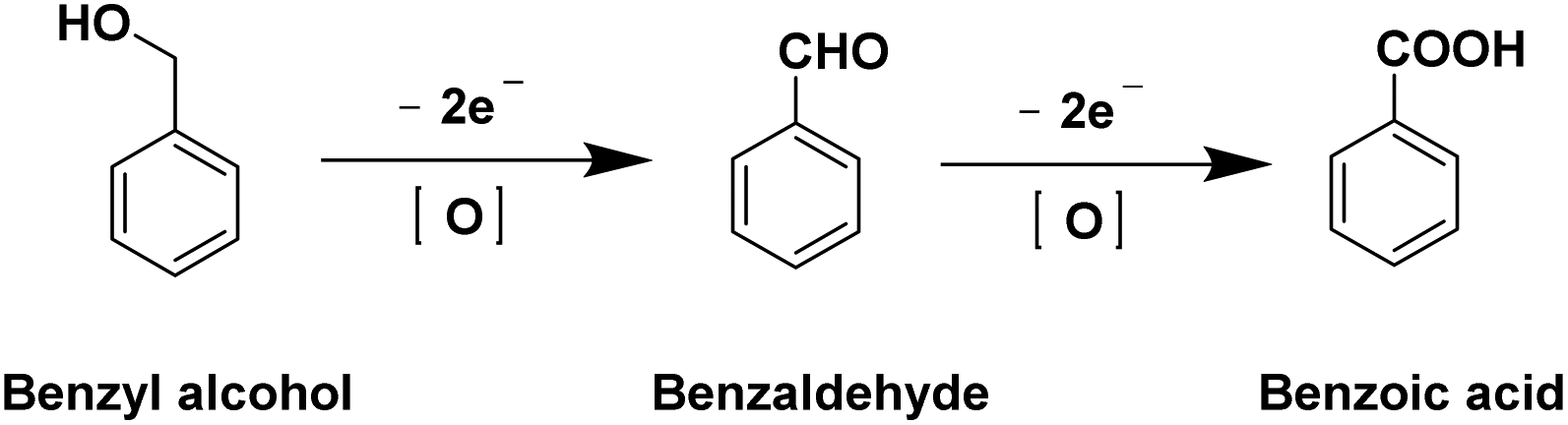 | ||
| Fig. 2 The reaction pathways for the oxidation of benzyl alcohol. | ||
Recent studies have focused on developing well-designed catalysts for benzyl alcohol oxidation reaction (BOR). Wang et al.86 introduced a self-supported layered porous NC@CuCo2Nx/CF catalyst, as depicted in Fig. 3a–d. This catalyst demonstrated high efficiency as a bifunctional catalyst for BOR and hydrogen evolution in alkaline electrolyte solutions (pH 14). Fig. 3e shows that, without benzyl alcohol, over 1.46 V vs. RHE was needed to achieve a current density of 10 mA cm−2. However, this requirement dropped to 1.25 V vs. RHE with the addition of benzyl alcohol, indicating a preference for BOR over oxygen evolution. In a bifunctional electrolytic system (HER and BOR), NC@CuCo2Nx/CF showed excellent BOR performance with higher FE compared to other catalysts (Fig. 3f). The good performance of NC@CuCo2Nx/CF is attributed to its unique layered porous structure, which enhances surface area and ion diffusion. Additionally, the cooperative effect of Cu3N and Co5.47N optimizes the adsorption energy of key intermediates, improving reaction kinetics and electrocatalytic activity.
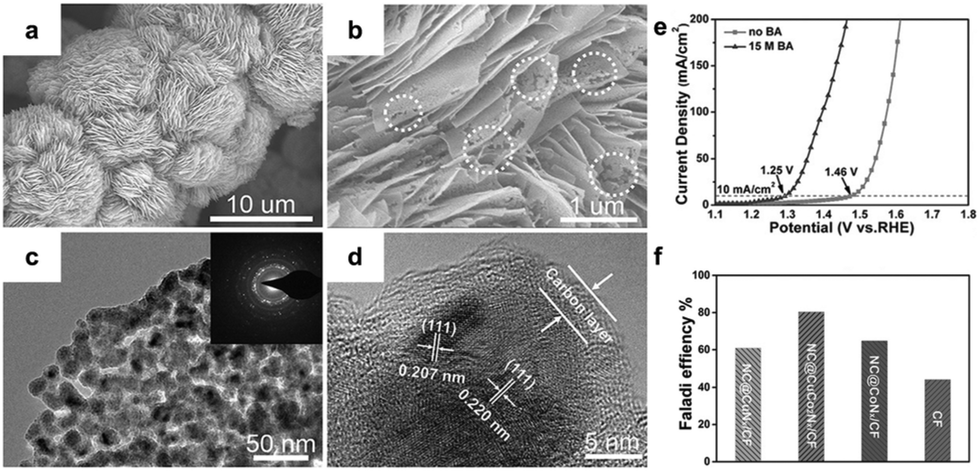 | ||
| Fig. 3 (a–d) Structural characterization of NC@CuCo2Nx/CF. (e) LSV curves of OER and BOR. (f) faradaic efficiency of BOR using different catalysts. Reproduced with permission from ref. 86. Copyright 2017 Wiley-VCH. | ||
Qiu et al.22 used a hydrothermal synthesis method to create Ni and Co hydroxide nanosheets on a Ni foam substrate (A–Ni–Co–H/NF), shown in Fig. 4a and b. This material offers a large active surface area and lower charge transfer resistance. Without benzyl alcohol, achieving an OER current density of 100 mA cm−2 required a voltage of 1.60 V. This decreased to 1.35 V with the addition of benzyl alcohol, enabling the attainment of an industrial-scale current density above 400 mA cm−2 for electrocatalytic oxidation of benzyl alcohol without undergoing OER. In contrast, the NF substrate could not reach 100 mA cm−2 even with benzyl alcohol (BA) (Fig. 4c). The stability of the catalyst was investigated through five repeated amperometric tests at a consistent potential of 1.5 V. As depicted in Fig. 4e and f, the conversion of BA remained consistently above 99%, with the yield of Ph-COOH maintaining a level of over 95% throughout the five trials. Additionally, the FE for Ph-COOH production was consistently maintained at 90–95%.
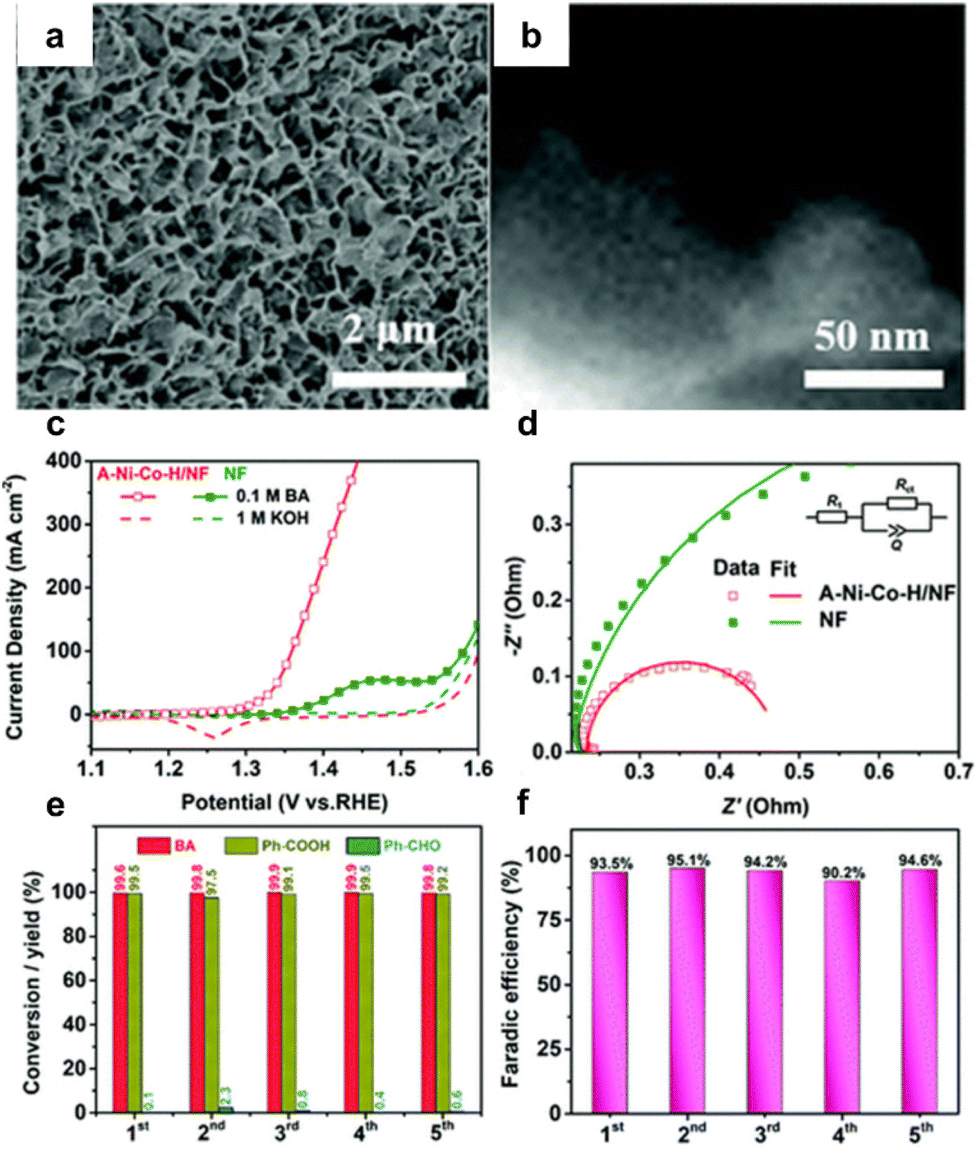 | ||
| Fig. 4 (a and b) Structural characterization of A–Ni–Co–H/NF, (c) LSV curves of A–Ni–Co–H/NF and NF in 1 M KOH electrolyte with and without 0.1 M benzyl alcohol, (d) electrochemical impedance spectroscopies of A–Ni–Co–H/NF and NF, (e) conversion and yield of Ph-COOH after five successive electrolysis cycles, and (f) faradaic efficiency of Ph-COOH corresponding to panel (e). Reproduced with permission from ref. 22. Copyright 2020 Royal Society of Chemistry. | ||
 | ||
| Fig. 5 The HMF electrooxidation reaction pathway. | ||
Nickel-based materials have garnered significant interest as electrocatalysts for HMFOR, owing to their high activity and cost-effectiveness.100 In a study by Sun et al.,101 a bimetallic Ni–Cu catalyst supported on Ni foam (Ni–Cu/NF) was developed for efficient HMFOR. Structural analysis indicated a non-uniform distribution of the Ni–Cu catalyst on the Ni foam surface. Additionally, the catalyst was in an amorphous state, with Ni, Cu, and O elements evenly dispersed, as shown in Fig. 6a–d. The Ni–Cu/NF catalyst exhibited efficient performance, with a low onset potential for HMFOR (approximately 1.35 V vs. RHE) and a high current density of 1000 mA cm−2 at 1.50 V. Notably, the potential required to achieve current densities of 200, 500, and 1000 mA cm−2 was over 200 mV lower than that for the oxygen evolution reaction (OER), as demonstrated in Fig. 6e and f. The Ni–Cu/NF catalyst also showed remarkable stability over 70 consecutive cycles, maintaining consistent performance without significant degradation, as depicted in Fig. 6g. During the stability evaluation, FEs and yields of FDCA remained above 95%, indicating the efficient utility of the catalyst for biomass upgrading and its substantial practical application value.
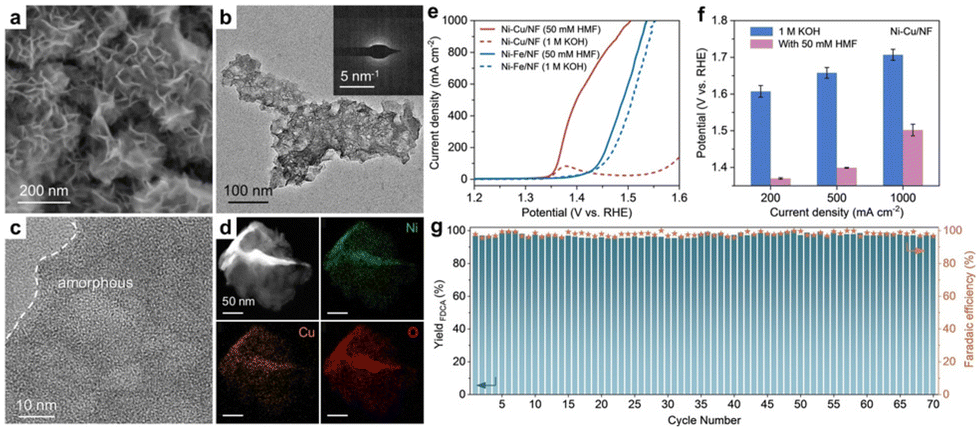 | ||
| Fig. 6 (a–d) Structural characterization of Ni-Cu/NF. (e) LSV curves of Ni–Cu/NF and Ni–Fe/NF (reference sample) compared with and without the presence of 50 mM HMF in a 1 M KOH solution, with 100% iR compensation. (f) Potential required by Ni–Cu/NF to achieve different current densities in the presence or absence of 50 mM HMF. (g) Stability, yield, and faradaic efficiency of FDCA for Ni–Cu/NF with the addition of 50 mM HMF over 70 consecutive cycles at 1.45 V vs. RHE, with each cycle lasting for 60 min. Reproduced with permission from ref. 101. Copyright 2023 Wiley-VCH. | ||
Wang et al.102 explored an electrocatalyst known as Mn0.2NiS/GF, demonstrating promising HMFOR to FDCA conversion. This electrocatalyst showed an industrial-level current density (500 mA cm−2), high FE (94.2%), and robust stability. Furthermore, other notable Ni-based electrocatalysts such as Co0.4NiS@NF,103 NiSx/β-Ni(OH)2/Ni,104 Co9S8@Ni3S2/NF,105 and NiMoO-Ni106 also exhibited high FDCA conversion rates. These findings underscore the significant potential of Ni-based electrocatalysts in HMFOR-to-FDCA conversion and offer valuable insights for developing highly efficient electrocatalysts.
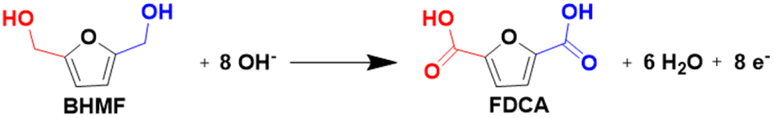
2,5-Bis(hydroxymethyl)furan (BHMF) has a structural similarity to HMF and can be obtained by the selective hydrogenation of HMF.107 It can be further oxidized to FDCA through electrocatalysis (Fig. 7), which has recently attracted significant attention due to its higher stability in electrolytes compared to HMF.108 A study by Chen et al.109 introduced a NiCo/CF electrocatalyst for the efficient electrooxidation of BHMF, demonstrating a substantial current density of 1981.7 mA cm−2 at 2.293 V vs. RHE. This electrocatalyst also exhibited high yields and faradaic efficiencies of FDCA production, reaching 95.4% and 96.5% respectively, at a relatively low potential of 1.4 V vs. RHE. Moreover, Zhang et al.108 reported the use of CoOOH/Ni electrodes for the effective electrooxidation of BHMF, achieving complete conversion and a high FDCA yield of 90.2%. These findings suggest that BHMF is a promising candidate for the production of FDCA through mild electrocatalysis.
 | ||
| Fig. 7 The BHMF electrooxidation reaction pathway. | ||
2.2 Polyols
 | ||
| Fig. 8 Schematic illustration of glycerol production from biomass. | ||
| Product | Application |
|---|---|
|
Used in cosmetic and food industries,111 pharmaceutical intermediate,112 synthon in organic chemistry,113 monomer in polymeric biomaterials114 |
|
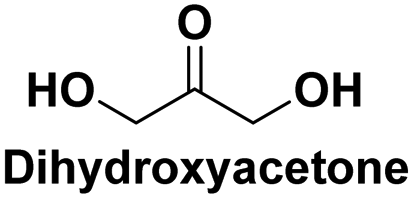
Used in medicine: as a metabolite in the glycolysis cycle, as an intermediate in the synthesis of aminoacids,115 acting as base materials for functional surfactants and monomers for oligoesters or polymers,116 used for treatment of skin diseases114 |
|
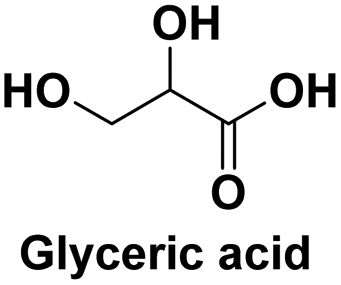
Used as a pharmaceutical for weight loss and prevention of coronary heart disease,117 used as a pharmaceutical and anti-corrosive protective agent, as well as for the monomer of biopolymers118 |
|
Organic synthon,113 anti-HIV agent,119 salt form isused in the treatment of diabetes119 |
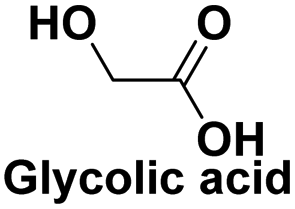
|
Degreasing and rust removal,120 as a major raw material in the fields of fine chemistry and pharmacy121 |
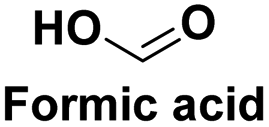
|
Chemical raw material widely used in the tanning, pharmaceutical, pesticide,122 fuel in fuel cell123 |
Compared to traditional industrial production, using electrocatalytic oxidation involves milder reaction conditions and effectively resolves the issues that may arise in traditional industrial glycerol oxidation, and the reaction pathways of glycerol electrooxidation as shown in Fig. 9. Recent research efforts have been centered on the development of highly efficient and cost-effective electrocatalysts for glycerol oxidation reaction (GOR). A notable advancement in this area is the use of bimetallic electrocatalysts, exemplified by NiCo2O4/NF, investigated by Shi et al.110 This electrocatalyst, synthesized through hydrothermal and calcination processes, featured a distinctive 3D nanoneedle array structure, as shown in Fig. 10a and b. In GOR, NiCo2O4/NF, along with other tested samples, demonstrated significantly lower onset potential and higher current density than those in the OER. Specifically, NiCo2O4/NF's onset potential for GOR was about 400 mV lower than that of OER, underlining its superior performance in GOR applications. Chronoamperometry (CA) tests at various potentials showed that the formic acid production's FE peaked at 89.9% at 1.40 V vs. RHE. This result underscores NiCo2O4/NF's efficacy as an electrocatalyst for GOR, suggesting its potential in advancing the development of more effective and economical catalysts for these processes.
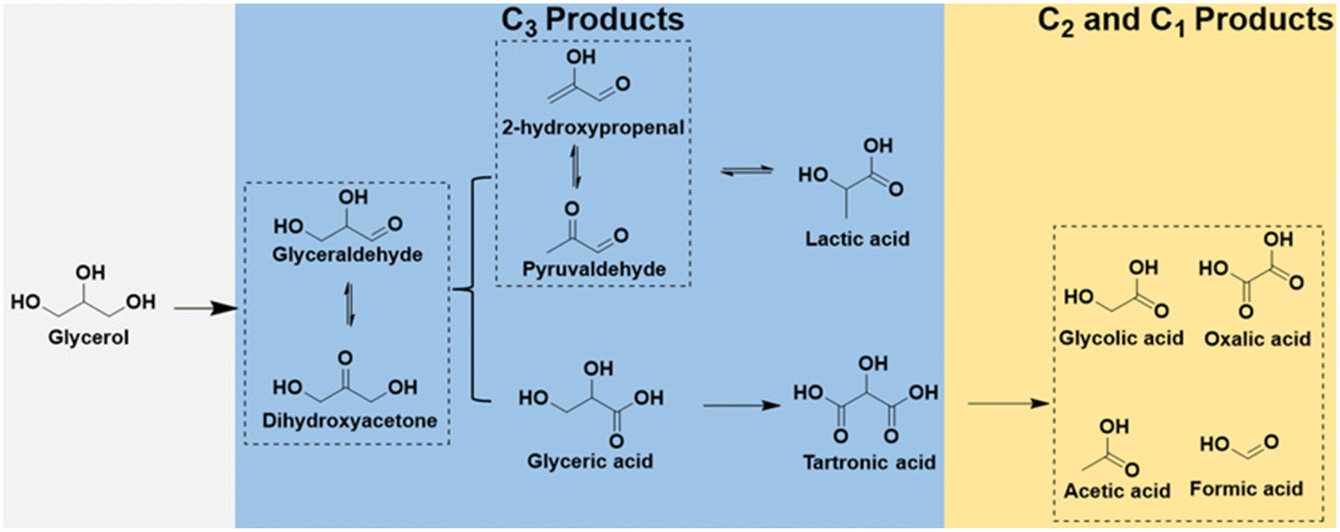 | ||
| Fig. 9 Schematic illustration of electrooxidation pathway of glycerol. | ||
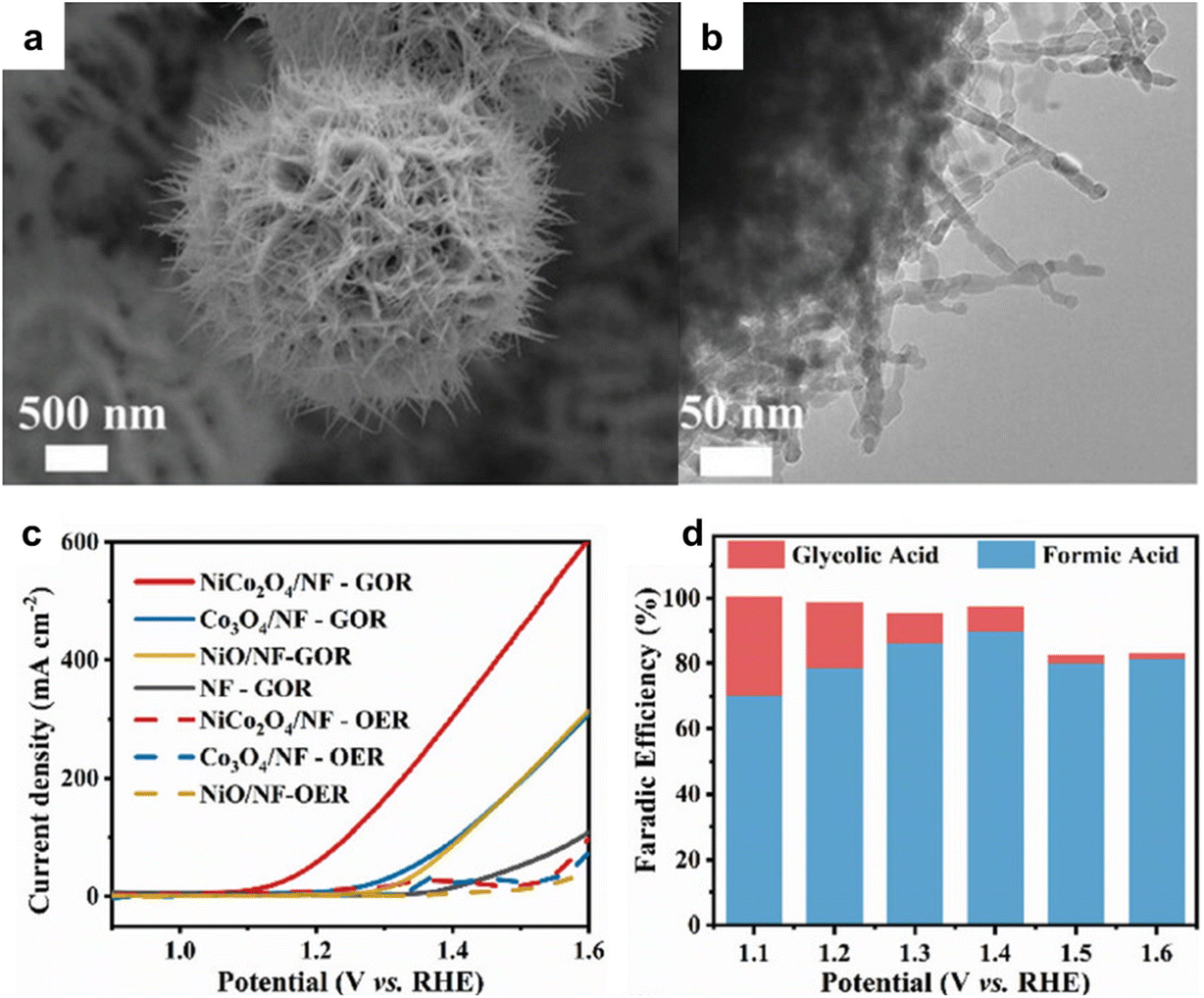 | ||
| Fig. 10 (a) SEM (b) TEM images of NiCo2O4/NF. (c) LSV curves of NiCo2O4/NF and other reference samples in 1 M KOH with and without 0.1 M glycerol. (d) faradaic efficiency of products obtained by conducting CA tests at different potentials. Reproduced with permission from ref. 110. Copyright 2023 Wiley-VCH. | ||
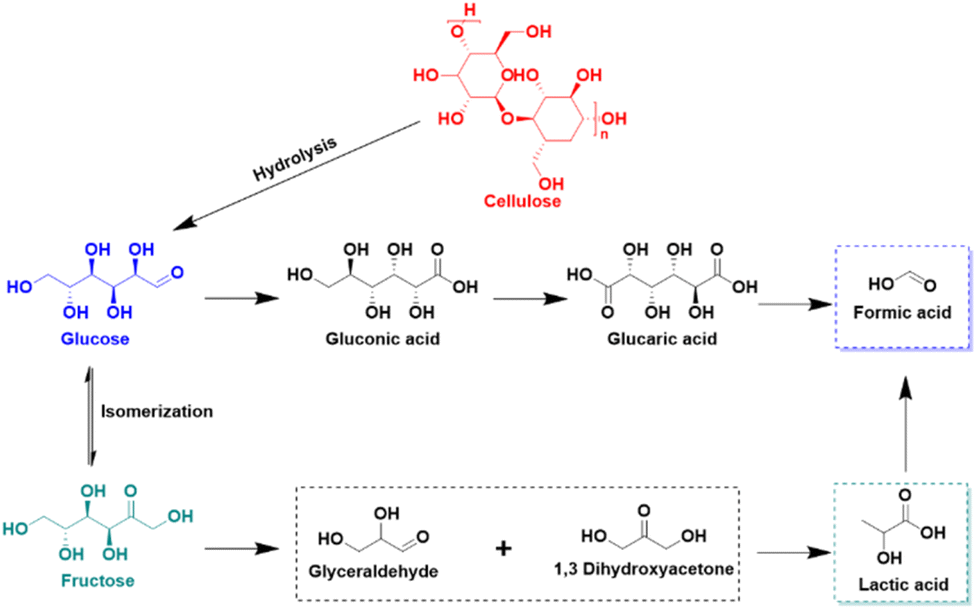 | ||
| Fig. 11 Glucose production from biomass hydrolysis and electrooxidation pathway of glucose. | ||
For glucose electrooxidation, Duan et al.129 reported an efficient electrocatalyst, CoOOH/CC, synthesized using an electrochemical method. SEM and HAADF–STEM analyses confirmed its interconnected array structure (Fig. 12a and b). Fig. 12c showed that, with the addition of 0.1 M glucose, the onset potential for glucose oxidation was 200 mV lower than that for OER. This indicates easier glucose oxidation with more favorable dynamics and thermodynamics compared to water oxidation. CoOOH/CC displayed significant performance in converting glucose to formate across a broad potential window of 1.3–1.7 V vs. RHE, achieving 60–70% FE, as demonstrated in Fig. 12d (reactions were conducted in a custom/upgraded reactor).
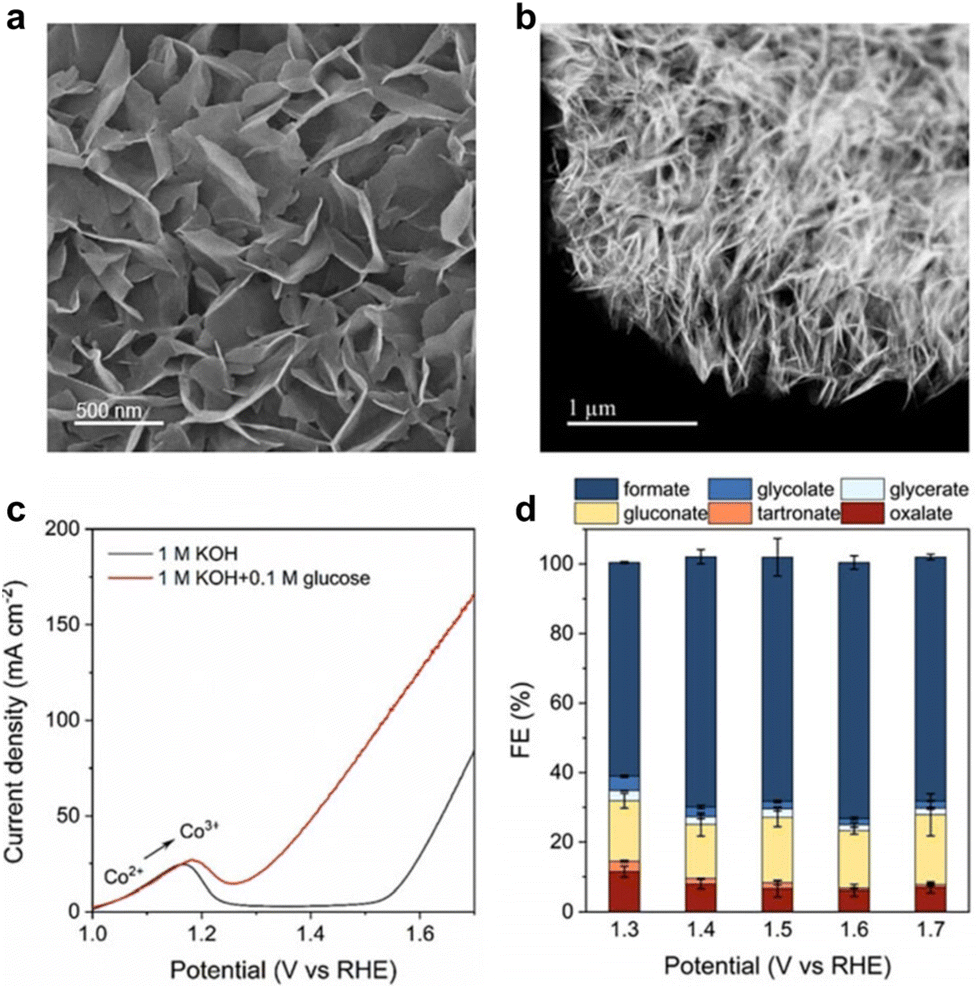 | ||
| Fig. 12 (a) SEM (b) HAADF–STEM images of CoOOH/CC. (c) LSV curves of CoOOH/CC in 1 M KOH with 0.1 M glucose and without 0.1 M glucose. (d) faradaic efficiency of products at different applied potentials. Reproduced with permission from ref. 129. Copyright 2023 Wiley-VCH. | ||
Additional research has identified several other electrocatalysts for glucose oxidation. For instance, CuO/CF130 exhibited a high FE of 95.34% ± 2.20% for formate production at 1.4 V vs. RHE in an alkaline solution. Moreover, other electrocatalysts have shown capabilities in producing various high-value chemicals. For example, Fe0.1–CoS2131 reached a low onset potential of 1.22 V vs. RHE for 10 mA cm−2 and achieved an 86.7% FE for gluconate production, while oxidized Cu132 resulted in a 23.3% yield for lactate production, both conducted in alkaline solutions.
2.3 Selectivity and stability of AOR's catalyst
The above analysis primarily focuses on catalyst activity. However, selectivity and stability are also two critical factors for catalyst design. Achieving incomplete oxidation products, such as aldehydes and ketones, is of utmost importance due to their higher value compared to fully oxidized products like acids. Fine-tuning the catalyst selectivity remains a challenge, but valuable insights have already been gained. For instance, in a study by Bitter et al.,133 they controlled the selectivity of glucose electrooxidation by manipulating the Pt oxidation state. They found that Pt0 promoted glucose dialdehyde formation, exhibiting higher activity for primary alcohol dehydrogenation compared to aldehyde oxidation. In contrast, PtOx accelerated gluconate formation, enhancing the selectivity of electrocatalytic dehydrogenation/oxidation at the anomeric carbon, albeit at the cost of primary alcohol dehydrogenation. Furthermore, the selectivity of AOR can also be adjusted through heteroatom doping. Song et al.134 reported an electrocatalyst, Au1+n-NiAl-LDH, which consists of both Au single atoms and Au nanoparticles on NiAl-LDH, achieving a high selectivity for the electrooxidation of benzyl alcohol to benzaldehyde (BAD). While pure NiAl-LDH displayed approximately 56% selectivity in the production of BAD, Au1+n-NiAl-LDH exhibited a significantly higher selectivity of 91%. This demonstrates that the selectivity of AOR can be effectively modified through heteroatom doping. Another method to adjust selectivity is by transitioning from acids to aldehydes through the addition of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) into the electrolyte. For instance, research by Wang et al.57 showcased an electrocatalyst (1.0 h-Ni(OH)2) with a selectivity of 92% for benzoic acid without the use of TEMPO. Upon the addition of TEMPO, the selectivity shifted to >94% for benzaldehyde. These findings provide valuable insights into adjusting selectivity in AOR processes.Regarding catalyst stability, Shi's group81 reported a catalyst (MnO2/CP) for acidic glycerol electrooxidation with a demonstrated stabilization of over 865 hours. Mechanistic investigations revealed that long-term stability can be attributed to stabilizing the manganese oxidation state and reduced excessive oxygen vacancies, accomplished through protective interactions with glycerol. Furthermore, Duan et al.135 reported that a cooperative catalyst (Au/CoOOH) exhibited significantly higher stability for the BOR compared to pure Au. By employing an intermittent potential (IP) strategy, the current density could be maintained at elevated levels (250–400 mA cm−2) over a 24-hour period. The improved stability of Au/CoOOH over Au was attributed to the stronger adsorption of BA at the Au/CoOOH interface through the σ–π interaction between benzyl alkoxide and Au. Despite the potential risk of deactivation over prolonged reactions due to the formation of AuOx, the IP strategy effectively reduced AuOx back to Au, thereby enhancing the overall stability of Au/CoOOH.
3. Mechanism analysis for AOR
A comprehensive understanding of the mechanisms underlying AOR systems is vital for designing and synthesizing efficient catalysts. The advancement of in situ technologies such as infrared (IR) spectroscopy, Raman spectroscopy, and X-ray absorption spectrum (XAS) has facilitated the tracking of active sites and reaction intermediates during AOR. These technologies enable a more detailed understanding of reaction pathways and aid in a thorough analysis of the reaction mechanism. Concurrently, DFT calculations are instrumental in elucidating the AOR mechanism, offering guidelines for electrocatalyst design. DFT calculations allow for the exploration of reaction energetics and thermodynamics, electronic structure, and reactivity of active sites, aiding in predicting and optimizing catalyst materials and properties. The integration of in situ technologies and DFT calculations significantly enhances the understanding of the AOR mechanism, guiding the development of more efficient electrocatalysts. This synergistic approach not only provides deep insights into reaction pathways and intermediates but also informs the design of catalysts with improved performance.3.1 In situ characterization
There are four main working modes of in situ IR spectroscopy for electrocatalytic reactions: infrared reflection absorption spectroscopy (IRAS), attenuated total reflection (ATR), transmission (TIR), and diffuse reflectance Fourier transform IR (DRFTIR).139,140 IRAS and ATR are particularly prevalent due to their shorter IR path and reduced susceptibility to external interferences. For example, Wang et al.141 employed in situ ATR-IR spectroscopy to explore the mechanism of the MOR on NiPx-R surfaces. The intensity of the adsorbed peak at 2920 cm−1, indicative of CH3O* species formation, slightly increased from potentials of 1.35 V to 1.65 V (Fig. 13a). In situ IRAS spectra (Fig. 13b) revealed two broad bands at 1700 to 1200 cm−1 becoming more pronounced with increasing potential. These findings led to a proposed MOR mechanism on NiPx-R (Fig. 13c), involving the initial adsorption of OH* and CH3OH molecules, their subsequent reaction, and the final oxidation of CH3O* to produce formate (alkaline medium).
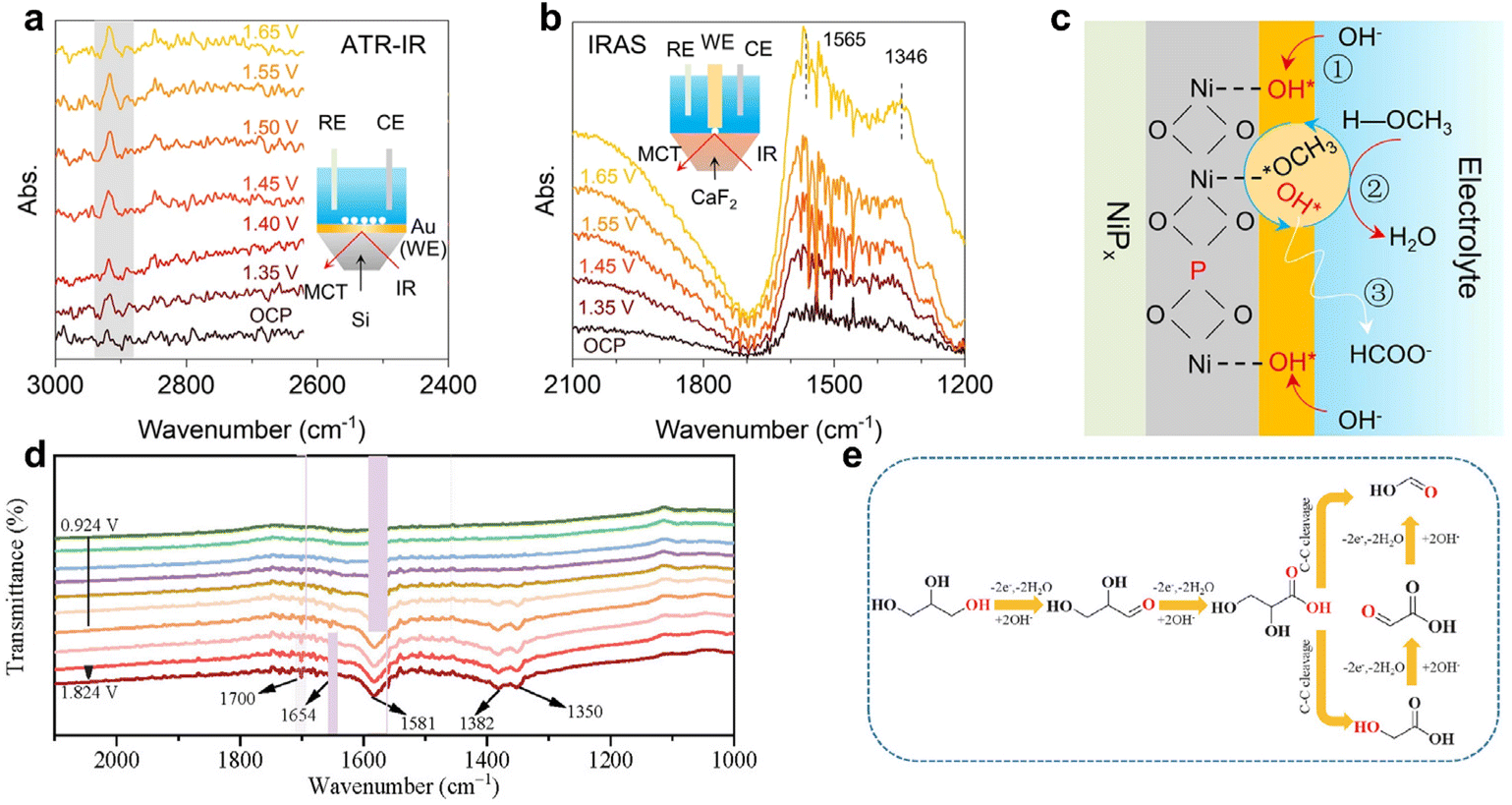 | ||
| Fig. 13 (a and b) In situ ATR-IR and IRAS spectra on the NiPx-R surface for the MOR process at various potentials. (c) Schematic illustration of the MOR mechanism on the NiPx-R surface. (a–c) Reproduced with permission from ref.141 Copyright 2022 Springer Nature. (d) In situ ATR-FTIR spectra for GOR on the CuCoN0.6/CP surface. (e) Proposed reaction pathway for producing formate from GOR on CuCoN0.6/CP in an alkaline medium. (d and e) Reproduced with permission from ref. 142. Copyright 2023 ELSEVIER. | ||
In another study by Shi et al.,142in situ ATR-IR spectroscopy was utilized to investigate the GOR pathways on CuCoN0.6/CP. ATR-FTIR spectra at different potentials (ranging from 0.924 to 1.824 V) were obtained to monitor reaction intermediates (Fig. 13d). Key observations included a downward peak at 1700 cm−1, signifying the formation of carbonyl group-containing species like aldehydes and carboxylic acids, indicative of initial glycerol oxidation. The persistent peak at 1654 cm−1 across all potentials, attributed to OH bending vibration from the electrolyte, suggested glycerate formation. Additionally, the intensifying bands at 1382 and 1581 cm−1 with increasing potential indicated further conversion of glycerol to carboxylic acids. These ATR-IR findings, combined with NMR results, led to the proposed GOR pathway on CuCoN0.6/CP (Fig. 13e). This pathway starts with glycerol converting to glyceraldehyde, then glyceric acid, followed by the oxidation of glyceric acid (first C–C cleavage) to form formate and glycolic acid. Through the second C–C cleavage, glycolic acid yields two formate molecules, allowing one glycerol molecule to produce up to three formate molecules, thereby maximizing carbon atom utilization.
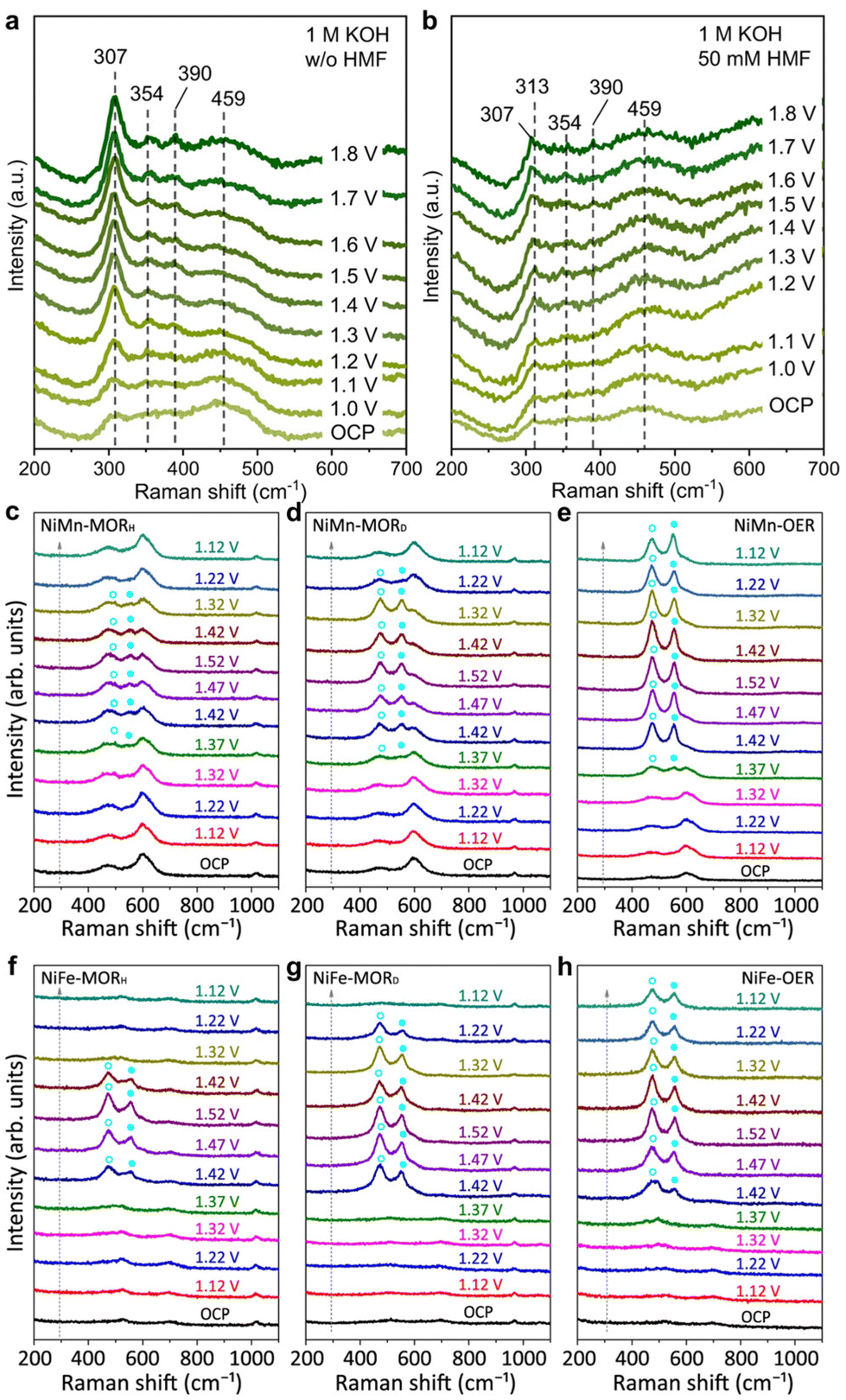 | ||
| Fig. 14 In situ Raman spectra of InOOH-Ov during (a) OER and (b) HMFOR processes. (a and b) Reproduced with permission from ref. 96. Copyright 2023 Springer Nature. Along with the spectra of (c) NiMn and (d) NiFe-LDHs under optimal conditions for MOR (the subscript H or D denotes CH3OH/H2O or CD3OD/D2O solution) and OER at various potentials. The blue circles highlight the characteristic bands of NiIII–OOH. (c–h) Reproduced with permission from ref. 21. Copyright 2023 Springer Nature. | ||
Feng et al.21 used in situ Raman spectroscopy to examine electrocatalyst structural evolution during MOR. They observed three Raman bands at 468, 533, and 600 cm−1 under open circuit potential (OCP), attributed to NiII–O and MnIII–O. During OER, characteristic bands at 473 and 551 cm−1 emerged at 1.37 V vs. RHE, indicating NiIII–O in NiIII–OOH (Fig. 14e). These bands were prominent between 1.42–1.52 V vs. RHE, suggesting NiIII–OOH stability as the source of OER. In the MOR process, NiIII–O bands also appeared at 1.37 V vs. RHE but were less pronounced compared to the original NiII–O bands within 1.37–1.52 V vs. RHE (Fig. 14c). The NiIII–O bands diminished and vanished when the potential decreased from 1.52 to 1.12 V vs. RHE. Conversely, the MnIII–O peak at 600 cm−1 remained noticeable throughout the potential sweep, indicating Mn ions in NiMn-LDH did not participate in redox transitions, while Ni ions did. Further confirmation of the transition from NiII–(OH)2 to NiIII–OOH was obtained using in situ Raman spectroscopy in deuterium media, showing clearer NiIII–O bands between 1.42–1.52–1.32 V vs. RHE (Fig. 14d). This suggests the methanol-induced reduction of NiIII–OOH was impeded due to stronger O–D or C–D bonds with the adsorbate. Similar outcomes were observed with NiFe-LDH as the anode catalyst (Fig. 14f–h). These in situ Raman spectroscopy findings indicate the transient and limited formation of NiIII–OOH in NiM-LDHs under optimal MOR conditions, likely as the origin of MOR catalysis.
Duan et al.130 explored the evolution of a high valent Cu mediated region in the electrochemical oxidation of glucose on CuO using in situ Cu K edge XANES. The absorption edge energies for CuO–KOH–OCP, CuO–KOH-1.0 V, and CuO–KOH-1.5 V were found between the reference states of Cu2O and CuO, suggesting a positively charged Cuδ+ state (1 < δ < 2) in these samples (Fig. 15a). This positive charge might originate from oxygen vacancies created during synthesis. Notably, even at a high potential of 1.5 V vs. RHE, the valence state of Cu in CuO–KOH-1.5 V was lower than the CuO reference. This is attributed to the formation of CuOOH on CuO's surface, with most of the remaining Cu being defective CuO. These findings, combined with other results, confirmed that the presence of Cu3+ species (CuOOH) was due to surface reconstruction in the Cu3+ mediated region, not complete reconstruction. Additionally, introducing 0.1 M glucose into the electrolyte decreased the adsorption energy, indicating a reduced valence state of Cu in the samples (Fig. 15b). This reduction was further supported by Cu K edge FT-EXAFS spectra of the Cu2+ (Fig. 15c) and Cu3+ (Fig. 15d) regions, confirming the reduction of Cu(OH)2 and CuOOH through interaction with glucose. The observed decrease in Cu oxidation state was attributed to the formation of lower-valent Cu species during glucose oxidation, resulting from the reduction of Cu(OH)2 and CuOOH.
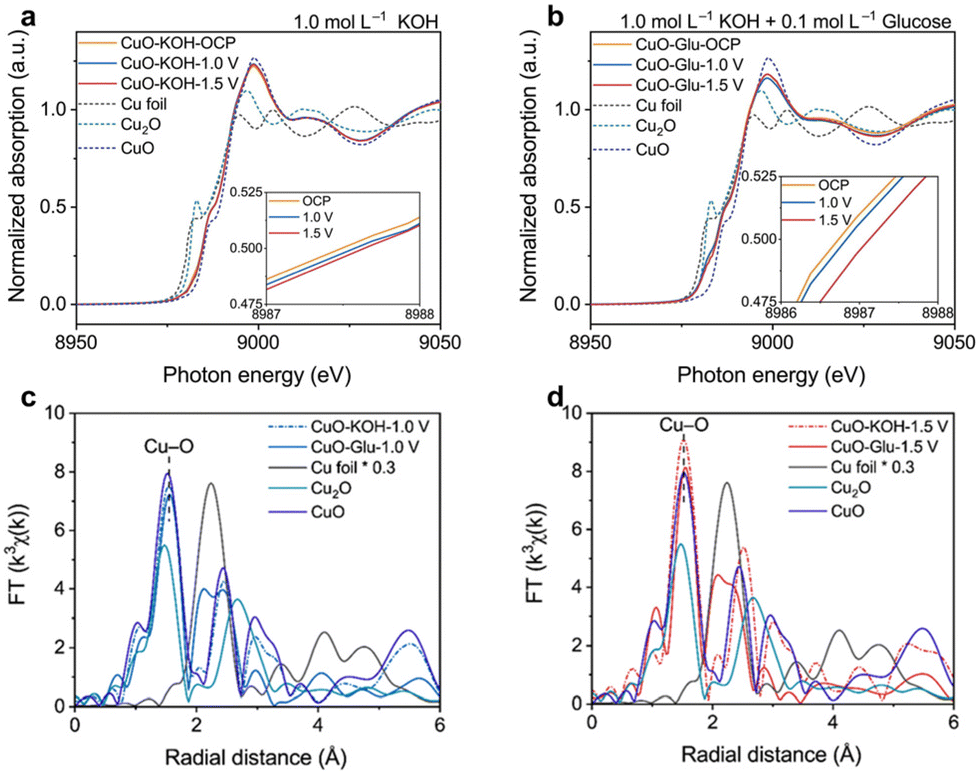 | ||
| Fig. 15 In situ XANES of CuO/CC in electrolytes of (a) 1.0 mol L−1 KOH + 10 mmol L−1 glucose and (b) 1.0 mol L−1 KOH. In situ FT-EXAFS spectra of (c) CuO–KOH-1.0 V, CuO–Glu-1.0 V and (d) CuO–KOH-1.5 V, CuO–Glu-1.5 V. Reproduced with permission from ref. 130. Copyright 2023 ELSEVIER. | ||
These findings underscore the significance of in situ XAS spectroscopy in elucidating the dynamic alterations of target elements and their electronic structures during electrochemical reactions, offering valuable insights into catalyst behavior and reaction mechanisms.
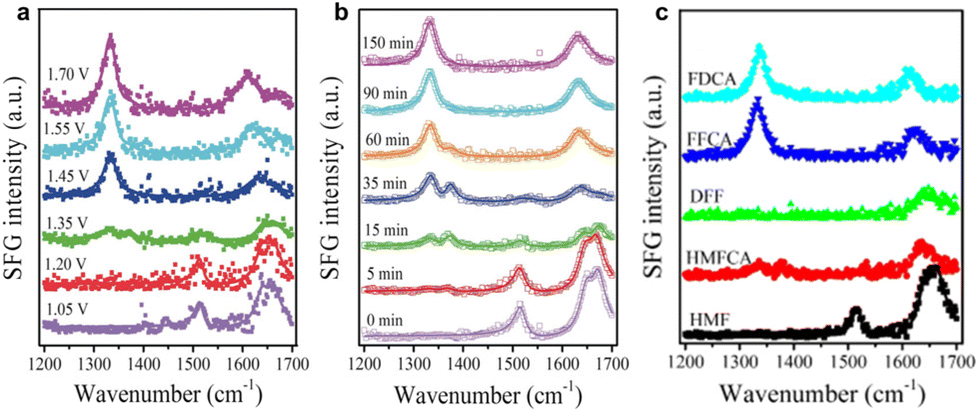 | ||
| Fig. 16 In situ SFG spectra (a) after operating at different potentials for 90 min and (b) at different times during the CA process at a potential of 1.45 V vs. RHE. (c) Standard SFG spectra of pure chemicals (FDCA, FFCA, DFF, HMFCA, and HMF). Reproduced with permission from ref. 145. Copyright 2019 Wiley-VCH. | ||
Zou et al.146 utilized in situ electrochemical impedance spectroscopy (EIS) to examine the electrochemical behavior of CuO and CuO–PdO interfaces during HMFOR catalysis. Inflection points (marked by an orange circle) in the low-frequency region of Bode plots at 1.5 V vs. RHE indicated OER occurrence at CuO and CuO–PdO interfaces (Fig. 17a and b). CuO exhibited a higher phase angle in the high-frequency region than CuO–PdO, suggesting slower reaction kinetics. During HMFOR, inflection points for CuO and CuO–PdO emerged at lower potentials of 1.2 V and 1.1 V vs. RHE, respectively (Fig. 17c and d). This indicated that the CuO–PdO interface required a lower oxidation potential for HMFOR compared to CuO and displayed faster reaction kinetics. CuO–PdO also showed lower charge transfer resistance and a faster reaction rate (Fig. 17e). These analyses conclude that the CuO–PdO interface enhances HMF initial adsorption and accelerates reaction kinetics (Fig. 17f), exhibiting superior catalytic performance towards HMFOR with increased activity and rate.
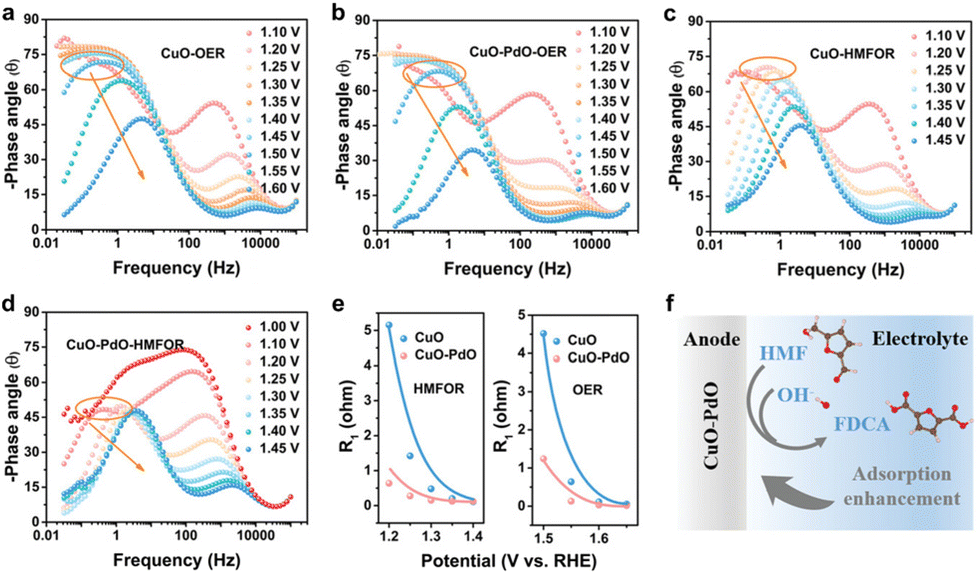 | ||
| Fig. 17 In situ analysis for CuO and CuO–PdO interfaces. (a–d) Bode plots of CuO and CuO–PdO during OER and HMFOR. (e) Fitting R1 values of CuO and CuO–PdO during OER and HMFOR. (f) Diagram of the initial reaction over CuO–PdO interface in the inner Helmholtz layer. Reproduced with permission from ref.146 Copyright 2022 Wiley-VCH. | ||
3.2 DFT calculations
Understanding the adsorption and desorption behavior of alcohol molecules and OHad is crucial for designing AOR electrocatalysts.50 DFT calculations are instrumental in exploring adsorption behavior on electrocatalyst interfaces, providing theoretical insights for electrocatalyst design. Peng et al.34 engineered the NiO/Fe2O3 electrocatalyst for MOR, calculating methanol's adsorption energy using DFT (Fig. 18a). Comparisons between the single NiO interface and NiO/Fe2O3 interface revealed lower adsorption energy for methanol on NiO/Fe2O3, suggesting that electronic interaction between the two metal oxides optimizes alcohol molecule adsorption and improves AOR reaction kinetics. Additionally, Zou and co-workers147 applied DFT to analyze the synergetic relationship of Pt and Ni in understanding HMF and OHad adsorption during HMFOR. The calculated adsorption energies of HMF on Pt, Pt26Ni74 nanowires (NWs), and NiO indicated strong adsorption on Pt, moderate on Pt26Ni74 NWs, and weak on NiO (Fig. 18b). Following the Sabatier principle, Pt26Ni74 NWs demonstrated optimal HMF adsorption, as overly strong adsorption can lead to active site poisoning, while weak adsorption may reduce catalytic activity. Moreover, OHad adsorption energies on these interfaces showed superior OHad adsorption on Pt26Ni74 NWs due to the Pt–O–Ni bond interaction (Fig. 18c). The HMFOR activity is thus enhanced synergistically, with Ni modulating Pt's d-band to alter HMF adsorption, while Pt facilitates OHad adsorption on Ni. These findings emphasize the importance of DFT calculations in understanding and optimizing molecular adsorption behavior at electrocatalyst interfaces for efficient AOR.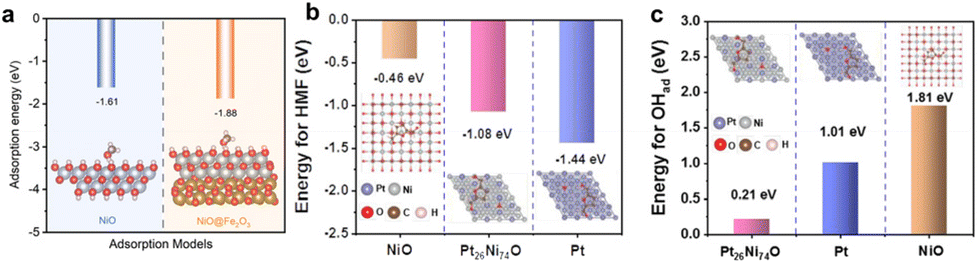 | ||
| Fig. 18 (a) Calculated absorption energy of methanol on Ni (111) and NiO/Fe2O3. (a) Reproduced with permission from ref. 34. Copyright 2023 Royal Society of Chemistry. (b and c) Calculated absorption energy of HMF and OHad on Pt, NiO, and Pt26Ni74 NWs. (b, c) Reproduced with permission from ref. 147. Copyright 2022 American Chemical Society. | ||
Another vital aspect of DFT calculations is the exploration of the Gibbs free energy of a reaction. Chen et al.17 investigated the electrocatalyst NiCo hydroxide for GOR efficiency. To elucidate the high GOR efficiency in NiCo hydroxide, DFT calculations were performed to unveil the GOR mechanism on Ni, Co, and NiCo hydroxide. Fig. 19a shows the (de)intercalation of protons and oxygen anions in hydroxide as actively involved in elementary reaction steps, including initial electrochemical-driven deintercalation of protons from the electrocatalyst lattice (IS → 0) and deintercalation of oxygen anions during desorption of reaction products (3 → A1 → A2, 7A → 7B, B1 → B2, and 9B → FS). The first dehydrogenation process of adsorbed intermediates (1 → 2, 2 → 3, 4 → 5) was spontaneous, with NiCo hydroxide exhibiting the most negative Gibbs free energy changes compared to Ni and Co hydroxide (Fig. 19b). In the desorption steps (3 → A1 → A2, 7A → 7B, B1 → B2, and 9B → FS), Co hydroxide showed the lowest energy barrier, facilitating product desorption from the catalyst's surface and resulting in diverse products, as confirmed by electrochemical tests. For the 1st C–C bond cleavage steps, similar energy barriers were observed among these catalysts, but NiCo exhibited the lowest energy barrier in the 2nd C–C cleavage step, expediting formic acid formation as GOR's final main product. This aligns with the high efficiency and selectivity of formate production in electrochemical tests from a theoretical perspective.
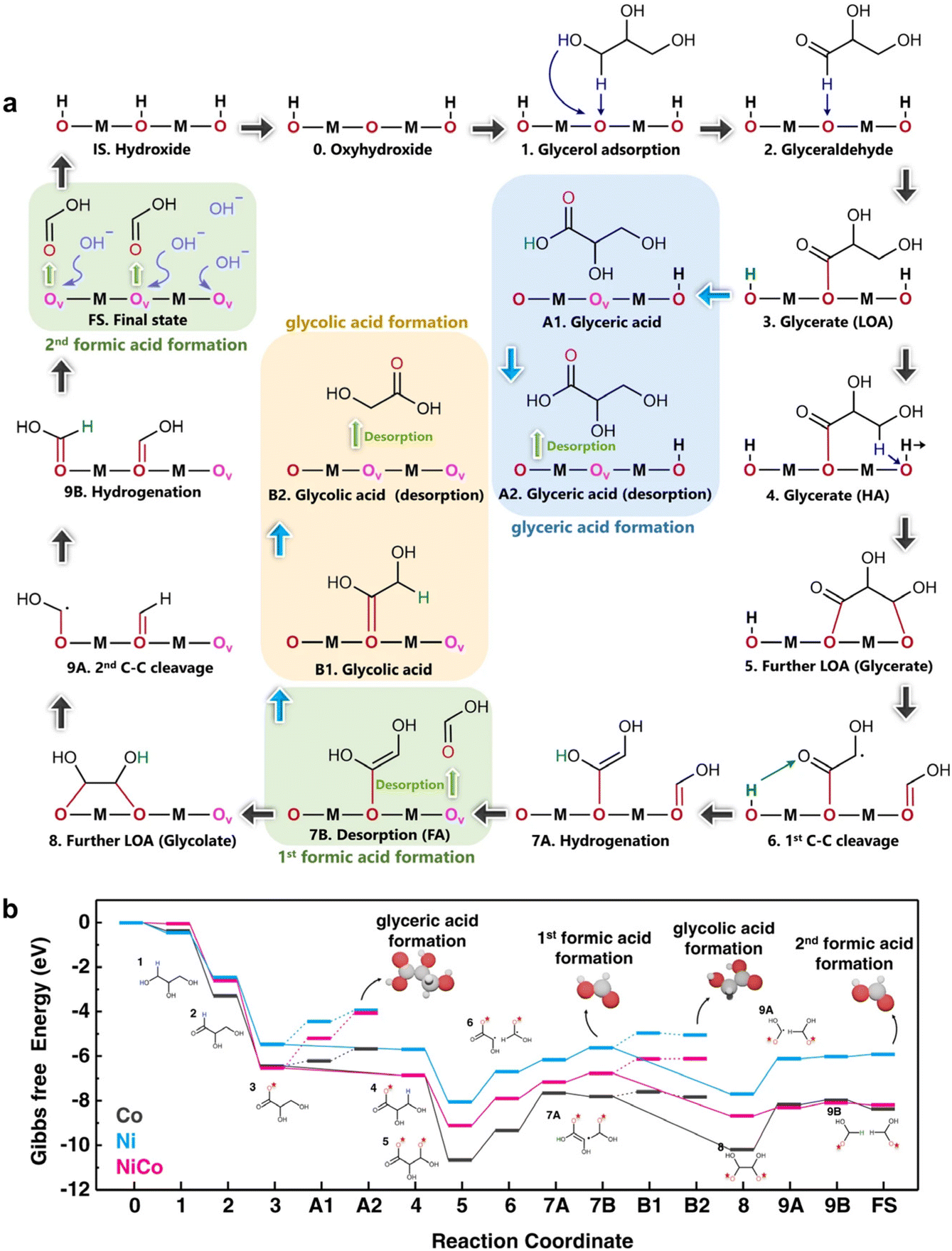 | ||
| Fig. 19 (a) Schematic illustration of the GOR pathway (b) calculated Gibbs free energy diagrams for GOR on Ni, NiCo, and Co hydroxide. Reproduced with permission from ref. 17. Copyright 2022 Springer Nature. | ||
4. Conclusion and prospect
Recent years have seen considerable interest in the electrochemical conversion of biomass-derived alcohols, driven by the diverse sources of substrates, potential for high-value added products, and milder reaction conditions compared to traditional production methods. In AOR electrocatalysis, advanced non-noble electrocatalysts have been developed and applied across various AOR systems, including monohydric alcohols and polyols oxidation. These non-noble electrocatalysts demonstrate high AOR activity and economic viability compared to noble metal catalysts, which is crucial for transitioning from lab-scale production to practical applications.Understanding AOR mechanisms is vital for designing efficient electrocatalysts. in situ characterization techniques have significantly contributed to this understanding by allowing researchers to observe the structural evolution of electrocatalysts during AOR. This aids in identifying real active sites and reaction intermediates, offering insights for optimizing AOR efficiency. Additionally, theoretical calculations like DFT are employed to study reactant adsorption and desorption properties and determine AOR reaction Gibbs free energy. These calculations guide electrocatalyst design and optimization and aid in screening catalytic materials before experimentation, conserving time and resources. The synergy of advanced electrocatalysts, in situ characterization techniques, and theoretical calculations significantly advances the development and optimization of electrochemical methods for biomass-derived alcohol conversion.
While significant progress has been made in the field, several challenges persist: (I) the stability of non-noble metal catalysts for AOR remains inadequate. (II) Relative low selectivity for incomplete oxidation products. (III) Transition metal-based catalysts are mainly focused on the electrooxidation of simple alcohols and lack attention on complex alcohols such as asymmetric alcohols. (IV) Insufficient in situ cells can negatively impact test results, affecting the study of reaction mechanisms.
In order to address these challenges, the following aspects should be considered: (I) non-noble metal catalysts offer cost advantages over noble metal electrocatalysts, but their stability for AOR is still lacking. While many studies have focused on enhancing electrocatalyst activity, there has been relatively less emphasis on strategies to improve catalyst stability. Therefore, it is crucial to dedicate more effort to exploring catalyst stability, as it plays a vital role in determining their practical applicability. (II) While most electrocatalysts are typically designed for carboxylic acid product synthesis, there is significant value in incomplete oxidation, which can lead to the production of ketones or aldehydes. It is important to concentrate efforts on adjusting the selectivity of incomplete oxidation products. (III) The majority of research in this field has concentrated on the electrooxidation of simple alcohols, rather than more complex alcohols like asymmetric alcohols. However, the oxidation products of these complex alcohols, such as chiral compounds, hold great importance in various areas such as drug synthesis, materials production, and other useful applications. Utilizing transition metal-based catalysts for the electrooxidation of asymmetric alcohols can provide a green and sustainable method for producing chiral compounds. (IV) Optimal design of in situ reaction cells is crucial for obtaining valuable insights into the reaction process, including tracking intermediates and identifying genuine active sites. Enhancing the design of in situ reaction cells is essential to improve signal quality. For example, in certain in situ Raman spectroscopic measurements, a thick electrolyte layer between the objective and the target catalyst surface can directly impact signal quality. It is recommended to incorporate an adjustable-height transmission window to reduce the liquid layer thickness or use an immersion lens to enhance signal quality. Additionally, bubbles generated during high potential or high current density conditions from OER can disrupt signal quality, highlighting the importance of researching methods to eliminate these bubbles.
Author contributions
Yuguo Zhao: conceptualization, investigation, visualization, writing – original draft, and writing – review and editing. Emma M. Björk: investigation, supervision and writing – review and editing. Yong Yan: investigation, funding acquisition, supervision and writing – review and editing. Peter Schaaf: supervision and writing – review and editing. Dong Wang: investigation, supervision and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was funded by the National Natural Science Foundation of China (No. 22278012) and the Swedish Energy Agency (No. 2022-00909).References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- P. Prabhu, Y. Wan and J.-M. Lee, Matter, 2020, 3, 1162–1177 CrossRef.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- R. Ge, J. Li and H. Duan, Sci. China Mater., 2022, 65, 3273–3301 CrossRef CAS.
- C. Tang, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 19572–19590 CrossRef CAS PubMed.
- H. C. Fu, X. H. Chen, B. Yang, Y. H. Luo, T. Li, X. H. Wang, Q. Zhang, X. L. Li, N. B. Li and H. Q. Luo, Appl. Catal., B, 2023, 332, 122739 CrossRef CAS.
- S. Liu, H. Li, J. Zhong, K. Xu, G. Wu, C. Liu, B. Zhou, Y. Yan, L. Li, W. Cha, K. Chang, Y. Y. Li and J. Lu, Sci. Adv., 8, eadd6421.
- Z. Liang, N. Kong, C. Yang, W. Zhang, H. Zheng, H. Lin and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 12759–12764 CrossRef CAS PubMed.
- Y. Li, F.-M. Li, X.-Y. Meng, S.-N. Li, J.-H. Zeng and Y. Chen, ACS Catal., 2018, 8, 1913–1920 CrossRef CAS.
- X. Wang, D. Wu, S. Liu, J. Zhang, X.-Z. Fu and J.-L. Luo, Nano-Micro Lett., 2021, 13, 125 CrossRef CAS PubMed.
- H. Y. Zhou, Y. B. Qu, J. C. Li, Z. L. Wang, C. C. Yang and Q. Jiang, Appl. Catal., B, 2022, 305, 121023 CrossRef CAS.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS PubMed.
- T. Dou, J. He, S. Diao, Y. Wang, X. Zhao, F. Zhang and X. Lei, J. Energy Chem., 2023, 82, 497–506 CrossRef CAS.
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS PubMed.
- F. W. S. Lucas, R. G. Grim, S. A. Tacey, C. A. Downes, J. Hasse, A. M. Roman, C. A. Farberow, J. A. Schaidle and A. Holewinski, ACS Energy Lett., 2021, 6, 1205–1270 CrossRef CAS.
- G. Bharath and F. Banat, ACS Appl. Mater. Interfaces, 2021, 13, 24643–24653 CrossRef CAS PubMed.
- Z. He, J. Hwang, Z. Gong, M. Zhou, N. Zhang, X. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 3777 CrossRef CAS PubMed.
- M. Yang, Z. Yuan, R. Peng, S. Wang and Y. Zou, Energy Environ. Mater., 2022, 5, 1117–1138 CrossRef CAS.
- C.-J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed.
- S. Xu, X. Ruan, M. Ganesan, J. Wu, S. K. Ravi and X. Cui, Adv. Funct. Mater., 2024, 2313309 CrossRef.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Nat. Commun., 2023, 14, 1686 CrossRef CAS PubMed.
- H. Huang, C. Yu, X. Han, H. Huang, Q. Wei, W. Guo, Z. Wang and J. Qiu, Energy Environ. Sci., 2020, 13, 4990–4999 RSC.
- Y. Yang and T. Mu, Green Chem., 2021, 23, 4228–4254 RSC.
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed.
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- L. Chen and J. Shi, J. Mater. Chem. A, 2018, 6, 13538–13548 RSC.
- B. Rausch, M. D. Symes and L. Cronin, J. Am. Chem. Soc., 2013, 135, 13656–13659 CrossRef CAS PubMed.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- K.-C. Cheung, W.-L. Wong, D.-L. Ma, T.-S. Lai and K.-Y. Wong, Coord. Chem. Rev., 2007, 251, 2367–2385 CrossRef CAS.
- R. Li, K. Xiang, Z. Peng, Y. Zou and S. Wang, Adv. Energy Mater., 2021, 11, 2102292 CrossRef CAS.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef PubMed.
- B. Zhao, J. Liu, C. Xu, R. Feng, P. Sui, L. Wang, J. Zhang, J.-L. Luo and X.-Z. Fu, Adv. Funct. Mater., 2021, 31, 2008812 CrossRef CAS.
- Y. Hao, D. Yu, S. Zhu, C.-H. Kuo, Y.-M. Chang, L. Wang, H.-Y. Chen, M. Shao and S. Peng, Energy Environ. Sci., 2023, 16, 1100–1110 RSC.
- X. Peng, S. Xie, X. Wang, C. Pi, Z. Liu, B. Gao, L. Hu, W. Xiao and P. K. Chu, J. Mater. Chem. A, 2022, 10, 20761–20769 RSC.
- K. Zhang, Y. Deng, Y. Wu, L. Wang and L. Yan, J. Colloid Interface Sci., 2023, 647, 246–254 CrossRef CAS PubMed.
- S. Jiang, T. Xiao, C. Xu, S. Wang, H.-Q. Peng, W. Zhang, B. Liu and Y.-F. Song, Small, 2023, 19, 2208027 CrossRef CAS PubMed.
- S. Bhat, J. Banday and M. Wahid, Energy Fuels, 2023, 37, 6012–6024 CrossRef CAS.
- K. Zhang, Y. Han, J. Qiu, X. Ding, Y. Deng, Y. Wu, G. Zhang and L. Yan, J. Colloid Interface Sci., 2023, 630, 570–579 CrossRef CAS PubMed.
- M. Khan, M. I. Abdullah, A. Samad, Z. Shao, T. Mushiana, A. Akhtar, A. Hameed, N. Zhang, U. Schwingenschlögl and M. Ma, Small, 2023, 19, 2205499 CrossRef CAS PubMed.
- D. Wu, W. Zhang and D. Cheng, ACS Appl. Mater. Interfaces, 2017, 9, 19843–19851 CrossRef CAS PubMed.
- H. Wang, L. Cui, S. Yin, H. Yu, K. Deng, Y. Xu, X. Wang, Z. Wang and L. Wang, J. Mater. Chem. A, 2022, 10, 18889–18894 RSC.
- X.-T. Gao, Y.-F. Wang, L. Fu, R.-X. Zhang, R.-M. Li, Z.-H. Gao, Z.-F. Yan, Y.-M. Liu, W. Huang, L. Liu and Z.-J. Zuo, Int. J. Hydrogen Energy, 2023, 48, 32408–32419 CrossRef CAS.
- J. Wang, B. Zhang, W. Guo, L. Wang, J. Chen, H. Pan and W. Sun, Adv. Mater., 2023, 35, 2211099 CrossRef CAS PubMed.
- H. Xie, S. Chen, J. Liang, T. Wang, Z. Hou, H.-L. Wang, G. Chai and Q. Li, Adv. Funct. Mater., 2021, 31, 2100883 CrossRef CAS.
- F. Galvanin, M. Sankar, S. Cattaneo, D. Bethell, V. Dua, G. J. Hutchings and A. Gavriilidis, Chem. Eng. J., 2018, 342, 196–210 CrossRef CAS.
- W. Gao, Y. Song, W. Jiao and Y. Liu, J. Taiwan Inst. Chem. Eng., 2019, 103, 1–6 CrossRef CAS.
- J. Li, R. Wei, X. Wang, Y. Zuo, X. Han, J. Arbiol, J. Llorca, Y. Yang, A. Cabot and C. Cui, Angew. Chem., Int. Ed., 2020, 59, 20826–20830 CrossRef CAS PubMed.
- J. Li, C. Xing, Y. Zhang, T. Zhang, M. C. Spadaro, Q. Wu, Y. Yi, S. He, J. Llorca, J. Arbiol, A. Cabot and C. Cui, Small, 2021, 17, 2006623 CrossRef CAS PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- W. Wang, Y.-B. Zhu, Q. Wen, Y. Wang, J. Xia, C. Li, M.-W. Chen, Y. Liu, H. Li, H.-A. Wu and T. Zhai, Adv. Mater., 2019, 31, 1900528 CrossRef PubMed.
- Y. Zhang, W. Zhu, J. Fang, Z. Xu, Y. Xue, D. Liu, R. Sui, Q. Lv, X. Liu, Y. Wang, W. Chen and Z. Zhuang, Nano Res., 2022, 15, 2987–2993 CrossRef CAS.
- A. Shekhawat, R. Samanta, S. Panigrahy and S. Barman, ACS Appl. Energy Mater., 2023, 6, 3135–3146 CrossRef CAS.
- H. Wang, A. Guan, J. Zhang, Y. Mi, S. Li, T. Yuan, C. Jing, L. Zhang, L. Zhang and G. Zheng, Chin. J. Catal., 2022, 43, 1478–1484 CrossRef CAS.
- J. Li, X. Wang, C. Xing, L. Li, S. Mu, X. Han, R. He, Z. Liang, P. Martinez, Y. Yi, Q. Wu, H. Pan, J. Arbiol, C. Cui, Y. Zhang and A. Cabot, Chem. Eng. J., 2022, 440, 135817 CrossRef CAS.
- J. Wan, X. Mu, Y. Jin, J. Zhu, Y. Xiong, T. Li and R. Li, Green Chem., 2022, 24, 4870–4876 RSC.
- X. Chen, X. Zhong, B. Yuan, S. Li, Y. Gu, Q. Zhang, G. Zhuang, X. Li, S. Deng and J.-g. Wang, Green Chem., 2019, 21, 578–588 RSC.
- R. Li, P. Kuang, L. Wang, H. Tang and J. Yu, Chem. Eng. J., 2022, 431, 134137 CrossRef CAS.
- L. Ming, X.-Y. Wu, S.-S. Wang, W. Wu and C.-Z. Lu, Green Chem., 2021, 23, 7825–7830 RSC.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- X. Song, X. Liu, H. Wang, Y. Guo and Y. Wang, Ind. Eng. Chem. Res., 2020, 59, 17348–17356 CrossRef CAS.
- H. Wang, C. Li, J. An, Y. Zhuang and S. Tao, J. Mater. Chem. A, 2021, 9, 18421–18430 RSC.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- B. Zhang, H. Fu and T. Mu, Green Chem., 2022, 24, 877–884 RSC.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. J. J. Timmer, M. S. G. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS PubMed.
- L. Gao, Z. Liu, J. Ma, L. Zhong, Z. Song, J. Xu, S. Gan, D. Han and L. Niu, Appl. Catal., B, 2020, 261, 118235 CrossRef.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- R. Ge, Y. Wang, Z. Li, M. Xu, S.-M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200211 CrossRef CAS PubMed.
- L. Zeng, Y. Chen, M. Sun, Q. Huang, K. Sun, J. Ma, J. Li, H. Tan, M. Li, Y. Pan, Y. Liu, M. Luo, B. Huang and S. Guo, J. Am. Chem. Soc., 2023, 145, 17577–17587 CrossRef CAS PubMed.
- W. Jia, B. Liu, R. Gong, X. Bian, S. Du, S. Ma, Z. Song, Z. Ren and Z. Chen, Small, 2023, 19, 2302025 CrossRef CAS PubMed.
- Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Appl. Catal., B, 2022, 312, 121400 CrossRef CAS.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. C. M. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300094 CrossRef CAS PubMed.
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, 2300841 CrossRef CAS PubMed.
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS.
- L. Dong, G.-R. Chang, Y. Feng, X.-Z. Yao and X.-Y. Yu, Rare Met., 2022, 41, 1583–1594 CrossRef CAS.
- X. Yu, R. B. Araujo, Z. Qiu, E. Campos dos Santos, A. Anil, A. Cornell, L. G. M. Pettersson and M. Johnsson, Adv. Energy Mater., 2022, 12, 2103750 CrossRef CAS.
- Y. Wang, Y.-Q. Zhu, Z. Xie, S.-M. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, ACS Catal., 2022, 12, 12432–12443 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, Z. Zhang, L. Yi, K. Chen, X. Liu and Z. Wen, J. Energy Chem., 2022, 72, 424–431 CrossRef CAS.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 21464–21472 CrossRef CAS PubMed.
- Q. Qian, X. He, Z. Li, Y. Chen, Y. Feng, M. Cheng, H. Zhang, W. Wang, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 CrossRef CAS PubMed.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- D. Li, Y. Huang, Z. Li, L. Zhong, C. Liu and X. Peng, Chem. Eng. J., 2022, 430, 132783 CrossRef CAS.
- Y. Zhang, B. Zhou, Z. Wei, W. Zhou, D. Wang, J. Tian, T. Wang, S. Zhao, J. Liu, L. Tao and S. Wang, Adv. Mater., 2021, 33, 2104791 CrossRef CAS PubMed.
- J. Zheng, X. Chen, X. Zhong, S. Li, T. Liu, G. Zhuang, X. Li, S. Deng, D. Mei and J.-G. Wang, Adv. Funct. Mater., 2017, 27, 1704169 CrossRef.
- G. Han, Y.-H. Jin, R. A. Burgess, N. E. Dickenson, X.-M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CrossRef CAS PubMed.
- Y. Gao, L. Ge, H. Xu, K. Davey, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 13, 11204–11231 CrossRef CAS.
- G. Zhao, G. Hai, P. Zhou, Z. Liu, Y. Zhang, B. Peng, W. Xia, X. Huang and G. Wang, Adv. Funct. Mater., 2023, 33, 2213170 CrossRef CAS.
- G. Chen, X. Li and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202209014 CrossRef CAS PubMed.
- C. Chen, Z. Zhou, J. Liu, B. Zhu, H. Hu, Y. Yang, G. Chen, M. Gao and J. Zhang, Appl. Catal., B, 2022, 307, 121209 CrossRef CAS.
- H. Zhou, Y. Ren, B. Yao, Z. Li, M. Xu, L. Ma, X. Kong, L. Zheng, M. Shao and H. Duan, Nat. Commun., 2023, 14, 5621 CrossRef CAS PubMed.
- R. Zhong, P. Wu, Q. Wang, X. Zhang, L. Du, Y. Liu, H. Yang, M. Gu, Z. C. Zhang, L. Huang and S. Ye, Green Chem., 2023, 25, 4674–4684 RSC.
- K. A. P. Payne, S. A. Marshall, K. Fisher, M. J. Cliff, D. M. Cannas, C. Yan, D. J. Heyes, D. A. Parker, I. Larrosa and D. Leys, ACS Catal., 2019, 9, 2854–2865 CrossRef CAS PubMed.
- D. Xiao, X. Bao, D. Dai, Y. Gao, S. Si, Z. Wang, Y. Liu, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Adv. Mater., 2023, 35, 2304133 CrossRef CAS PubMed.
- F. Ye, S. Zhang, Q. Cheng, Y. Long, D. Liu, R. Paul, Y. Fang, Y. Su, L. Qu, L. Dai and C. Hu, Nat. Commun., 2023, 14, 2040 CrossRef CAS PubMed.
- S. Fan, B. Zhu, Y. Zhong, S. Shi, J. Zhang and C. Chen, Chem. Eng. J., 2023, 474, 145905 CrossRef CAS.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778–3786 RSC.
- D. Bonincontro, A. Lolli, A. Villa, L. Prati, N. Dimitratos, G. M. Veith, L. E. Chinchilla, G. A. Botton, F. Cavani and S. Albonetti, Green Chem., 2019, 21, 4090–4099 RSC.
- S. Fan, B. Zhu, X. Yu, Y. Gao, W. Xie, Y. Yang, J. Zhang and C. Chen, J. Energy Chem., 2024, 92, 1–7 CrossRef CAS.
- D. Chen, Y. Ding, X. Cao, L. Wang, H. Lee, G. Lin, W. Li, G. Ding and L. Sun, Angew. Chem., Int. Ed., 2023, 62, e202309478 CrossRef CAS PubMed.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- Y. Sun, J. Wang, Y. Qi, W. Li and C. Wang, Adv. Sci., 2022, 9, 2200957 CrossRef CAS PubMed.
- C. Liu, X.-R. Shi, K. Yue, P. Wang, K. Zhan, X. Wang, B. Y. Xia and Y. Yan, Adv. Mater., 2023, 35, 2211177 CrossRef CAS PubMed.
- R. Zhang, F. Gao, C. Yang, Y. Bian, G. Wang, K. Xue, J. Zhang, C. Wang and X. Gao, Mater. Today Nano, 2023, 23, 100373 CrossRef CAS.
- J. Wu, Z. Zhai, T. Yu, X. Wu, S. Huang, W. Cao, Y. Jiang, J. Pei and S. Yin, J. Energy Chem., 2023, 86, 480–489 CrossRef CAS.
- Z. Li, L. Huai, P. Hao, X. Zhao, Y. Wang, B. Zhang, C. Chen and J. Zhang, Chin. J. Catal., 2022, 43, 793–801 CrossRef CAS.
- B. Zhu, C. Chen, L. Huai, Z. Zhou, L. Wang and J. Zhang, Appl. Catal., B, 2021, 297, 120396 CrossRef CAS.
- J. Liu, B. Zhu, Y. Zhong, S. Fan, L. Huai, H. Hu, Y. Yang, J. Zhang and C. Chen, Chem. Eng. J., 2023, 472, 144877 CrossRef CAS.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2024, 34, 2306995 CrossRef CAS.
- D. Liu, J. C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef PubMed.
- Z. He, X. Ning, G. Yang, H. Wang, Y. Cao, F. Peng and H. Yu, Catal. Today, 2021, 365, 162–171 CrossRef CAS.
- S. Bagheri, N. M. Julkapli and W. A. Yehye, Renewable Sustainable Energy Rev., 2015, 41, 113–127 CrossRef CAS.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- Y. Li and F. Zaera, J. Catal., 2015, 326, 116–126 CrossRef CAS.
- H. Yan, S. Yao, W. Liang, S. Zhao, X. Jin, X. Feng, Y. Liu, X. Chen and C. Yang, J. Catal., 2020, 381, 248–260 CrossRef CAS.
- T. Zhan, W. Liu, J. Teng, C. Yue, D. Li, S. Wang and H. Tan, Chem. Commun., 2019, 55, 2620–2623 RSC.
- J. Cai, H. Ma, J. Zhang, Z. Du, Y. Huang, J. Gao and J. Xu, Chin. J. Catal., 2014, 35, 1653–1660 CrossRef CAS.
- L. Xin, Z. Zhang, Z. Wang and W. Li, ChemCatChem, 2012, 4, 1105–1114 CrossRef CAS.
- M. Sankar, N. Dimitratos, D. W. Knight, A. F. Carley, R. Tiruvalam, C. J. Kiely, D. Thomas and G. J. Hutchings, ChemSusChem, 2009, 2, 1145–1151 CrossRef CAS PubMed.
- Y. Zhan, W. Hou, G. Li, Y. Shen, Y. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2019, 7, 17559–17564 CrossRef CAS.
- K. Kong, D. Li, W. Ma, Q. Zhou, G. Tang and Z. Hou, Chin. J. Catal., 2019, 40, 534–542 CrossRef CAS.
- Y. Xiong, J. Dong, Z. Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C. R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS PubMed.
- Y.-B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- W. Deng, Q. Zhang and Y. Wang, Catal. Today, 2014, 234, 31–41 CrossRef CAS.
- P. Pal, R. Kumar and S. Banerjee, Chem. Eng. Process., 2016, 104, 160–171 CrossRef CAS.
- T. N. Smith, K. Hash, C.-L. Davey, H. Mills, H. Williams and D. E. Kiely, Carbohydr. Res., 2012, 350, 6–13 CrossRef CAS PubMed.
- Y.-Q. Zhu, H. Zhou, J. Dong, S.-M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li, M. Shao and H. Duan, Angew. Chem., Int. Ed., 2023, 62, e202219048 CrossRef CAS PubMed.
- Y. Wang, M. Xu, X. Wang, R. Ge, Y.-Q. Zhu, A.-Z. Li, H. Zhou, F. Chen, L. Zheng and H. Duan, Sci. Bull., 2023, 68, 2982–2992 CrossRef CAS PubMed.
- D. Zheng, J. Li, S. Ci, P. Cai, Y. Ding, M. Zhang and Z. Wen, Appl. Catal., B, 2020, 277, 119178 CrossRef CAS.
- L. Ostervold, S. I. Perez Bakovic, J. Hestekin and L. F. Greenlee, RSC Adv., 2021, 11, 31208–31218 RSC.
- M. P. J. M. van der Ham, E. van Keulen, M. T. M. Koper, A. A. Tashvigh and J. H. Bitter, Angew. Chem., Int. Ed., 2023, 62, e202306701 CrossRef CAS PubMed.
- Z. Song, T. Shen, Y. Hu, G. Liu, S. Bai, X. Sun, S.-M. Xu and Y.-F. Song, Nanoscale, 2023, 15, 11867–11874 RSC.
- Z. Li, Y. Yan, S.-M. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 147 CrossRef CAS PubMed.
- J.-T. Li, Z.-Y. Zhou, I. Broadwell and S.-G. Sun, Acc. Chem. Res., 2012, 45, 485–494 CrossRef CAS PubMed.
- Y. He, S. Liu, M. Wang, Q. Cheng, H. Ji, T. Qian and C. Yan, Energy Environ. Mater., 2022, 6, e12552 CrossRef.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- R. Mendelsohn, G. Mao and C. R. Flach, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 788–800 CrossRef CAS PubMed.
- T. Petit and L. Puskar, Diamond Relat. Mater., 2018, 89, 52–66 CrossRef CAS.
- S. Li, R. Ma, J. Hu, Z. Li, L. Liu, X. Wang, Y. Lu, G. E. Sterbinsky, S. Liu, L. Zheng, J. Liu, D. Liu and J. Wang, Nat. Commun., 2022, 13, 2916 CrossRef CAS PubMed.
- K. Shi, D. Si, X. Teng, L. Chen and J. Shi, Chin. J. Catal., 2023, 53, 143–152 CrossRef CAS.
- H.-Q. Chen, L. Zou, D.-Y. Wei, L.-L. Zheng, Y.-F. Wu, H. Zhang and J.-F. Li, Chin. J. Catal., 2022, 43, 33–46 CrossRef CAS.
- T. Zhao, Y. Wang, S. Karuturi, K. Catchpole, Q. Zhang and C. Zhao, Carbon Energy, 2020, 2, 582–613 CrossRef CAS.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- P. Zhou, X. Lv, S. Tao, J. Wu, H. Wang, X. Wei, T. Wang, B. Zhou, Y. Lu, T. Frauenheim, X. Fu, S. Wang and Y. Zou, Adv. Mater., 2022, 34, 2204089 CrossRef CAS PubMed.
- J. Wu, Z. Kong, Y. Li, Y. Lu, P. Zhou, H. Wang, L. Xu, S. Wang and Y. Zou, ACS Nano, 2022, 16, 21518–21526 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |