
The role of the terminal cysteine moiety in a metallopeptide mimicking the active site of the NiSOD enzyme†
Dóra
Bonczidai-Kelemen
ab,
Klaudia
Tóth
a,
István
Fábián
 a and
Norbert
Lihi
*a
a and
Norbert
Lihi
*a
aHUN-REN–UD Mechanisms of Complex Homogeneous and Heterogeneous Chemical Reactions Research Group, Department of Inorganic and Analytical Chemistry, Faculty of Science and Technology, University of Debrecen, H-4032 Debrecen, Hungary. E-mail: lihi.norbert@science.unideb.hu
bDoctoral School of Chemistry, University of Debrecen, Debrecen H-4032, Hungary
First published on 12th December 2023
Abstract
Superoxide dismutase (SOD) enzymes are pivotal in regulating oxidative stress. In order to model Ni containing SOD enzymes, the results of the thermodynamic, spectroscopic and SOD activity studies on the complexes formed between nickel(II) and a NiSOD related peptide, CysCysAspLeuProCysGlyValTyr–NH2 (wtCC), are reported. Cysteine was introduced to replace the first histidine residue in the amino acid sequence of the active site of the NiSOD enzyme. The novel peptide exhibits 3 times higher metal binding affinity compared to the native NiSOD fragment. This is due to the presence of the first cysteine in the coordination sphere of nickel(II). At physiological pH, the (NH2,S–,S–,S–) coordinated complex is the major species. This coordination mode is altered when one thiolate group is replaced by an amide nitrogen of the peptide backbone above pH 7.5. The nickel complexes of wtCC exhibit similar SOD activity to that of the complex formed with the active site fragment of the native NiSOD. The reaction between the complexes and the superoxide anion was studied by the sequential stopped-flow method. These studies revealed that the nickel(II) complex is always in excess over the nickel(III) complex during the dismutation process. However, the nickel(III) species is also involved in a relatively fast degradation process. This unambiguously proves that a protective mechanism must be operative in the NiSOD enzyme which prevents the oxidation of the sulfur atom of cysteine in the presence of O2−. The results provide new possibilities for the use of NiSOD mimics in bio- and industrial catalytic processes.
Introduction
Reactive oxygen species (ROS) form during the incomplete reduction of molecular oxygen and are essential byproducts of cellular respiration in aerobic organisms.1 Although ROS are cytotoxic, the cells require their presence at a specific concentration level since they are involved in different signaling processes.1–3 In order to regulate the concentration of ROS, biological systems accommodate several enzymes (catalases, peroxidases, superoxide dismutases and reductases) to convert them into harmless products.4 The absence of these enzymes leads to elevated ROS concentration levels, and oxidative stress occurs that triggers pathological episodes including inflammatory disease, neurodegenerative disorder or cancer.5–7 Consequently, the development of novel antioxidant systems capable of regulating the level of ROS has received considerable interest in recent years.The superoxide anion radical is one of the most toxic ROS, and its dismutation is catalyzed by superoxide dismutases (SODs).8,9 The decomposition leads to the formation of molecular oxygen and hydrogen peroxide, which degrades through different pathways, yielding harmful products.10 All SOD enzymes possess a redox-active metal ion cofactor,11 and the dismutation reaction is essentially based on the redox cycling of the metal center as shown in eqn (1) and (2).12
| Mred + O2˙− + 2H+ = Mox + H2O2 | (1) |
| Mox + O2˙− = Mred + O2 | (2) |
A recently isolated SOD enzyme contains nickel in the active center, which is expressed by the sodN gene and found in several soil bacteria and cyanobacteria.13–15 The NiSOD enzyme is a homohexamer and each nickel center is catalytically active. X-ray crystallography revealed that the active sites of the enzyme are located at the N-terminal part of the identical peptide chains where the nickel ions are in a unique coordination environment. In the reduced form of the enzyme, nickel(II) is coordinated via the terminal amino group, the first peptide nitrogen as well as the two thiolate groups of cysteine residues in a square-planar coordination environment. Oxidation of nickel(II) to nickel(III) results in the binding of the imidazole–N of the terminal histidine in the apical position, leading to the formation of a square–pyramidal coordination environment. This coordination motif exhibits a loop which is termed a NiSOD binding hook. During the dismutation cycle, NiSOD cycles between NiII and NiIII oxidation states and dismutation occurs through a proton-coupled electron-transfer process. The rate constant of dismutation approaches the diffusion-controlled limit. Earlier studies have revealed that at least six amino acid residues are required in the peptide sequence to reproduce the key spectroscopic and structural features as well as the catalytic activity of NiSOD enzymes.16
Since NiSODs exhibit a rather unusual active site, significant efforts have been made to understand the fundamental spectroscopic and catalytic features of these compounds. Accordingly, several Ni-based metallopeptides17–21 and low-molecular-weight synthetic nickel complexes22–25 have been studied. Our work focused on how the stability of NiII and NiIII complexes as well as the catalytic activity can be tuned by altering the primary coordination sphere of nickel. This may provide deeper insights into the mechanism of superoxide dismutation by NiSOD.
Our results and previous literature studies confirmed the essential role of cysteine residues in effective NiSOD mimics.20,26 By using a combination of pH-potentiometric and spectroscopic methods, it was confirmed that the cysteine in the secondary position of the peptide chain is crucial to induce spin pairing, leading to the formation of a square-planar nickel(II) complex, while the distant cysteine affects the redox potential of the NiII/NiIII redox couple.20 Incorporation of penicillamine into the NiSOD enzyme fragments yielded different spectroscopic and redox features. The in-depth analysis of the dismutation process clearly proved that the NiIII complex rapidly accumulates in this system.26 This is due to the presence of electron donating substituents of penicillamine. The NiIII complex is also involved in a fast self-degradation process, and thus the catalytic dismutation ceases. A plausible explanation of this phenomenon is that the bulky methyl substituents of penicillamine hinder the formation of a hydrogen bond network surrounding the catalytically active center and thus promote the degradation of the complex. Therefore, we have proposed an alternative or secondary role of the hydrogen bond network which can be responsible for the stabilization of the NiIII oxidation state against the redox degradation processes. However, not only the cysteinyl residues alter the catalytic activity of NiSOD models, but also the terminal histidine is important in the dismutation reaction. In this respect, contradictory conclusions were reached on the role of the axial ligands.27,28 It is obvious from the model systems and theoretical calculations that the terminal histidine plays an essential role in optimizing the rate of the disproportionation of the superoxide ion.29 Computational studies concluded that the imidazole moiety of histidine remains coordinated throughout the catalysis.30 Nevertheless, metallopeptides without the terminal histidine residues are capable of assisting the decomposition of the superoxide anion. So far, only weakly coordinating amino acids (glutamine, alanine, aspartic acid) have been introduced into the first position of NiSOD related peptides.29 These results demonstrated that the nickel complexes possess moderate SOD activity, but the lack of the axially coordinated ligand does not fully eliminate the SOD activity.
With the aim of designing novel NiSOD mimics and studying the role of axial coordination in the presence of a strongly coordinating amino acid, now we report through equilibrium, spectroscopic and SOD activity studies on a new Ni-containing metallopeptide. In this case, the terminal histidine of the NiSOD binding site was replaced by cysteine yielding a new peptide (CysCysAspLeuProCysGlyValTyr–NH2) denoted as wtCC throughout the article (Scheme 1). The fundamental thermodynamic, spectroscopic and catalytic features are compared with those of the native NiSOD enzyme fragment (wtNiSOD) and further NiSOD related peptides (Scheme S1†).
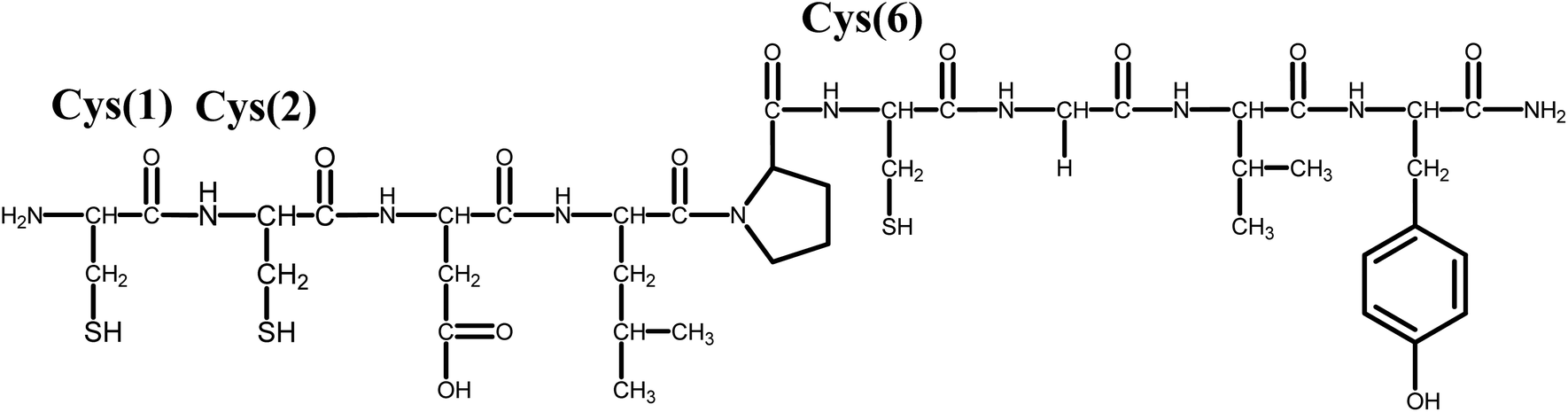 | ||
| Scheme 1 Structural formula of the NiSOD related peptide (wtCC) investigated in this work. | ||
The results contribute to the interpretation of the correlation between the catalytic activity and coordination chemical features of SOD enzymes and their mimics.
Experimental
Materials
The peptide, wtCC, was purchased from Synpeptide Co. (Shanghai, China) and used without further purification. The purity of the peptide was determined using the HPLC-MS technique while the concentration of its stock solution (ca. 2 mM) was determined by pH-potentiometric titrations. NiCl2 stock solution was prepared from anhydrous, highest available grade NiCl2 (>99.95%, VWR Int., USA), and its concentration was determined by complexometric titration with standardized Na2H2EDTA in the presence of a murexide indicator at pH 8.0. Xanthine, xanthine oxidase (0.5 U mg−1), nitro blue tetrazolium chloride (NBT), DMSO, and 18-crown-6 were purchased from Sigma-Aldrich and KO2 was obtained from Acros Organics. In all solution equilibrium and spectroscopic studies, doubly deionized and ultrafiltered water was used (ELGA Purelab Classic system).Thermodynamic studies
The protonation constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ki) of the ligands and the overall stability constants of the nickel(II) complexes (logβpqr) were determined by the pH-potentiometric titration method using carbonate ion-free KOH solution. The carbonate contamination (less than 0.10%) was determined using the appropriate Gran functions.31 In these titrations, 3 mL aliquots of the ligands (ca. 2.0 mM) were titrated either in the absence or in the presence of a metal ion at 1:1 ratio (I = 0.2 M KCl, T = 298 K). The headspace over the sample was purged with argon to ensure the absence of oxygen and carbon dioxide. The samples were stirred using a VELP scientific magnetic stirrer and the pH measurements were made using a MOLSPIN pH-meter equipped with a 6.0234.110 combined glass electrode (Metrohm) and a MOL-ACS microburette controlled by a computer. The pH reading was converted into hydrogen ion concentration as described by Irving et al.32 The ionic product of water was also determined and found to be 13.756 under the experimental conditions. Protonation constants of the ligand, KHi = [HiL]/[Hi−1][H+], and the overall stability constants of the nickel(II) complexes, βpqr = [NipHqLr]/[Ni]p[H]q[L]r, were estimated by using the designated computational programs, SUPERQUAD33 and PSEQUAD.34 The concentration distribution curves of the complexes formed between nickel(II) and wtCC as well as the pNi values (pNi = −log[Ni(II)free]) were calculated using the computational program MEDUSA.
Ki) of the ligands and the overall stability constants of the nickel(II) complexes (logβpqr) were determined by the pH-potentiometric titration method using carbonate ion-free KOH solution. The carbonate contamination (less than 0.10%) was determined using the appropriate Gran functions.31 In these titrations, 3 mL aliquots of the ligands (ca. 2.0 mM) were titrated either in the absence or in the presence of a metal ion at 1:1 ratio (I = 0.2 M KCl, T = 298 K). The headspace over the sample was purged with argon to ensure the absence of oxygen and carbon dioxide. The samples were stirred using a VELP scientific magnetic stirrer and the pH measurements were made using a MOLSPIN pH-meter equipped with a 6.0234.110 combined glass electrode (Metrohm) and a MOL-ACS microburette controlled by a computer. The pH reading was converted into hydrogen ion concentration as described by Irving et al.32 The ionic product of water was also determined and found to be 13.756 under the experimental conditions. Protonation constants of the ligand, KHi = [HiL]/[Hi−1][H+], and the overall stability constants of the nickel(II) complexes, βpqr = [NipHqLr]/[Ni]p[H]q[L]r, were estimated by using the designated computational programs, SUPERQUAD33 and PSEQUAD.34 The concentration distribution curves of the complexes formed between nickel(II) and wtCC as well as the pNi values (pNi = −log[Ni(II)free]) were calculated using the computational program MEDUSA.
Spectroscopic measurements
UV-visible spectra of the ligand and its nickel(II) complexes were recorded with an Agilent Technologies Cary 60 UV-VIS spectrophotometer in the 200–800 nm wavelength range. The circular dichroism spectra were obtained with a Jasco J-810 spectropolarimeter using 1 mm and/or 1 cm cells in the 250–800 nm wavelength range. The individual spectra of the nickel(II) complexes were calculated by solving the overdetermined linear equation system with Matlab35 using the estimated stability constants. CW-EPR spectra were recorded with a BRUKER EleXsys E500 spectrometer (microwave frequency 9.45 GHz, microwave power 13 mW, modulation amplitude 5 G, modulation frequency 100 kHz). A 0.2 mL aliquot of Ni(II) sample solution was introduced into a quartz EPR tube, and then 0.1 mL DMSO solution containing KO2 was added for in situ oxidation. Frozen solution EPR spectra were recorded using a Dewar container filled with liquid nitrogen at 77 K.SOD activity studies
The SOD activity of the nickel(II) complexes was tested using the xanthine/xanthine oxidase/NBT model system36 and also by the sequential stopped-flow method. The xanthine/xanthine oxidase system produces constantly O2− which reduces the para-nitro blue tetrazonium chloride (NBT) to NBT diformazan with characteristic molar absorptivity at 560 nm. Upon addition of a SOD mimic to the system, the reduction of NBT by O2− is inhibited, and the indicator reaction becomes slower. The assay was carried out in phosphate buffer (0.05 M) containing NBT (5 × 10−5 M) and xanthine (2 × 10−4 M). The reaction was initiated by adding an appropriate amount of xanthine oxidase to reach an absorbance change of around ΔA560 nm = 0.025–0.028 min−1. First, the reaction was monitored with the blank sample (without any nickel(II) complex) for 3–4 minutes, then the nickel(II) complex was added to the sample and the absorbance change was followed for another 4 min. The extent of inhibition was estimated by comparing the rates of the absorbance change, i.e., the slopes of the absorbance vs. the time profile, in the two phases of the experiment. The SOD-activity was expressed by the IC50 value which is the concentration of the SOD mimic causing 50% inhibition.The catalytic effect of the nickel complexes on the decomposition of O2− was also studied using an Applied Photophysics SX-20 stopped-flow instrument equipped with a photomultiplier tube as the detector. The kinetic traces were collected using 2 mm optical path length at 25 °C. These measurements were carried out in a 1:1 DMSO–water mixture, and the instrument was used in sequential mode to circumvent the relatively slow homogenization of the reaction mixture when the reactants dissolved in pure water and DMSO are mixed. The first syringe was filled with water, the second one with KO2 in DMSO, and the third one with the complex dissolved in 1:1 DMSO and aqueous HEPES buffer (50 mM). In the first phase of these experiments, the aging loop was filled with a 1:1 mixture of the first and second solutions to produce a KO2 reagent in the 1:1 water–DMSO solvent. To avoid spectral interference due to slow homogenization, this mixture was incubated for 40 s and subsequently was reacted with the solution of the nickel(II) complexes in the third syringe. The progress of the reaction was monitored at 260 nm.
The O2− solutions were freshly prepared before each experiment by dissolving solid KO2 in vigorously stirred DMSO containing 18-crown-6. The concentration of the superoxide stock solution was determined by the stopped-flow method. In the absence of the catalyst, the initial absorbance values were used to determine the concentration using the molar absorbance of the superoxide anion at 260 nm, εO2− = 2686 M−1 cm−1.37
Results and discussion
Protonation equilibria
The acid dissociation constants of the ligand were determined by pH-potentiometry and UV-vis spectroscopy and the data are reported in Table 1. In accordance with the structure of the ligand, the terminal amino group, the carboxyl group of aspartic acid, the aromatic hydroxyl group of tyrosine and the three thiolate groups of Cys(1), Cys(2) and Cys(6) residues are involved in acid–base equilibria. Although some of these stepwise processes significantly overlap, the 6 acid dissociation constants could be estimated by fitting the titration curve to the appropriate equilibrium model. It is reasonable to assume that the lowest logKi value is assigned to the carboxylate group of aspartic acid. The next deprotonation process is assigned mainly to the deprotonation of the terminal ammonium function of the first cysteinyl residues. According to earlier literature studies, this group exhibits enhanced acidity (pKa = 6.50 for CSSACS–NH2) when cysteine is placed in the first position of the N-terminal part of the peptide, and thus the same scenario is expected in the H+/wtCC system.38 To clarify the deprotonation sequence of the ligand, UV-vis spectra were recorded as a function of pH (Fig. 1).
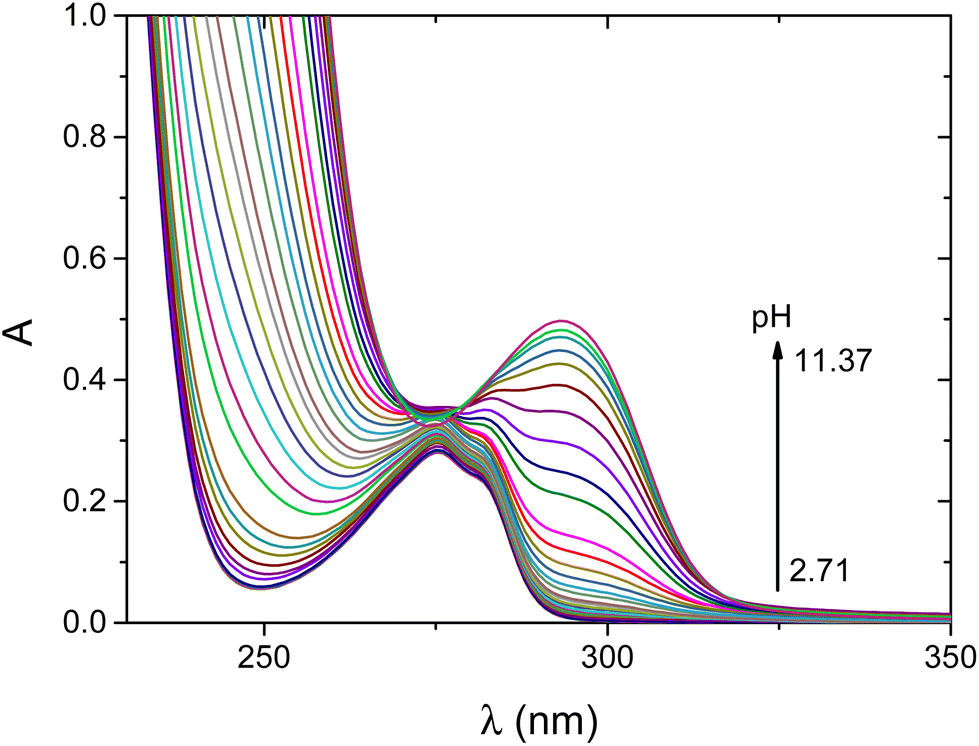 | ||
| Fig. 1 pH-dependent UV-vis spectra recorded for the H+/wtCC system. cwtCC = 0.99 mM, I = 0.2 M KCl, T = 25 °C, and l = 2 mm. | ||
Ki) of the NiSOD related peptidesa
| Species | wtCC | wtNiSOD |
|---|---|---|
| a 3σ standard deviations are indicated in parentheses. I = 0.2 M KCl and T = 298 K. b Data are taken from ref. 19. | ||
| [H6L]+/2+ | 3.49(9) | 3.58 |
| [H5L]0/+ | 6.10(9) | 5.48 |
| [H4L]−/0 | 7.86(6) | 7.20 |
| [H3L]2−/− | 8.98(5) | 8.25 |
| [H2L]3−/2− | 9.36(4) | 8.92 |
| [HL]4−/3− | 10.25(4) | 9.88 |
| ∑logKi |
46.04 | 43.31 |
| Fitted pH range | 3.0–11.0 | |
NiSOD related peptides containing the tyrosine moiety feature a characteristic absorption maximum at 294 nm which corresponds to the absorption of the deprotonated phenolic side chain of tyrosine.19,26 Therefore, the deprotonation of this group can selectively be followed (Fig. 2). The concentration distributions of the different protonated forms of wtCC together with the absorbance at 294 nm as a function of pH are shown in Fig. S1.† Upon increasing the pH, the absorbance steadily increases, and the maximum is reached above pH = 11 where the ligand is fully deprotonated. This confirms that the phenolic OH becomes fully deprotonated in the last step. However, the absorbance is not strictly proportional to the equilibrium concentration of L5−. This is most likely the consequence of microspeciation. The last two deprotonation steps of the ligand partly overlap and HL4− is present as the equilibrium mixture of the two forms in which either the last thiol group or the phenolic OH group is deprotonated. The latter form of HL4− is also expected to contribute to the absorbance at 294 nm.
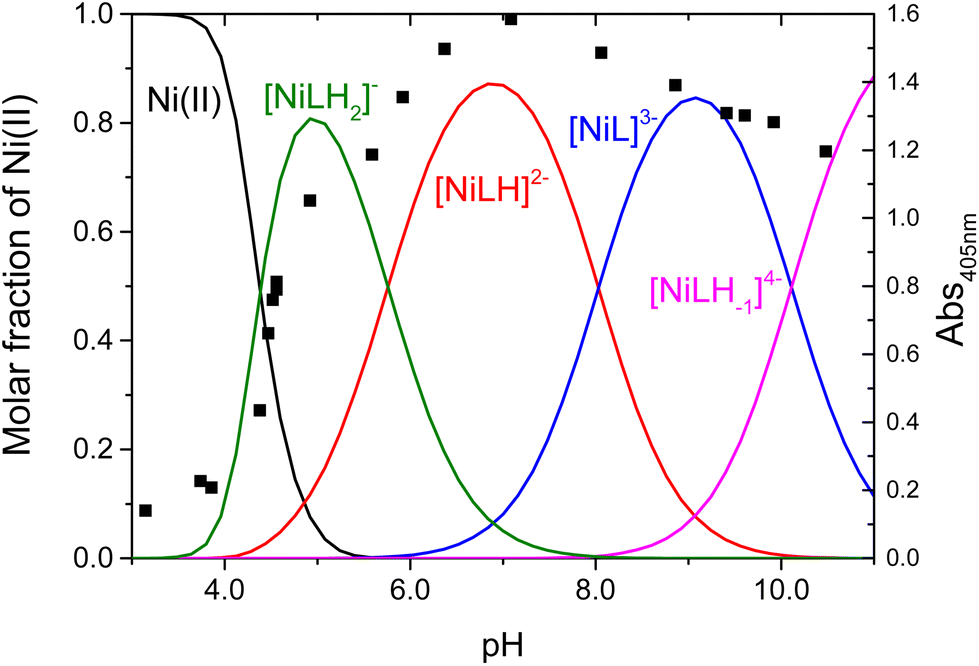 | ||
| Fig. 2 Concentration distribution of the complexes formed between Ni(II) and wtCC (continuous lines) and the absorbance at 405 nm (black square) as a function of pH. cwtCC = 2.08 mM, cNi(II) = 1.96 mM, I = 0.2 M KCl, and T = 25 °C. | ||
The overall cumulative basicity of wtCC (∑logKi, Table 1) is higher than that of wtNiSOD which is due to the replacement of the terminal histidine with the cysteine residue that may influence the stability of the nickel(II) complexes (Table 2).
βpqr) of the complexes formed between nickel(II) and the NiSOD related peptidesa
The pH-potentiometric data can be fitted well by considering the formation of [NiLH2]−, [NiLH]2−, [NiL]3− and [NiLH−1]4− complexes according to eqn (S1)–(S3).† The complex formation reaction starts in the acidic pH range with the [NiLH2]− complex. In this pH range, the carboxyl group of aspartic acid is deprotonated (pKa = 3.49), and there is no spectroscopic or thermodynamic evidence that the carboxylate group is involved in the coordination of nickel(II). N-terminally free peptides containing cysteine in the first position favor the (NH2,S−) coordination mode.38 The same is expected in this complex; however, the calculated equilibrium constant for the formation of this species (eqn (3)) is higher than the corresponding equilibrium constant for the formation of the (NH2,S−) coordination mode (logK = 7.94 calculated in the Ni(II):CysGly system39). Thus, it is reasonable to assume that nickel(II) is coordinated by the (NH2,S−,S−) donor atoms.
| logK (NH2,S−,S−) = logβ (NiLH2) − pK (HL) − pK (H2L) = 12.78. | (3) |
This binding mode is further confirmed by monitoring the spectral changes both in the UV-vis (Fig. 3) and CD spectra (Fig. S2†). The calculated individual UV-vis spectra of different nickel(II) complexes are shown in Fig. S3.† For the [NiLH2]− complex, the corresponding spectrum exhibits two significant transitions: one belongs to the LMCT band of the S− → Ni(II) transition (λ = 308 nm, ε = 4989 M−1 cm−1) and the second corresponds to a d–d transition characteristic for square-planar nickel(II) complexes (λ = 416 nm, ε = 594 M−1 cm−1). The CD spectra provide further information on the structure of the complex. It is obvious that nickel(II) coordinates to wtCCvia the (NH2,S−) donors of Cys(1); however, both Cys(2) or Cys(6) may be involved in the coordination of the metal ion (Scheme S2†). The CD spectra exhibit a band at 650 nm, which is characteristic of the d–d transition when the distant cysteinyl residue binds to nickel(II).40 Consequently, the (NH2,S−) binding mode is supported by macrochelation with the distant cysteinyl (Cys(6)) residue in the [NiLH2]− complex.
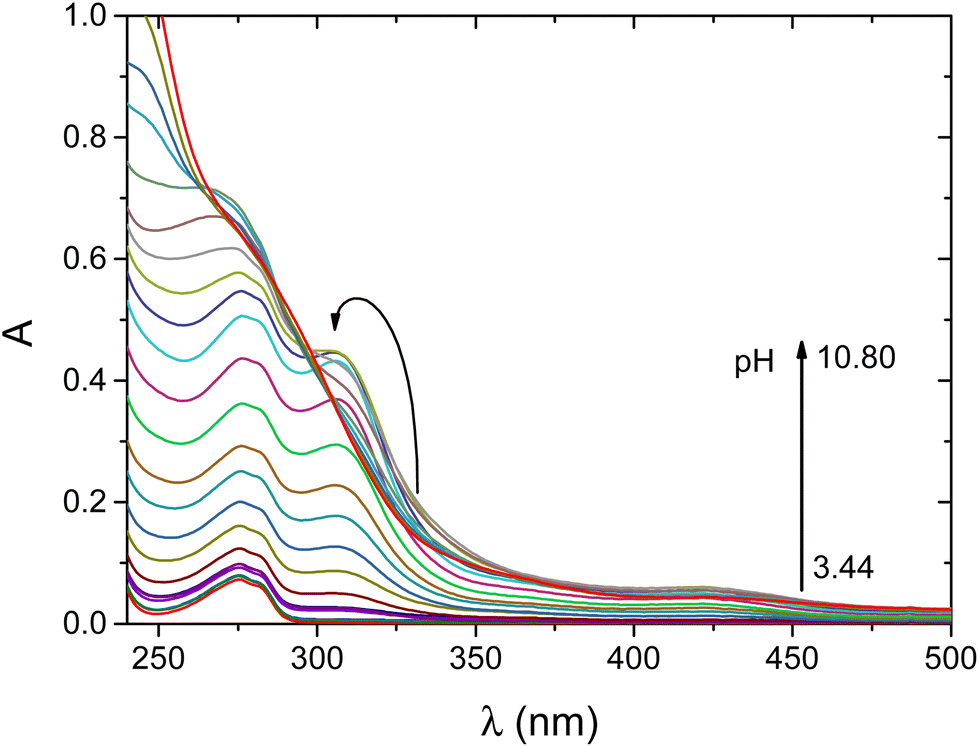 | ||
| Fig. 3 pH-dependent UV-vis spectra recorded for the Ni(II)/wtCC system at a 0.9:1 metal to ligand ratio. cwtCC = 0.52 mM, cNi(II) = 0.51 mM, I = 0.2 M KCl, T = 25 °C, and l = 1 mm. | ||
By increasing the pH, an additional base consumption process yields the [NiLH]2− complex. The corresponding deprotonation constant of the complex is logK = 5.76; thus, this process can be assigned either to the binding of an additional thiol group or to the metal induced deprotonation and coordination of a peptide-N. The formation of this species is accompanied by characteristic changes both in the UV-vis and CD spectra. The absorbance at the characteristic LMCT band of the S− → Ni(II) transition increases by increasing the equilibrium concentration of this complex (ε = 9474 M−1 cm−1). Thus, we expect that an additional thiolate group is involved in the metal ion coordination leading to the formation of the (NH2,S−,S−,S−) donor set around nickel(II). The intensive d–d transition at 421 nm (ε = 1340 M−1 cm−1) indicates that the complex possesses a square-planar geometry (Scheme S2†). This complex is dominant in the physiological pH range. Accordingly, the presence of cysteine in the first position of the peptide leads to a different coordination mode compared to the native NiSOD enzyme fragment.
The additional base consumption process corresponds to the metal induced ionization and coordination of the peptide-N atom, logK = 8.03. Since nickel(II) has square-planar geometry and only four donor atoms can be involved in the chelation of the metal ion, the coordination sphere around nickel(II) is reorganized and the thiolate group of the terminal cysteine becomes non-coordinating. The reorganization of the coordination sphere can readily be followed by UV-vis spectroscopy. The calculated individual spectrum of the complex exhibits three significant transitions; two LMCT bands are assigned to the N− → Ni(II) (λ = 270 nm, ε = 14320 M−1 cm−1) and S− → Ni(II) transitions (λ = 310 nm, ε = 6195 M−1 cm−1), while the d–d transition is observed at 425 nm (ε = 987 M−1 cm−1). The relatively small molar absorptivity of the S− → Ni(II) LMCT also confirms that one of the thiolate groups is not involved in the coordination of nickel(II). This coordination mode results in a (5,5)-membered chelate system via the (NH2,N−,S−) donor atoms which is supported by macrochelation with the distant cysteinyl residue (Scheme S2†). Consequently, the formation of the [NiL]3− complex provides the reduced form of the NiSOD enzyme; however, its formation is shifted into the alkaline pH-range. This is due to the high thermodynamic stability of the [NiLH]2− complex (logβ = 18.60) with the (NH2,S−,S−,S−) donor set which hinders the subsequent deprotonation process, i.e., the ionization and coordination of the peptide N-atom.
In alkaline solution, a further base consumption process yields the [NiLH−1]4− complex. Neither the UV-vis nor the CD spectra exhibit significant changes, and thus, the coordination sphere remains intact. Consequently, the deprotonation of the non-coordinating tyrosine residue yields this species. The corresponding deprotonation constant of the complex, logK = 9.75, is close to the logKi of the side chain of the hydroxyl group of tyrosine. This provides strong support to the assumption that tyrosine is not coordinated to the metal center.
The main conclusion is that the presence of cysteine in the first position of the peptide chain significantly increases the nickel binding ability of the ligand; however, the formation of the active center of the NiSOD enzyme is shifted to the alkaline pH range. The metal binding ability of wtCC is compared to a set of NiSOD related peptides as shown in Fig. 4. The pNi values were calculated at physiological pH and clearly show that the pNi of wtCC is about 3 units higher than that of the native NiSOD enzyme fragment (pNi = 13.66 for wtCC and 11.27 for wtNiSOD). In other words, the concentration of free nickel(II) is 1000 times lower in the Ni(II)/wtCC system than that of wtNiSOD, if the ligands are present in equal concentrations. The noted difference is explained by the higher cumulative basicity of wtCC and the different coordination modes of the metallopeptides (cf. Scheme S1†).
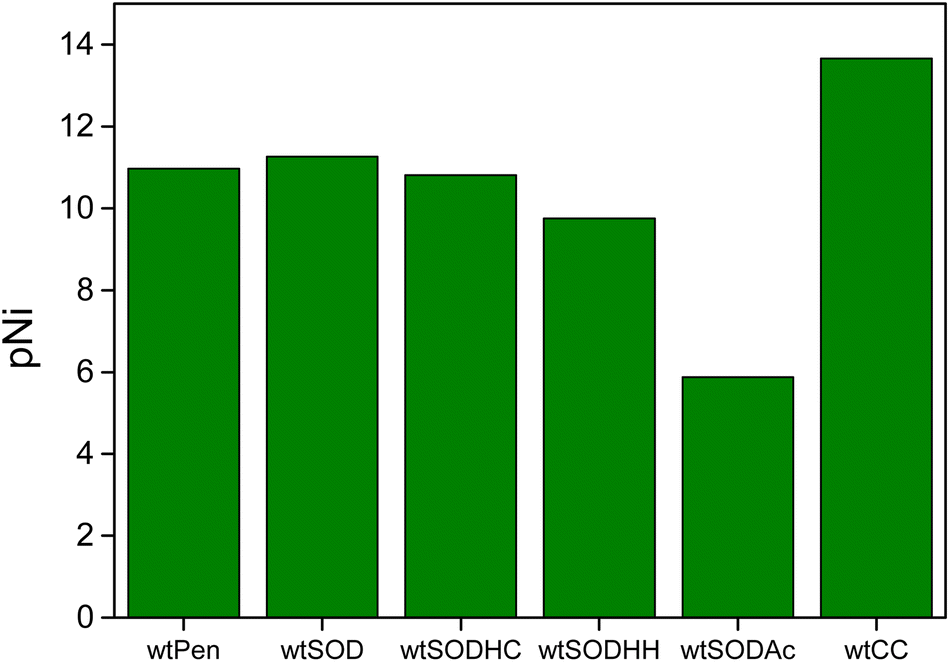 | ||
| Fig. 4 Calculated pNi values of NiSOD related peptides (cf. Scheme S1†) at pH 7.6. cNi(II) = 10 μM, cligand = 50 μM, and pNi = −log[Ni(II)free]. | ||
Catalytic activity
The catalytic activity of the nickel(II)–wtCC system was thoroughly studied at physiological pH. KO2 was used to oxidize the nickel(II) complexes in situ. In an attempt to trap the transient species, the reaction mixture was frozen using liquid nitrogen and studied by EPR spectroscopy (Fig. S4†). The EPR spectra clearly show that a substantial amount of superoxide is consumed; however, neither the formation of nickel(III) transient species nor the generation of radical species could be observed. There are two plausible possibilities for the interpretation of these observations: (i) the reactions between the superoxide anion and the nickel(II) complexes yield nickel(III) transient species, however their concentration is too low or their lifetime is too short to be detected by EPR; (ii) the reaction between the superoxide anion and the nickel complexes leads to the degradation of the peptide.The SOD activity of the nickel(II)–wtCC system was tested by using the xanthine/xanthine oxidase/NBT assay. Preliminary experiments confirmed that the peptide itself does not exhibit any SOD activity. The dismutation reaction was primarily monitored at pH 7.8. At lower pH, the self-decomposition of O2− is fast and the catalyst has a minor role in the overall process. In a designated set of experiments, the deceleration of the catalytic dismutation rate was observed upon increasing the pH (Table S1†). The species distributions were calculated for each nickel(II) concentration used in these experiments. Under the conditions applied, the predominant complex is [NiLH]2− and its equilibrium concentration decreases by increasing the pH. Thus, it is reasonable to assume that the major catalytically active form is the [NiLH]2− complex. However, [NiL]3− may also contribute to the dismutation of O2−. In this case, the dismutation reaction is expected to proceed via fully analogous parallel reaction paths.
The inhibition curve as a function of nickel(II) concentration is shown in Fig. 5. The IC50 value of this system was estimated to be 7 ± 2 μM, which is close to that of the wild-type fragment of NiSOD (IC50 = 3.9 μM), i.e., they show similar performance in the degradation of the superoxide anion.
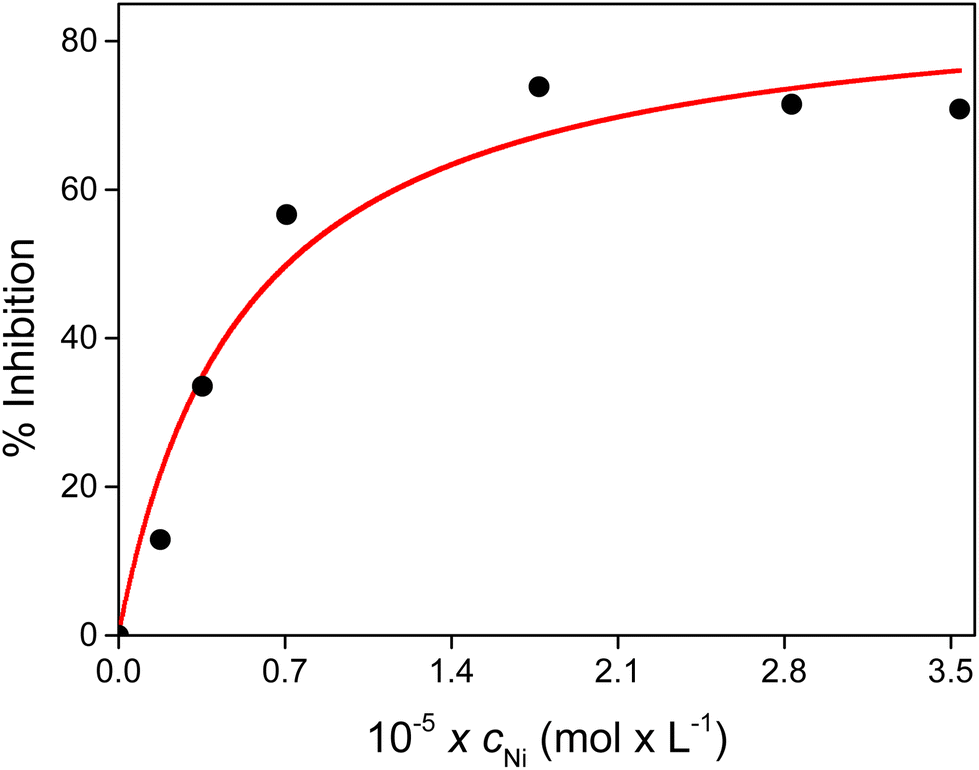 | ||
| Fig. 5 Inhibition percentage as a function of the total concentration of Ni(II) at pH 7.8. cxanthine = 200 μM, cNBT = 50 μM, and T = 25 °C. | ||
Sequential stopped-flow experiments were carried out to explore the catalytic effect of the Ni(II)/wtCC system in the decomposition of the superoxide anion under real catalytic conditions. The decay of the absorbance was monitored at 260 nm where the dominant absorbing species is the superoxide anion (Fig. 6). The nickel complexes exhibit excellent SOD activity because the spontaneous decomposition of O2− occurs on a considerably longer timescale. These kinetic traces cannot be fitted with a simple first-order expression that would be expected if only a catalytic cycle was operative in this system. The observations strongly suggest that the degradation of the catalyst also occurs in a kinetically coupled process. Such a degradation process has already been reported for several NiSOD mimics.41
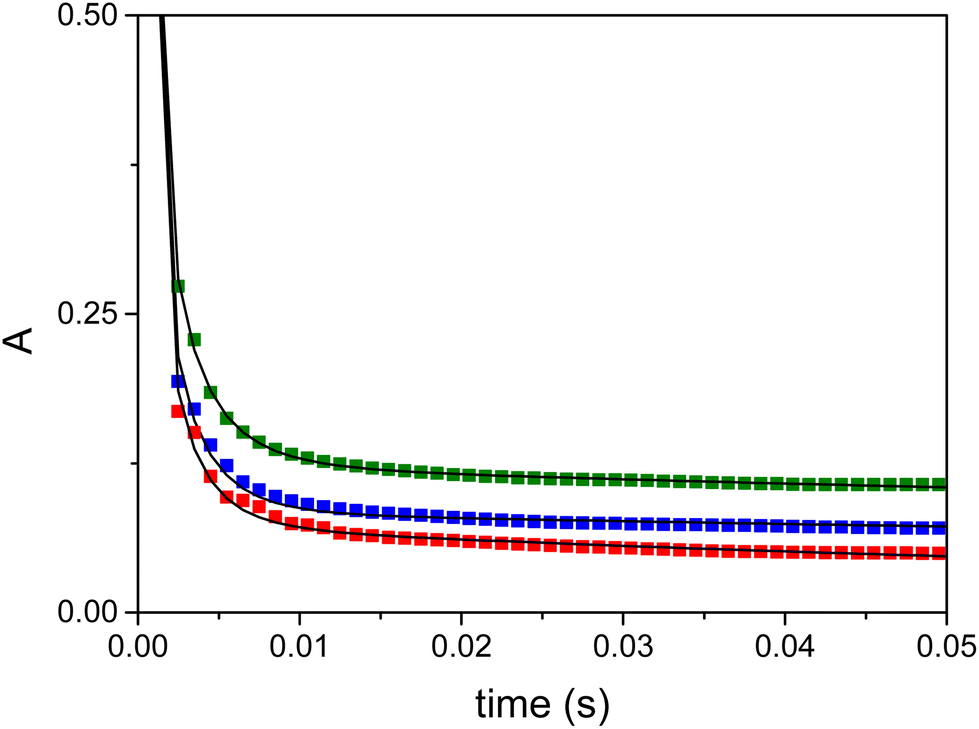 | ||
| Fig. 6 Decomposition of the superoxide anion recorded at 260 nm in the presence of the Ni/wtCC complexes (green: 9.86 μM, blue: 4.93 μM, and red: 1.98 μM). Solid lines represent the fitted kinetic traces based on the proposed kinetic model. Solvent 1:1 aqueous HEPES buffer (50 mM, pH 7.6)/DMSO mixture. c(O2−)0 = 1.61 mM, λ = 260 nm, T = 25 °C, and l = 2 mm. | ||
The kinetic traces were fitted on the basis of the kinetic model presented in Scheme 2.
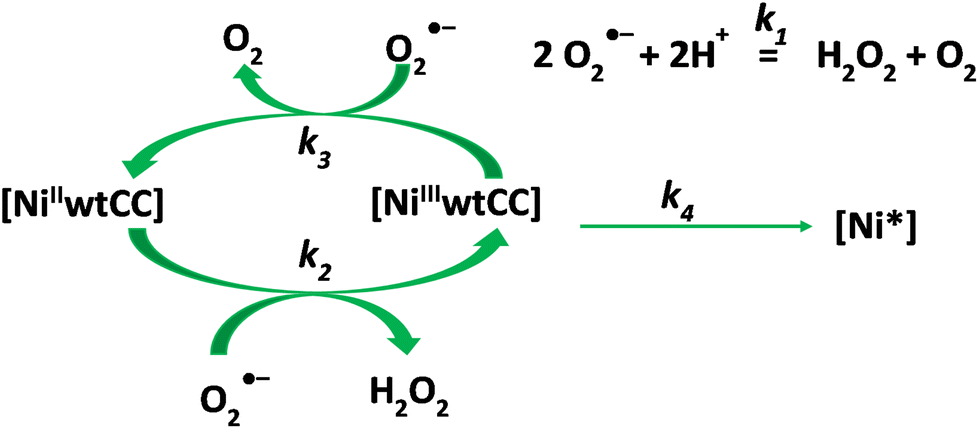 | ||
| Scheme 2 Postulated kinetic model of the decomposition of the superoxide anion in the presence of the Ni/wtCC complexes. | ||
The model includes the spontaneous degradation of the superoxide anion (k1) and the catalytic dismutation process (k2, k3). Since nickel(II) is stable in the absence of an oxidant, it is a plausible assumption that the catalytic activity is lost because of the degradation of the Ni(III) complex (k4). This reaction yields an unidentified nickel species (Ni*). The detailed kinetic model and the corresponding differential equation system are shown in the ESI (reactions (S4)–(S7) and eqn (S8)–(S12),† respectively). The parameters were estimated by numerically solving the differential equation system and fitting the absorbance (eqn (S13)†) using a non-linear least squares algorithm. The results are listed in Table 3. It is important to note that most of the fitted parameters are expected to be pH dependent and the reported values are applicable only at pH 7.6 in a 1:1 water/DMSO solvent mixture.
:1 aqueous HEPES buffer (50 mM, pH 7.6)/DMSO mixture
| Parameter | Ni–wtCC system | Ni–wtNiSODb system | Unit |
|---|---|---|---|
| a These values are known from independent experiments and were kept fixed during the fitting procedure. b Data are taken from ref. 26. | |||
| k 1 | 3.84 × 104 | M−1 s−1 | |
| k 2 | (6.6 ± 0.6) × 107 | 9.6 × 107 | M−1 s−1 |
| k 3 | (1.6 ± 0.4) × 108 | 1.72 × 108 | M−1 s−1 |
| k 4 | 1310 ± 90 | 258 | s−1 |
| ε Ni(II) | (1.33 ± 0.03) × 104 | 1.48 × 104 | M−1 cm−1 |
| ε Ni(III) | (1.7 ± 0.3) × 104 | 2.44 × 104 | M−1 cm−1 |
| ε Ni* | 6180 ± 70 | 1.8 × 104 | M−1 cm−1 |
|
ε
O2−a |
2686 | M−1 cm−1 | |
| ε H2O2 | 38 | M−1 cm−1 | |
The nickel–wtCC and nickel–native NiSOD enzyme fragment (wtNiSOD) systems show similarities in that the oxidation of the Ni(II) complex is slower than the reduction of the corresponding Ni(III) complex, k2 < k3. Consequently, Ni(II) is always present at a larger transient concentration compared to Ni(III) over the dismutation process (cf.Fig. 7). This common kinetic feature is believed to be a key factor in the relatively high dismutase activity of the wtCC and the native NiSOD enzyme fragment complexes compared to other NiSOD related metallopeptides. Recently, we have shown that a NiSOD mimic is less efficient when k2 > k3, i.e., Ni(III) is dominant over Ni(II).26
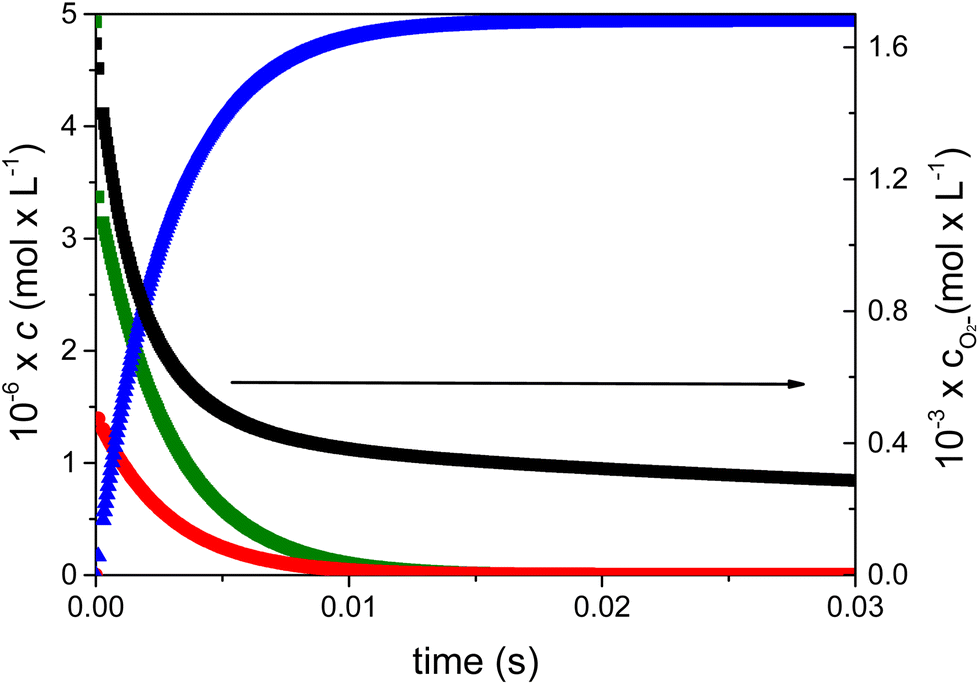 | ||
| Fig. 7 Calculated concentration profiles of each species as a function of time during the decomposition of the superoxide anion in the presence of the nickel complexes of wtCC in 1:1 aqueous HEPES buffer (50 mM, pH 7.6)/DMSO mixture. Green: NiII(wtCC), red: NiIII(wtCC), blue: Ni*(wtCC), and black: O2−. c(NiII/wtCC)0 = 9.86 μM and c(O2−)0 = 1.61 mM. | ||
In the nickel–wtCC system, the calculated concentration profiles confirm that about 75% of the initial amount of superoxide undergoes dismutation via the complex catalyzed pathway in the first 10 ms of the reaction (Fig. 7). However, the catalyst is quickly deactivated in a fast degradation process and the nickel-assisted dismutation ceases. The subsequent slow decay of O2− is due to the spontaneous decomposition of this species. The intramolecular redox degradation of the wtCC complex is faster than that observed in the case of the native NiSOD enzyme fragment system. In this process, an inactive Ni* species forms. Presumably, the thiolate group of cysteine in the first position of the peptide chain enhances the internal redox reaction leading to the oxidation of the ligand and, as a consequence, to the deactivation of the catalyst.
Conclusion
Incorporation of cysteine at the first position of the NiSOD peptide sequence yields a new peptide, wtCC. Due to the presence of cysteine in the vicinity of the metal center, this peptide exhibits high metal binding ability (pNi = 13.66), which is stronger than that obtained for the wild-type fragment of the NiSOD enzyme (pNi = 11.27). The main species has a (NH2,S−,S−,S−) donor set at physiological pH. Although this coordination motif differs from that of the active site of the NiSOD enzyme, the catalytic activities of these species are very similar. Presumably, this is due to the common feature of the two systems in that the Ni(II) state of the catalyst is dominant over the Ni(III) form in the catalytic cycle, i.e., the oxidation of the Ni(II) complex is slower than the reduction of the corresponding Ni(III) complex. However, the presence of a cysteine residue in the first position of the peptide chain increases the rate of the intramolecular redox degradation of the catalyst. This is 5 times faster in the nickel–wtCC system compared to that of the nickel complex of the native NiSOD fragment. These results imply that a protective mechanism is operative in the NiSOD enzyme that prevents the oxidative damage of the active center. Perhaps the terminal histidine residue is also involved in such a protective mechanism. Systematic modifications of the model systems to improve the robustness of NiSOD related metallopeptides are in progress in our laboratory.Data availability
Experimental data are provided in the manuscript and in the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. L. and I. F. are grateful for the financial support of the Hungarian National Research, Development and Innovation Office (NKFIH K-139140). N. L. acknowledges the financial support of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences and the ÚNKP-23-5 New National Excellence Program of the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund. Project no. C1018348 has been implemented with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the KDP-2020 funding scheme.References
- M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453–R462 CrossRef CAS PubMed.
- J. Zuo, Z. Zhang, M. Luo, L. Zhou, E. C. Nice, W. Zhang, C. Wang and C. Huang, MedComm, 2022, 3, e127 CrossRef CAS.
- J. Nordberg and E. S. J. Arnér, Free Radicals Biol. Med., 2001, 31, 1287–1312 CrossRef CAS.
- M. Zámocký and F. Koller, Prog. Biophys. Mol. Biol., 1999, 72, 19–66 CrossRef.
- A. C. Maritim, R. A. Sanders and J. B. Watkins III, J. Biochem. Mol. Toxicol., 2003, 17, 24–38 CrossRef CAS.
- J. N. Moloney and T. G. Cotter, Semin. Cell Dev. Biol., 2018, 80, 50–64 CrossRef CAS.
- P. R. Angelova and A. Y. Abramov, FEBS Lett., 2018, 592, 692–702 CrossRef CAS PubMed.
- I. A. Abreu and D. E. Cabelli, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 263–274 CrossRef CAS.
- I. Fridovich, in Encyclopedia of Biological Chemistry, ed. M. D. Lane, Elsevier, New York, 2004, pp. 135–138. DOI:10.1016/B0-12-443710-9/00648-7.
- C. R. Kliment, J. M. Tobolewski, M. L. Manni, R. J. Tan, J. Enghild and T. D. Oury, Antioxid. Redox Signaling, 2008, 10, 261–268 CrossRef CAS.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- A.-F. Miller, Curr. Opin. Chem. Biol., 2004, 8, 162–168 CrossRef CAS.
- H.-D. Youn, E.-J. Kim, J.-H. Roe, Y. C. Hah and S.-O. Kang, Biochem. J., 1996, 318, 889–896 CrossRef CAS PubMed.
- J. Wuerges, J.-W. Lee, Y.-I. Yim, H.-S. Yim, S.-O. Kang and K. D. Carugo, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8569–8574 CrossRef CAS.
- P. A. Bryngelson, S. E. Arobo, J. L. Pinkham, D. E. Cabelli and M. J. Maroney, J. Am. Chem. Soc., 2004, 126, 460–461 CrossRef CAS.
- J. Shearer and L. M. Long, Inorg. Chem., 2006, 45, 2358–2360 CrossRef CAS.
- J. Shearer, Acc. Chem. Res., 2014, 47, 2332–2341 CrossRef CAS.
- N. Lihi and I. Fábián, in Advances in Inorganic Chemistry, ed. R. van Eldik and C. D. Hubbard, Academic Press, 2022, vol. 79, pp. 1–22 Search PubMed.
- N. Lihi, G. Csire, B. Szakács, N. V. May, K. Várnagy, I. Sóvágó and I. Fábián, Inorg. Chem., 2019, 58, 1414–1424 CrossRef CAS PubMed.
- N. Lihi, D. Kelemen, N. V. May and I. Fábián, Inorg. Chem., 2020, 59, 4772–4780 CrossRef CAS PubMed.
- D. Kelemen, N. V. May, M. Andrási, A. Gáspár, I. Fábián and N. Lihi, Chem. – Eur. J., 2020, 26, 16767–16773 CrossRef CAS.
- S. K. Chatterjee, R. C. Maji, S. K. Barman, M. M. Olmstead and A. K. Patra, Angew. Chem., Int. Ed., 2014, 53, 10184–10189 CrossRef CAS PubMed.
- J. Domergue, P. Guinard, M. Douillard, J. Pécaut, S. Hostachy, O. Proux, C. Lebrun, A. Le Goff, P. Maldivi, C. Duboc and P. Delangle, Inorg. Chem., 2023, 62, 8747–8760 CrossRef CAS.
- J. Domergue, P. Guinard, M. Douillard, J. Pécaut, O. Proux, C. Lebrun, A. Le Goff, P. Maldivi, P. Delangle and C. Duboc, Inorg. Chem., 2021, 60, 12772–12780 CrossRef CAS PubMed.
- E. P. Broering, P. T. Truong, E. M. Gale and T. C. Harrop, Biochemistry, 2013, 52, 4–18 CrossRef CAS PubMed.
- D. Bonczidai-Kelemen, G. Sciortino, N. V. May, E. Garribba, I. Fábián and N. Lihi, Inorg. Chem. Front., 2022, 9, 310–322 RSC.
- K. C. Ryan, A. I. Guce, O. E. Johnson, T. C. Brunold, D. E. Cabelli, S. C. Garman and M. J. Maroney, Biochemistry, 2015, 54, 1016–1027 CrossRef CAS.
- H. T. Huang, S. Dillon, K. C. Ryan, J. O. Campecino, O. E. Watkins, D. E. Cabelli, T. C. Brunold and M. J. Maroney, Inorg. Chem., 2018, 57, 12521–12535 CrossRef CAS PubMed.
- K. P. Neupane, K. Gearty, A. Francis and J. Shearer, J. Am. Chem. Soc., 2007, 129, 14605–14618 CrossRef CAS PubMed.
- V. Pelmenschikov and P. E. M. Siegbahn, J. Am. Chem. Soc., 2006, 128, 7466–7475 CrossRef CAS PubMed.
- G. Gran, Analyst, 1952, 77, 661–670 RSC.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- P. Gans, A. Sabatini and A. Vacca, J. Chem. Soc., Dalton Trans., 1985, 6, 1195–1200 RSC.
- I. Nagypál and L. Zékány, Computational Methods for the Determination of Formation Constants , Plenum Press, New York, NY, USA , 1985, 291–299 Search PubMed.
- T. M. MATLAB and Statistics Toolbox Release 2012b, Inc., Natick, Massachusetts, United States Search PubMed.
- W. F. Beyer and I. Fridovich, Anal. Biochem., 1987, 161, 559–566 CrossRef CAS.
- B. J. Bolann, H. Henriksen and R. J. Ulvik, Biochim. Biophys. Acta, Gen. Subj., 1992, 1156, 27–33 CrossRef CAS PubMed.
- N. Lihi, Á. Grenács, S. Timári, I. Turi, I. Bányai, I. Sóvágó and K. Várnagy, New J. Chem., 2015, 39, 8364–8372 RSC.
- H. Kozlowski, B. D.-L. Révérend, D. Ficheux, C. Loucheux and I. Sovago, J. Inorg. Biochem., 1987, 29, 187–197 CrossRef CAS.
- M. Raics, N. Lihi, A. Laskai, C. Kállay, K. Várnagy and I. Sóvágó, New J. Chem., 2016, 40, 5420–5427 RSC.
- D. Tietze, J. Sartorius, B. Koley Seth, K. Herr, P. Heimer, D. Imhof, D. Mollenhauer and G. Buntkowsky, Sci. Rep., 2017, 7, 17194 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt03638c |
| This journal is © The Royal Society of Chemistry 2024 |