
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanomaterials for light-mediated therapeutics in deep tissue
Chung Yin
Tsang
 a and
Yong
Zhang
*b
a and
Yong
Zhang
*b
aDepartment of Biomedical Engineering, College of Design and Engineering, National University of Singapore, Singapore 117583, Singapore. E-mail: e0974156@u.nus.edu
bDepartment of Biomedical Engineering, The City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong. E-mail: yozhang@cityu.edu.hk
First published on 24th January 2024
Abstract
Light-mediated therapeutics, including photodynamic therapy, photothermal therapy and light-triggered drug delivery, have been widely studied due to their high specificity and effective therapy. However, conventional light-mediated therapies usually depend on the activation of light-sensitive molecules with UV or visible light, which have poor penetration in biological tissues. Over the past decade, efforts have been made to engineer nanosystems that can generate luminescence through excitation with near-infrared (NIR) light, ultrasound or X-ray. Certain nanosystems can even carry out light-mediated therapy through chemiluminescence, eliminating the need for external activation. Compared to UV or visible light, these 4 excitation modes penetrate more deeply into biological tissues, triggering light-mediated therapy in deeper tissues. In this review, we systematically report the design and mechanisms of different luminescent nanosystems excited by the 4 excitation sources, methods to enhance the generated luminescence, and recent applications of such nanosystems in deep tissue light-mediated therapeutics.
 Chung Yin Tsang | Chung Yin Tsang completed his bachelor's degree in biomedical engineering from the Chinese University of Hong Kong in 2022. Currently, he is pursuing a PhD degree in biomedical engineering at National University of Singapore (NUS). His research focuses on the design, synthesis and therapeutic applications of luminescent nanomaterials. |
 Yong Zhang | Yong Zhang is currently the Chair Professor of Biomedical Engineering and Head of the Department of Biomedical Engineering at City University of Hong Kong. Professor Zhang has authored over 300 peer-reviewed research papers in international journals. His current research interests include nanobiophotonics, nanomedicine, and microfluidic devices. |
1. Introduction
Light-mediated therapeutics have emerged significantly over the past few decades due to their specificity, non-invasiveness and high therapeutic efficacy. Typically, a photosensitive molecule is delivered to the target tissue, while light is directed onto the target tissue to activate the molecule for specific therapy. For example, photodynamic therapy involves the light activation of a photosensitizer delivered into the target tissue. Upon activation, reactive oxygen species (ROS) such as singlet oxygen (1O2) or hydroxyl radicals (˙OH) are generated, damaging the target cells.1 However, most photosensitizers are only activated by UV or visible light.2 Another well-studied light-mediated therapy involves light-triggered drug release, which generally involves nanoparticles conjugated with drug molecules through photocleavable linkers or bonds. Light in the UV/visible spectrum usually has enough energy to cleave such linkers and release the drug into the target tissue.3 Light-based therapeutics ensure the activation of nanomaterials only in the target tissue, while having minimal effects in other healthy tissues.However, light in the UV/visible spectrum exhibits poor penetration of just a few millimetres in biological tissue and would not be suitable for deep tissue light-mediated therapy.4 Therefore, recently, certain nanosystems have been engineered to produce UV/visible light upon activation with excitation sources that exhibit deeper penetration in biological tissues. This enables the implementation of light-mediated therapies in deep tissue.
Near-infrared (NIR) light, possessing a tissue penetration depth of 1–2 cm, can produce visible light through upconversion processes through excitation with NIR-I (700–1000 nm) or NIR-II light (1000–1700 nm).5–7 In addition, NIR-II light can also be generated in other NIR-excited nanosystems through downconversion processes.8 The visible light produced is usually utilised for therapeutic purposes, while the NIR-II light serves as an imaging modality given its lower tissue attenuation.6,9 Ultrasound, with a tissue penetration depth exceeding 10 cm, could also activate certain nanomaterials to produce light in the visible range.10 Ultrasound generates luminescence either through sonoluminescence (cavitation of bubbles in a liquid medium) or through the deformation of mechanoluminescent nanomaterials.11,12 X-rays have the deepest penetration depth compared to NIR and ultrasound, with almost unlimited penetration depth in biological tissue.13 More importantly, X-rays can pass through very dense body structures like bone, enabling therapies in organs protected by bone such as the brain.14 X-rays also carry sufficient energy to cause direct band-to-band excitation in many nanosystems, resulting in the generation of strong light in the UV/visible range and the activation of most photosensitive molecules. Finally, certain nanomaterials could enhance chemiluminescence produced from chemical reactions without the need for external activation.15 This allows the therapy to be carried out in deep tissue regions without considering light scattering.
Therefore, it is of great essence that we summarise the strategies reported that enable the activation of light-mediated therapies in deep tissue. In this review, we systematically discuss the design and mechanism of different nanomaterials that enable emission of chemiluminescence, NIR-, X-ray- and ultrasound-excited luminescence (Fig. 1). In addition, we will also report methods for enhancing luminescence intensity, applications of the nanosystems in deep tissue therapy, as well as the comparison between strengths and weaknesses of the 4 excitation modes.
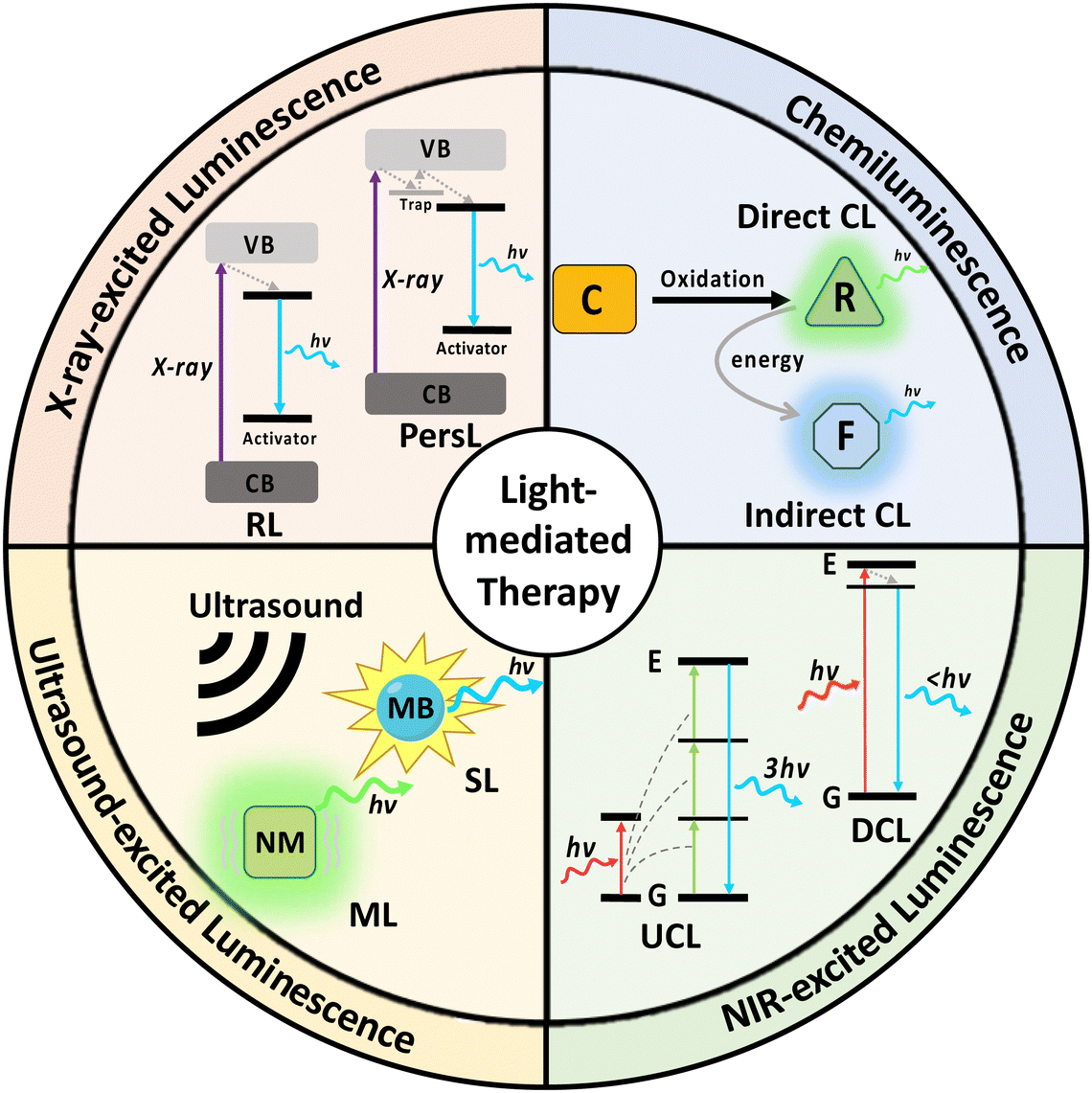 | ||
| Fig. 1 Schematic illustration of the different mechanisms of light generation with chemi-excitation, NIR-excitation, ultrasound-excitation and X-ray-excitation that support deep-tissue light-mediated therapeutics. (C: chemiluminophore; CL: chemiluminescence; DCL: downconversion luminescence; F: fluorophore; MB: microbubble; ML: mechanoluminescence; NM: nanomaterial; PersL: persistent luminescence; R: radical; RL: radioluminescence; SL: sonoluminescence; and UCL: upconversion luminescence). | ||
2. Chemiluminescence
Chemiluminescence is the phenomenon characterized by the generation of light through chemiexcitation occurring within a chemical reaction, without external light excitation.16 By harnessing the light emission generated through chemiluminescent reactions, it becomes possible to overcome the limitations of traditional light-mediated therapy with UV/visible light sources, such as low tissue penetration and potential damage to surrounding tissues. There are 2 major mechanisms of chemiluminescence: direct chemiluminescence and indirect chemiluminescence.17Direct chemiluminescence refers to the emission of light directly from the excited state of a chemical species formed during a chemical reaction (Fig. 2a). Examples of direct chemiluminescence involve the oxidation of chemiluminescent substrates (C), such as luminol, or its derivatives. They can be directly oxidized by reactive oxygen species, such as superoxide anions (O2−) and hydrogen peroxide (H2O2), resulting in the generation of excited radicals (R*). These radicals emit photons while returning to the ground state (R). A classic example of direct chemiluminescence is the reaction between luminol and hydrogen peroxide, producing light at 440 nm.18 In indirect chemiluminescence, the excited radicals do not return to the ground state directly. Instead, they interact with a nearby auxiliary species, such as fluorophores (F), to generate light. Indirect chemiluminescence relies on energy transfer processes between the chemiexcited species and the luminophore, normally achieved through chemiluminescence resonance energy transfer (CRET). Typical examples involve the oxidation of peroxyoxalate esters, certain ruthenium complexes or dioxetanes, generating high-energy radicals that can excite a nearby luminophore.
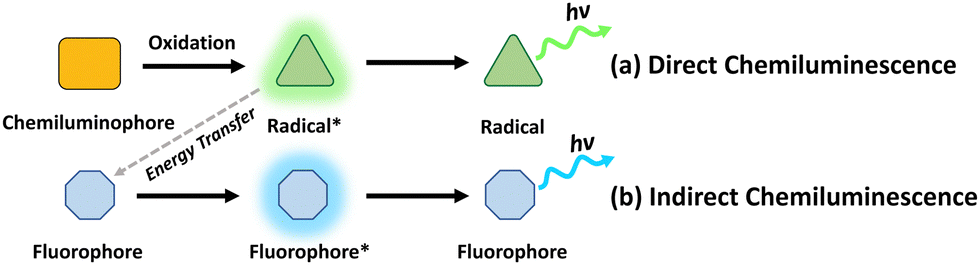 | ||
| Fig. 2 Schematic illustrations of the mechanisms of (a) direct chemiluminescence and (b) indirect chemiluminescence. | ||
Traditionally, chemiluminescence occurs randomly as a result of the interaction between reactants at low concentration. By leveraging nanomaterials, we can control the rate, location and enhance the intensity of chemiluminescence for therapeutic purposes. Nanomaterials, such as metal nanoparticles, quantum dots, and organic nanomaterials play important roles in the delivery and enhancement of chemiluminescence for deep tissue applications. Nanomaterials with high loading capacity and surface tunability, such as mesoporous silica nanoparticles (MSNs) or polymers, facilitate the targeted delivery of chemiluminophores or catalysts into deep tissues.19,20 On the other hand, other nanomaterials actively participate in chemiluminescence reactions by catalysing the necessary reactions or acting as a chemiluminophore.21,22 Through this strategic approach, nanomaterials pave the way for light generation in deep tissue regions for light-based therapeutics.
2.1 Nanomaterials as delivery agents
To generate chemiluminescence in specific areas, nanomaterials have been utilised as delivery agents of chemiluminophores, oxidizers, or catalysts that catalyse the chemiluminescence reaction. This enables control over the location of chemiluminescence.Guo et al. encapsulated luminol with the photosensitiser chlorin e6 (Ce6) in poly(ethylene glycol).18 Following the reaction between luminol and H2O2, light emission occurs at 440 nm, which overlaps with the absorption spectra of Ce6 for the initiation of photodynamic therapy. Apart from delivering chemiluminophores, delivering oxidizers could also enhance chemiluminescence. Certain Fe3+-containing nanomaterials could generate H2O2 endogenously through the Fenton reaction, which increases the rate of chemiluminophore oxidation. For example, the MnFe2O4 core was encapsulated in the Zr-based MOF, where Fe3+ acted as a catalyst to generate H2O2via the Fenton reaction.23 H2O2 then reacted with the encapsulated luminol, resulting in enhanced chemiluminescence, while the Zr-based MOF acted as a photosensitiser. The system exhibited significant enhancement in chemiluminescence and high antitumour efficacy in vivo.
Catalysts for chemiluminescence reactions can also be delivered by nanomaterials to enhance their catalytic activity. Ren et al. conjugated hemin, a catalyst of luminol–H2O2 reaction, onto polymer dots (hemin–Pdots), resulting in a 700-fold enhancement in chemiluminescence (CL) intensity and 20-fold prolonged emission.24 The surface of the nanomaterial could be further engineered for targeting. For example, the surface of a PEGylated chemiluminophore could be modified with folic acid (FA), allowing specific binding to cancer cells due to the overexpressed folate receptor (FR).20
To ensure specifically targeted chemiluminescence, Cao et al. developed hemin–MSN@DNA, incorporating DNA gates onto the pores of mesoporous silica nanoparticles (MSNs) loaded with hemin.25 Upon degradation of DNA gates by specific bacteria, hemin was released from the pores, enhancing the chemiluminescence signal (Fig. 3a). The authors assessed the CL intensity on E. coli and S. aureus, and the results revealed a strong correlation between bacterial concentration and CL intensity (Fig. 3b and c), confirming specific chemiluminescence generation. In addition, the DNA gating on the pores of MSNs allows for a hybridization chain reaction on their surface, enabling the formation of a hydrogel coating.26 This further improved stability and loading capacity compared to traditional gated mesoporous silica systems.
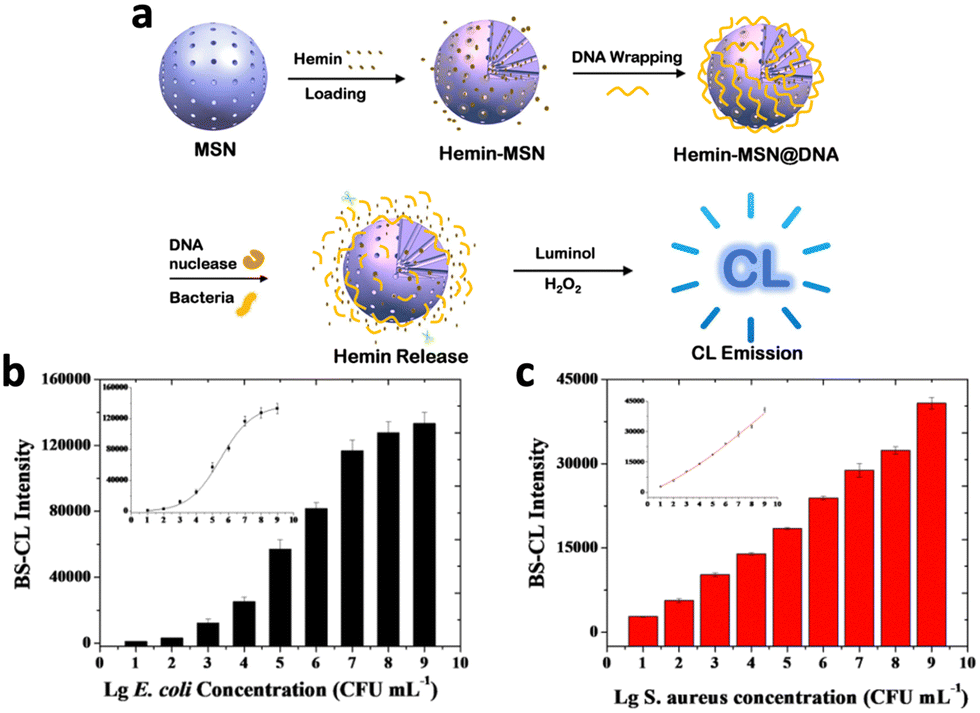 | ||
| Fig. 3 (a) Schematic representation of the mechanism of chemiluminescence generation from hemin–MSN@DNA. Correlation between chemiluminescence intensity of hemin-MSN@DNA and (b) E. coli and (c) S. aureus concentration. Reproduced with permission from ref. 25. Copyright 2019, American Chemical Society. | ||
2.2 Nanomaterials as catalysts for chemiluminescence enhancement
Instead of delivering the catalyst to the chemiluminescence reaction site, many metal-based nanomaterials offer intrinsic catalytic properties. In this section, we will discuss the nanomaterials that offer catalytic properties for different chemiluminescence reactions, including metal-based nanomaterials, organic nanomaterials and semiconducting nanomaterials.Chemiluminescence could also be increased through the aggregation of catalysts. For example, chemiluminescence was enhanced by aggregating Au NPs with complex DNA networks (CDNs).21 A hairpin (H1) exposed to the CDN is cleaved by a specific DNAzyme (BB′) in the network, forming a single-stranded hairpin (H1-1). Simultaneously, Au NPs that are tagged with nucleic acids complementary to H1-1(p,q) would be assembled by H1-1 to form an aggregated state (Fig. 4a). At a higher DNAzyme (BB′) level, the absorption band of the CDN decreased more quickly, indicating more efficient Au NP aggregation (Fig. 4b and c). In addition, aggregated Au NPs also produced stronger chemiluminescence compared to the un-aggregated ones (Fig. 4d). Finally, the TEM image validated the stronger aggregation of Au NPs in the CDN with a higher BB′ level (Fig. 4eIII and IV) as compared to a lower BB′ level (Fig. 4eI and II).
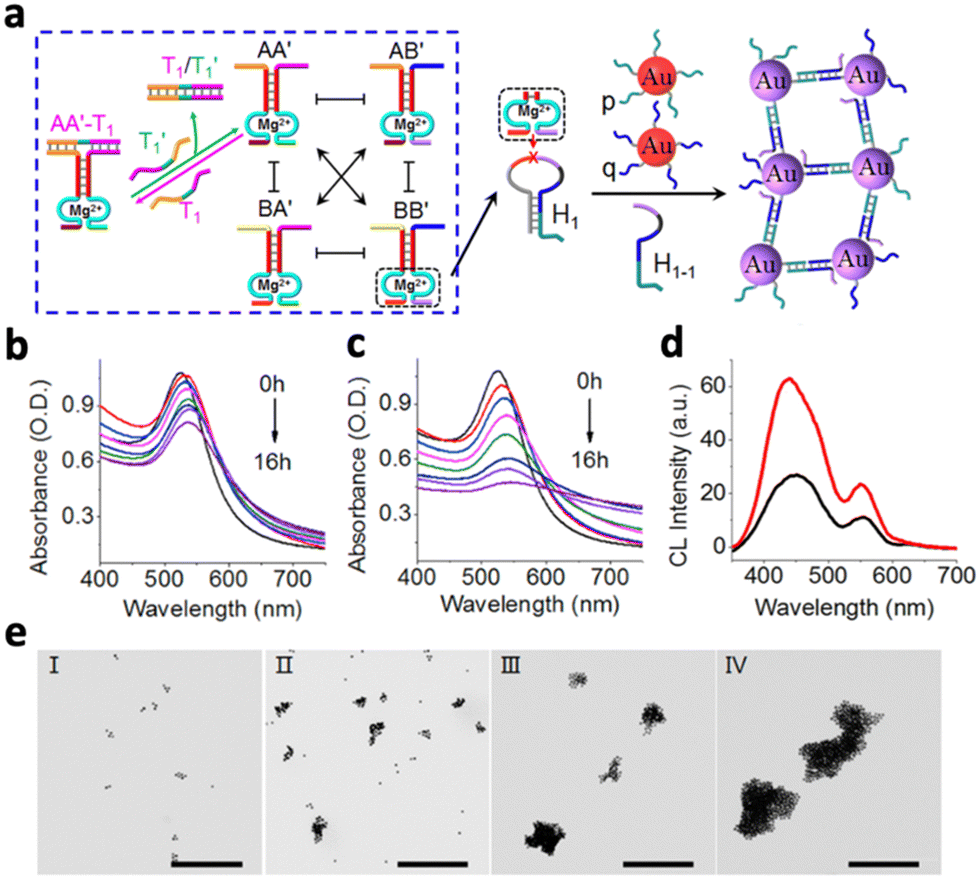 | ||
| Fig. 4 (a) Schematic illustration of the time-dependent aggregation of Au NPs. Time-dependent absorbance spectra of the complex DNA network with (b) low concentration of BB′ DNAzyme and (c) high concentration of BB′ DNAzyme. (d) Chemiluminescence emission spectra generated by the control (black curve) and the aggregated Au NPs (red curve). (e) TEM image corresponding to the aggregates of the Au NPs generated by CDN with a low BB′ level after 4 h (panel I) and 8 h (panel II) of aggragation and by CDN with a high BB′ level after 4 h (panel III) and 8 h (panel IV) of aggregation, respectively. Reproduced with permission from ref. 21. Copyright 2018, American Chemical Society. | ||
Silver and platinum nanoparticles also enhance the luminol–H2O2 reaction, with a similar catalytic mechanism to Au NPs.31,32 It was also found that the catalytic activity of silver nanoparticles (AgNPs) increased with the aggregation of small Ag NPs, due to the increase in the electron density in the Ag NPs' conduction bands.33 Other than gold, silver and platinum, copper(II) compounds like CuFe2O4 nanospheres could also act as efficient catalysts for luminol-based chemiluminescence reactions.34 This is because Cu2+ catalyses the formation of CL intermediates such as superoxide radicals (O2˙−) and hydroxyl radicals (˙OH).
Since metal–organic frameworks (MOFs) provide a high surface area for catalysis, constructing MOFs with catalytic metal ions would be more effective in enhancing chemiluminescence. For example, metal–organic framework (MOF) nanoparticles composed of Zr4+ and Cu2+ ions bridged by 2,2′-bipyridine-5,5′-dicarboxylic acid ligands provided almost 20-fold improvement in catalytic activity compared to Cu2+ ions alone.35 In addition, Fe-based MOFs modified with AuNPs showed around 110-fold enhancement in CL intensity compared to AuNPs alone.36
Instead of relying on radical generation, certain DNAzymes exhibit peroxidase-like activity and can be applied to catalyse the luminol–H2O2 reaction. For example, the microRNA (miRNA)-responsive DNAzyme system could enhance chemiluminescence intensity by 3 fold at 100 pm.38 The presence of a target microRNA triggered the assembly of the G4 DNAzyme following binding with specific hairpin probes, resulting in the specific catalysis of CL from the luminol–H2O2 reaction. However, a G4 DNAzyme has limited stability, as it is susceptible to many degradation pathways like nuclease attack. Instead of using DNA networks, DNAzymes can be conjugated on the surface of a gold nanoparticle (SNAzymes), protecting them from degradation.39 The SNAzymes not only displayed an improved resistance to nuclease degradation as compared to the G4 DNAzyme, but also improved the density of DNA on the nanoparticle (∼150 nucleic acids per particle). This eventually showed 100-fold CL signal enhancement compared to 1 molecule of G4 DNAzyme.
TGA-capped CdTe quantum dots (QDs) catalysed the luminol–KIO4 CL system by generating radicals.41 KIO4 is stongly oxidizing and injected a hole into the valence band of CdTe QDs, producing O2˙−. The presence of a reducer, hydroxide ions (OH−), injected an electron into the conduction band of CdTe QDs, producing ˙OH. These active oxygen-containing reactant intermediates accelerated the oxidation reaction of luminol and increased CL emission.
2.3 Nanomaterials as chemiluminophores
Certain nanomaterials can absorb energy from various CL systems to emit light. There are 2 major mechanisms: first, the nanomaterial could be injected with electrons and holes by the radicals in the CL system, where the electron–hole combination results in light emission. Otherwise, the nanomaterial accepts energy from excited radicals in the CL system through chemiluminescence resonance energy transfer (CRET).NaYF4:Yb3+/Er3+ nanoparticles, although being widely studied for their light upconversion properties, were shown to be able to absorb the energy from radicals in the NaHCO3–NH4OH–H2O2 CL system and emit light.52 The reaction between H2O2 and NaHCO3 yields reactive species such as ˙OH, ˙CO3−, and ˙O2− radicals, normally emitting luminescence at 441, 480, 580 and 634 nm (Fig. 5b). Following incubation with NaYF4:Yb3+/Er3+, Yb3+ sensitized the CL emission and transferred energy to the activator ion Er3+ (Fig. 5a). Relaxation of Er3+ resulted in enhanced CL emissions at 523, 544, and 653 nm, corresponding to the 2H11/2–4I15/2, 4S3/2–4I15/2, and 4F9/2–4I15/2 transitions of Er3+ (Fig. 5c). The disappearance of emission peaks from the original CL system indicated efficient energy transfer to NaYF4:Yb3+/Er3+, while CL intensity was enhanced by 334 fold.
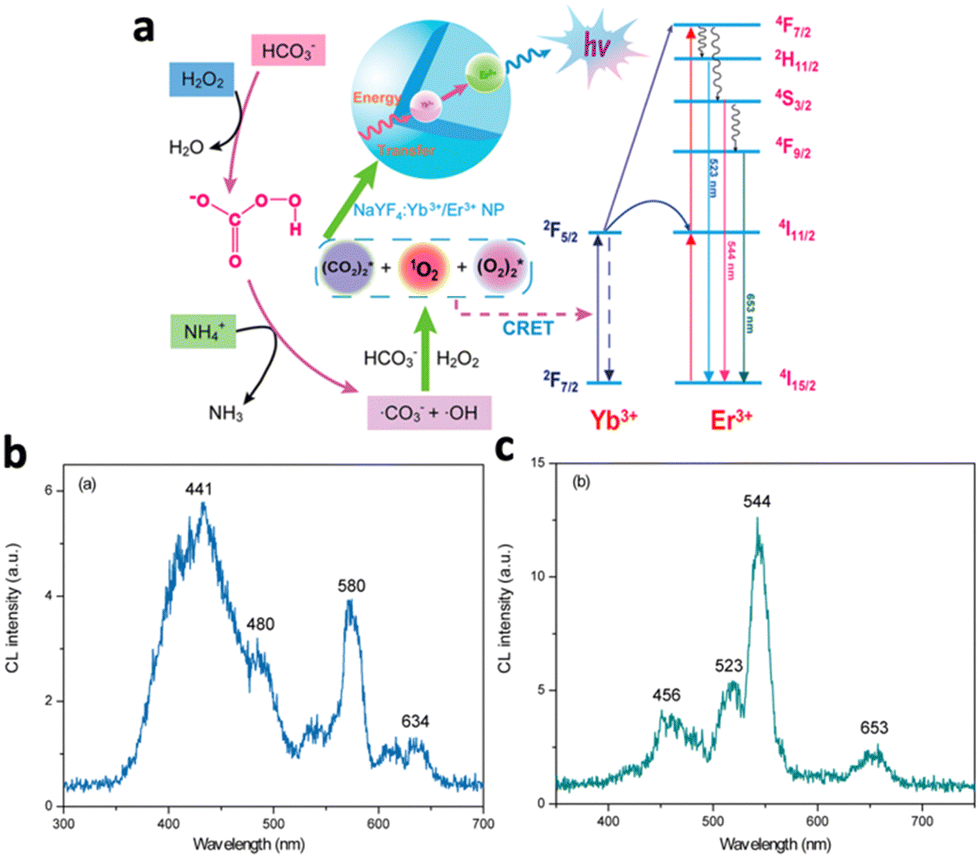 | ||
| Fig. 5 (a) Schematic illustration of the chemiluminescence mechanism of NaYF4:Yb3+,Er3+. Yb3+ absorbs energy from radicals and transfers energy to excite Er3+, which eventually relaxes and emits 3 characteristic wavelengths. (b) Emission spectrum of the NaHCO3–NH4OH–H2O2 system. (c) Emission spectrum of the NaHCO3–NH4OH–H2O2 system in the presence of NaYF4:Yb3+/Er3+. Reproduced with permission from ref. 52. Copyright 2012, American Chemical Society. | ||
2.4 Chemiluminescence-mediated light therapy
The ability of chemiluminescence to generate light endogenously without external light activation made many deep-tissue light-mediated therapies possible, including photodynamic therapy (PDT), photothermal therapy (PTT) and drug delivery. The efficacy of PDT is mainly based on the intensity of CL, which ultimately depends on the intracellular concentration of oxidizers like H2O2. However, intracellular H2O2 is limited and efforts have been undertaken to augment H2O2 levels to enhance the efficacy of PDT. For PTT and drug delivery, chemiluminescence is primarily employed for tracking the localization of photothermal agents or drugs. Therefore, current studies are primarily focused on the development of H2O2-generating nanoystems, nanosystems that emit near-infrared (NIR) chemiluminescence and “turn-on” nanoprobes that exclusively activate within the vicinity of the target tissue.In 2011, Jiang et al. produced a novel PDT system, which leveraged the chemiluminescence reaction between peroxalate ester and H2O2.53 They prepared polyoxometalate (POM) by encapsulating a peroxalate ester oligomer, a fluorescent dye (rubrene) and photosensitiser TPP within PEG–PCL copolymer micelles. Peroxalate in POMs reacted specifically with the enriched-H2O2 around the tumour area and formed 1,2-dioxetanedione, which emitted light to excite rubrene and TPP. This system was shown to exhibit significant anti-cancer efficacy against C6 and Lovo cells lines in vitro. To ensure more specific tumour targeting, Zhang et al. reported an active-targeting nanosystem (POCL) that uses folate as a targeting ligand.20 They co-delivered bis(2-carbopentyloxy-3,5,6-trichlorophenyl)oxalate (CPPO) and the photosensitizer tetraphenylporphyrin (TPP), where the CL from the reaction between CPPO and H2O2 excited TPP, generating 1O2. In vivo studies demonstrated a higher accumulation of this nanosystem in the tumour area, which was 3-fold greater compared to POCL without folate. After 21 days of treatment on tumour-bearing mice, the average tumour weight in the POCL group was significantly reduced by 85% compared to the untreated group.
However, the efficacy of CL-triggered PDT was still limited by the intracellular H2O2 level, which is often less than 100 μm. Hence, Li et al. proposed a CPPO-based nanoplatform (C1@M@C2G) that could self-generate H2O2 to improve treatment efficacy.54 The nanoplatform consisted of CPPO and the photosensitizer porphyrin encapsulated by Fe-MOF nanoparticles and glucose oxidase (GOD). GOD catalysed the decomposition of glucose from the tumour area, generating H2O2 and gluconic acid, which created a H2O2-rich and acidic microenvironment. A portion of the increased H2O2 underwent Fenton reaction catalysed by Fe in the MOF under a low pH environment, generating oxygen to combat hypoxia in the tumour area (Fig. 6a). The remaining H2O2 reacted with CPPO to produce CL that excited the porphyrin photosensitizers in the MOF, resulting in the generation of 1O2 (Fig. 6a). The incubation of the nanoplatform with more glucose resulted in higher H2O2 production, accompanied by a drop in pH from 6.8 to 2.6 after 10 minutes (Fig. 6b and c). In addition, 1O2 production was enhanced around 2-fold in the presence of glucose at 1 mg mL−1, accompanied by the greatest tumour inhibition compared to the control groups without GOD (Fig. 6d and e). Other than relying on glucose to generate H2O2, a recent study demonstrated the possibility to generate H2O2 by reacting with intracellular water.55 A nanosystem (mSCCC@SA) combining CaO2, CPPO and the photosensitiser Ce6 was introduced, where CaO2 reacted with intracellular water to generate H2O2 and O2, resulting in stronger CL and enhanced PDT efficacy. This nanosystem reduced the cell viability of HepG2 cells to 40% while the control groups without CaO2 maintained a cell viability close to 90%.
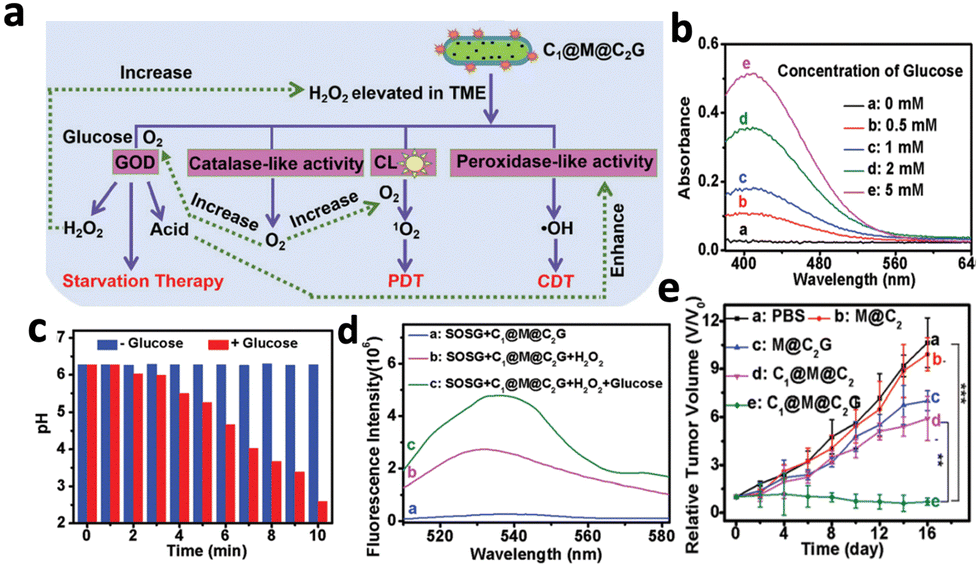 | ||
| Fig. 6 (a) Schematic illustrations of the enhanced PDT by C1@M@C2G through enhanced generation of H2O2 combined with other therapies. (b) Absorption spectra of C1@M@C2G particles and glucose, where higher absorbance refers to higher H2O2 content. (c) pH measurement of the C1@M@C2G particle following addition of glucose (1 mg mL−1) over time. (d) Fluorescence spectra of SOSG (1O2 indicator) added to C1@M@C2G after mixing with H2O2 and glucose, where higher fluorescence indicates higher 1O2 production. (e) In vivo tumour growth curves of mouse groups with different treatments. Reproduced with permission from ref. 54. Copyright 2021, Royal Society of Chemistry. | ||
Gold nanoparticles (AuNPs) have been applied for photothermal therapy due to their ability to generate heat upon NIR activation. However, these small AuNPs are easily excreted from the body, resulting in low tumour accumulation and poor therapeutic efficacy. A recent study by Shi et al. demonstrated the possibility of photo-cross-linking small Au NPs by CL to prevent rapid clearance from the tumour area.57 Small AuNPs (25 nm) were covalently conjugated with photolabile molecules and luminol (t&mAuNP/Lu). Under the reaction between luminol and H2O2, the luminescent nanoparticles emitted chemiluminescence and induced cross-linking of the AuNPs, forming covalently cross-linked AuNP aggregates (Fig. 7a). Tumours treated with t&mAuNP/Lu nanoparticles showed a local temperature increase to 55.4 °C following 10 minutes of 808 nm laser irradiation, while other treatment groups without luminol remained below 45 °C (Fig. 7b). In vivo studies with mice bearing 4T1 tumours showed significant growth inhibition following PTT with t&mAuNP/Lu (Fig. 7c). Apart from improving therapeutic efficacy, chemiluminescent nanoparticles could also provide image guidance to PTT. Li et al. developed a nanoplatform (ALPBs) that incorporates luminol and PCPDTBT, which is a photothermal agent with NIR emission.58 The CL generated from the luminol–H2O2 reaction activated PCPDTBT to produce NIR luminescence, which exhibited a strong intensity of several magnitudes higher than the CL in vivo. In addition, CL diminished after 5 hours while the NIR luminescence still exhibited high intensity after 7 hours. This enabled the monitoring of photothermal therapy in deep tissues using CL.
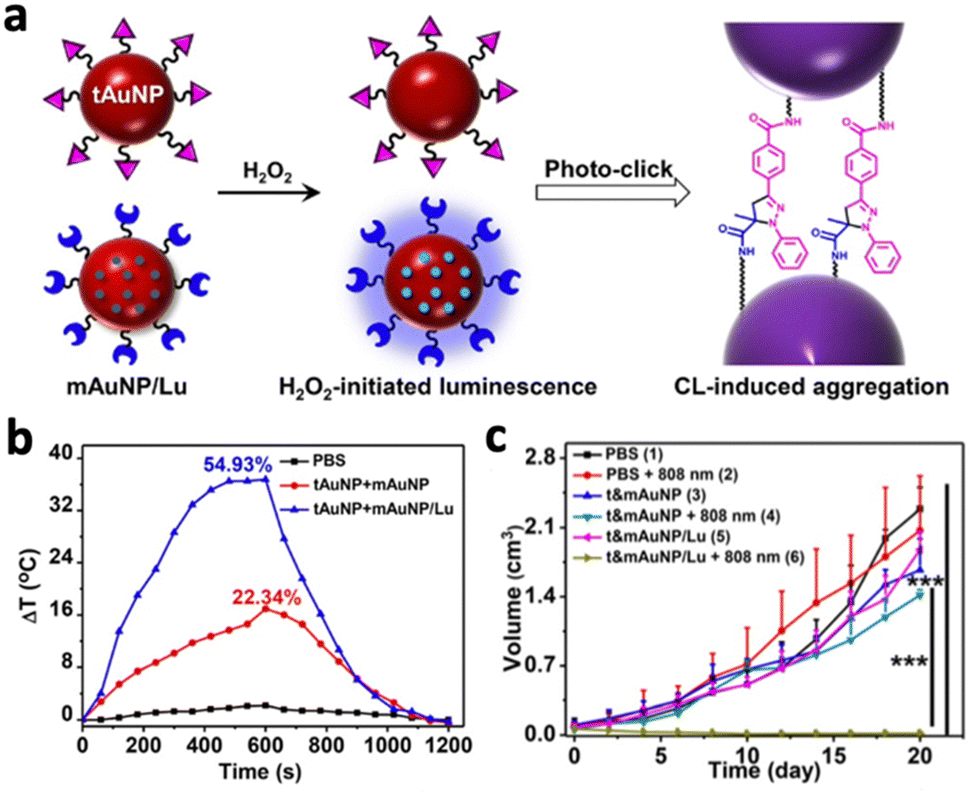 | ||
| Fig. 7 (a) Schematic illustration showing the chemiluminescence reaction between luminol on Au NPs that led to photoclicking of Au NPs. (b) Temperature changes following 808 nm irradiation on PBS, t&mAuNP and t&mAuNP/Lu. (c) Change in tumour volume of mice following 21 days of photothermal treatments with different systems. Reproduced with permission from ref. 57. Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Chemiluminescent nanomaterials could also trigger the release of light-sensitive drug for the treatment of inflammatory diseases. Wang et al. devised a ROS-responsive drug delivery system using covalently self-assembled polymer nanocapsules (Azo-NCs), which were formed through the cross-linking of macrocyclic cucurbit[6]urils with a photosensitive azobenzene derivative (Azo).59 Luminol was co-loaded into the Azo-NCs along with a therapeutic payload. Under inflammatory conditions, the upregulated H2O2 reacted with luminol to produce enhanced CL which induced photoisomerization of the Azo groups within the Azo-NCs, releasing the encapsulated payload. In vivo studies with a zebrafish model demonstrated a 5.5-fold higher CL intensity accompanied by enhanced drug release in the inflammatory regions compared to the healthy regions.
2.5 Strengths and weaknesses of chemiluminescence-mediated light therapy
Chemiluminescence-mediated light therapy has emerged as an intriguing therapeutic approach that offers deep tissue light-based therapies without the need for external light irradiation. Unlike traditional light-based therapies, light penetration depth is no longer a concern as it harnesses intrinsic chemiluminescent properties to generate light. Moreover, chemiluminescence-mediated light therapy could passively target inflammation-related diseases, including cancer, cardiovascular diseases, and autoimmune conditions. In most inflammatory conditions, reactive oxygen species like H2O2 are upregulated, which can react more rapidly with the chemiluminophores to generate stronger CL.However, there are several limitations to chemiluminescence that should be considered. For example, chemiluminescence-mediated light therapy lacks external control over CL generation. Unlike other light-based therapies excited with external sources, controlling the intensity and duration of chemiluminescent reactions is difficult. Furthermore, the therapeutic efficacy is highly dependent on the availability of intracellular ROS, which could be insufficient to generate a strong CL. Hence, the produced CL is usually too weak to serve as an effective therapy alone or to be detected in vivo. Although certain approaches have been developed to generate ROS endogenously, it highly depends on the reaction rate and kinetics. Moreover, the lack of NIR-emitting chemiluminophores also limits the imaging and tracking of deep tissue therapy.
3. NIR-excited luminescence
Using an external excitation source gives better control over the intensity and duration of the light generated in vivo. As mentioned before, both UV and visible light possess low tissue penetration and would not be suitable for deep tissue therapy. However, biological tissues possess an “optical transparency window” for light with wavelength ranging from 800–1000 nm, where they experience less light scattering and attenuation.60 Hence, studies have been conducted to engineer nanosystems that could be activated by NIR-I light (700–1000 nm), especially at 808 nm or 980 nm.61,62 Typically, NIR light excites nanomaterials to emit luminescence through 4 major mechanisms: two-photon excitation (TPE), energy transfer upconversion (ETU), downconversion (DC) and persistent luminescence (PersL) (Fig. 8).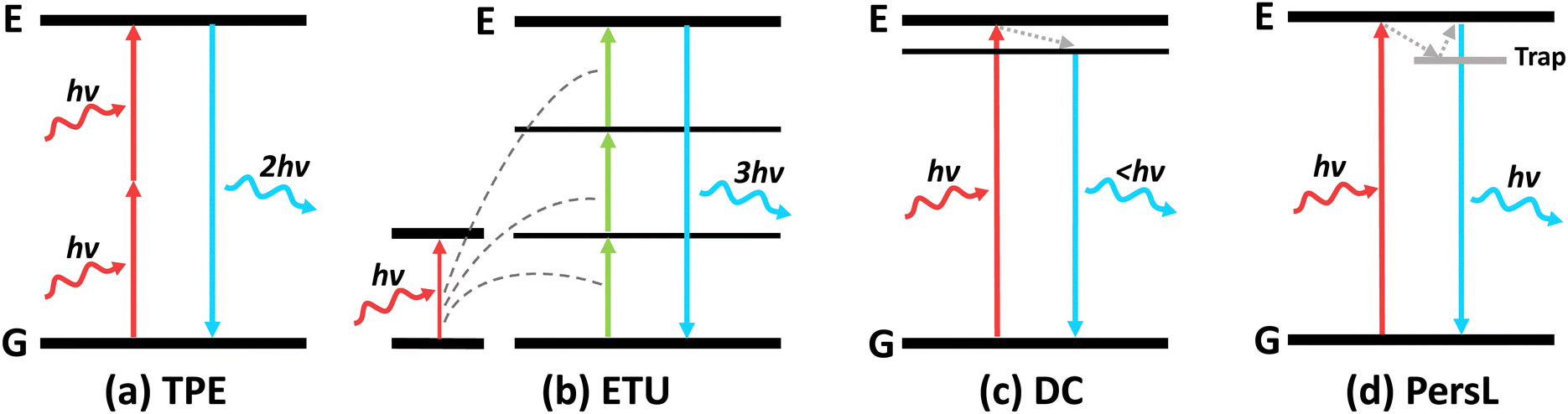 | ||
| Fig. 8 Schematic illustrations of the energy transfer mechanisms of (a) two-photon excitation (TPE), (b) energy transfer upconversion (ETU), (c) downconversion luminescence (DC) and (d) persistent luminescence (PersL) in NIR-excitable nanomaterials. | ||
Upconversion nanomaterials exhibit anti-Stokes behaviour and are activated by NIR light to emits light at a shorter wavelength.63 They emit in the UV/visible wavelength range to activate various light-sensitive molecules, such as photosensitizers, for light-mediated therapy. More recently, it was realised that NIR-II light (1000–1700 nm) possesses even less scattering in biological tissues and demonstrates deep tissue imaging ability at higher resolution than NIR-I light.9 Hence, downconversion nanomaterials were also engineered to be excited by NIR-I light to emit in the NIR-II region for deep tissue imaging and monitoring of therapeutic process.64,65 Due to the low tissue attenuation of NIR-II light, they were also used to excite certain upconversion nanosystems to increase the tissue depth for therapy.7
In this section, we will discuss the utilisation of NIR light located in the range of 700–1700 nm on activating upconversion or downconversion nanosystems and their application in deep tissue light-mediated therapy. Moreover, we will also summarise the strategies to enhance the luminescence produced by various nanosystems.
3.1 Upconversion luminescence
Upconversion luminescence typically refers to the anti-stoke shifts in emission, where the emission wavelength is always shorter than the excitation wavelength.63 Many mechanisms of upconversion luminescence exist, including two-photon excitation (TPE), frequency upconversion (FU) and energy transfer upconversion (ETU) (Fig. 8a–c).66–68 In the following section, we will review nanomaterials that exhibit upconversion luminescence and their corresponding mechanisms.However, the efficiency of two-photon excitation is low due to the low probability of 2 photons exciting the nanomaterial simultaneously. Hence, Peng et al. developed an organic upconversion nanomaterial that depends on single-photon excitation.68 The organic nanomaterial (FUCP-1) contains a rhodamine derivative (FUCP-1) and produces upconversion luminescence through frequency upconversion (Fig. 9a). FUCP-1 was first excited from the ground electronic state (S0) to thermally vibrational–rotational states (St) with the heat derived from the Boltzmann distribution of molecules (Fig. 9b). Therefore, a higher temperature enhances upconversion luminescence intensity (Fig. 9c). Following 808 nm excitation, FUCP-1 is excited from St to S1 and returns to the ground state, producing emission at 750 nm. FUCP-1 was shown to effectively sensitize O2 to 1O2 and presented superior inhibition of 4T1 cells. Although this nanosystem exhibited higher efficiency than most two-photon-excited nanomaterials, the emission wavelength was located near the excitation wavelength. This is due to the small energy level difference between St and S0, which limits its upconversion ability for application in deep tissue therapy.
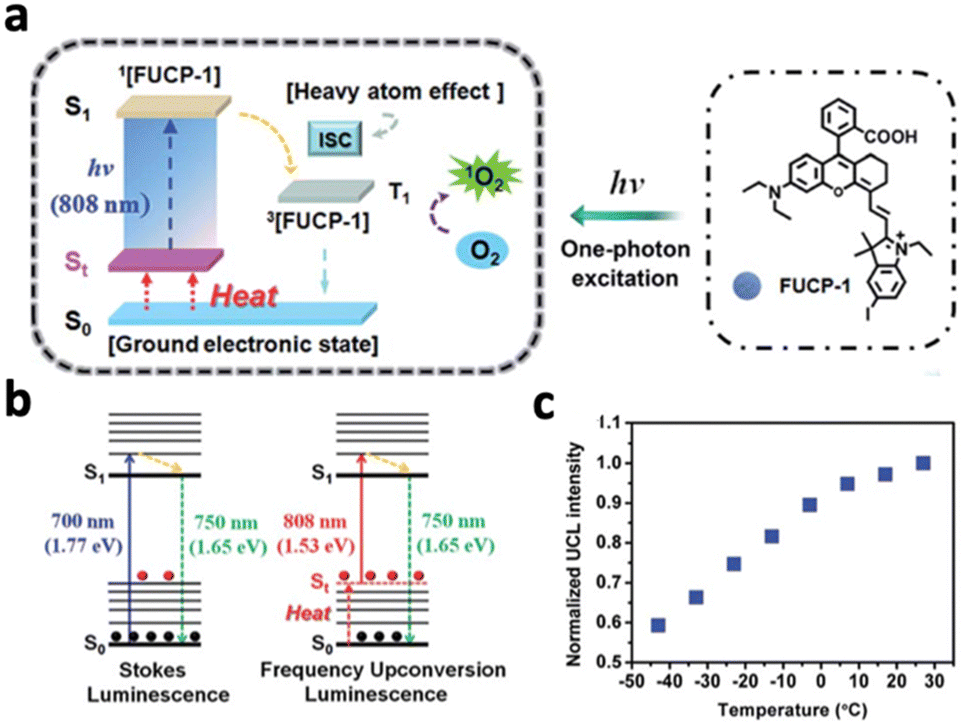 | ||
| Fig. 9 (a) Schematic diagram of upconversion luminescence and PDT mechanism of FUCP-1. (b) Schematic energy level demonstration of the mechanisms of traditional Stokes luminescence and frequency upconversion luminescence (FUCL). (c) Upconversion luminescence intensity changes of FUCP-1 at different temperatures. Higher temperature results in stronger UCL due to more St states. Reproduced with permission from ref. 68. Copyright 2019, Royal Society of Chemistry. | ||
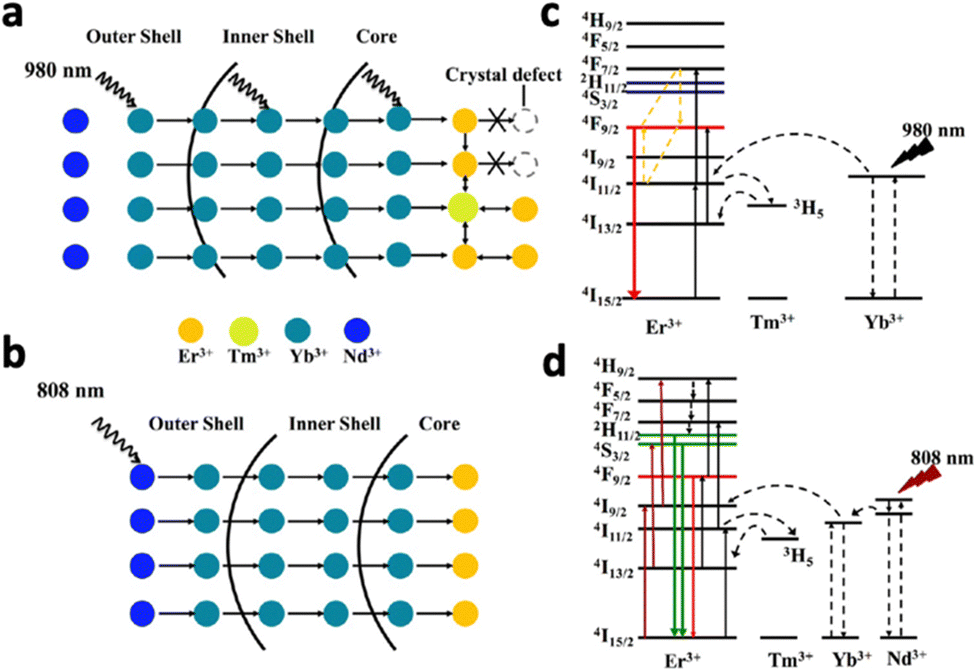
Upconversion NaYF4:20% Yb3+,2% Tm3+ nanocrystals were first reported by Chow et al. in 2006.76 Yb3+ acted as the sensitiser, absorbing NIR photons at 980 nm and transferring the energy to the activator Tm3+. Following 980 nm NIR excitation, NaYF4:20% Yb3+,2% Tm3+ exhibited blue fluorescence at 450.5 (1G4–3H6), 475 nm (1G4–3H6) and 800 nm. By changing the lanthanide dopants in the crystal, excitation and emission wavelengths can be altered. For example, substituting Tm3+ in NaYF4:Yb3+,Tm3+ with Nd3+ resulted in emission at 803 nm following 980 nm laser irradiation, due to the 2H9/2,4F5/2–4I9/2 transition of Nd3+.77 Exciting NaYF4:Yb3+,Tb3+ at 980 nm resulted in green upconversion emission at 480 nm, corresponding to Tb3+ transition at 5D4–7F6.75 In addition, doping Er3+ into NaYF4:Yb3+ resulted in visible emissions at 550 nm, 660 nm and 800 nm and 1000 nm, corresponding to 4S3/2–4I15/2, 4F9/2–4I15/2, 4I9/2–4I15/2 and 4I11/2–4I15/2 transitions of Er3+, respectively.76 Apart from acting as an activator, Er3+ could also act as a sensitizer by absorbing NIR-II light at 1500 nm due to its transition from 4I15/2 to 4I13/2.78 Therefore, exciting NaYF4:Er3+ at 1500 nm results in the same emissions at 550 nm, 660 nm and 800 nm and 1000 nm. The ability of Er3+ to be excited by NIR-II light is beneficial since NIR-II light exhibits higher tissue penetration depth than NIR-I light, allowing the nanosystems to be activated in deep tissue. However, despite its ability to be excited at 1500 nm, Er3+ is not efficient in transferring its energy to other activators due to its highly efficient upconversion (Fig. 10a). Therefore, Tian et al. constructed a multi-layered nanocrystal that was composed of a Yb3+-rich core doped with activators (NaYbF4:Ln, Ln = Ho and Tm), with the Er3+-doped NaYF4 coated on top.79 Upon excitation at 1550 nm, the localized enrichment of Yb3+ enables efficient energy transfer from Er3+ to Yb3+ ions via inter-ion energy transfer, effectively suppressing the multiphoton upconversion of Er3+ (Fig. 10b). Enhanced emissions at 1200 nm for Ho3+ and 1470 nm for Tm3+ were observed (Fig. 10c and d). Given that Ho3+ and Tm3+ also have emission peaks in the visible spectrum, this nanosystem was able to emit both visible and NIR-II photons following NIR-II activation, which would be beneficial for the simultaneous activation of phototherapeutic systems and imaging modes.
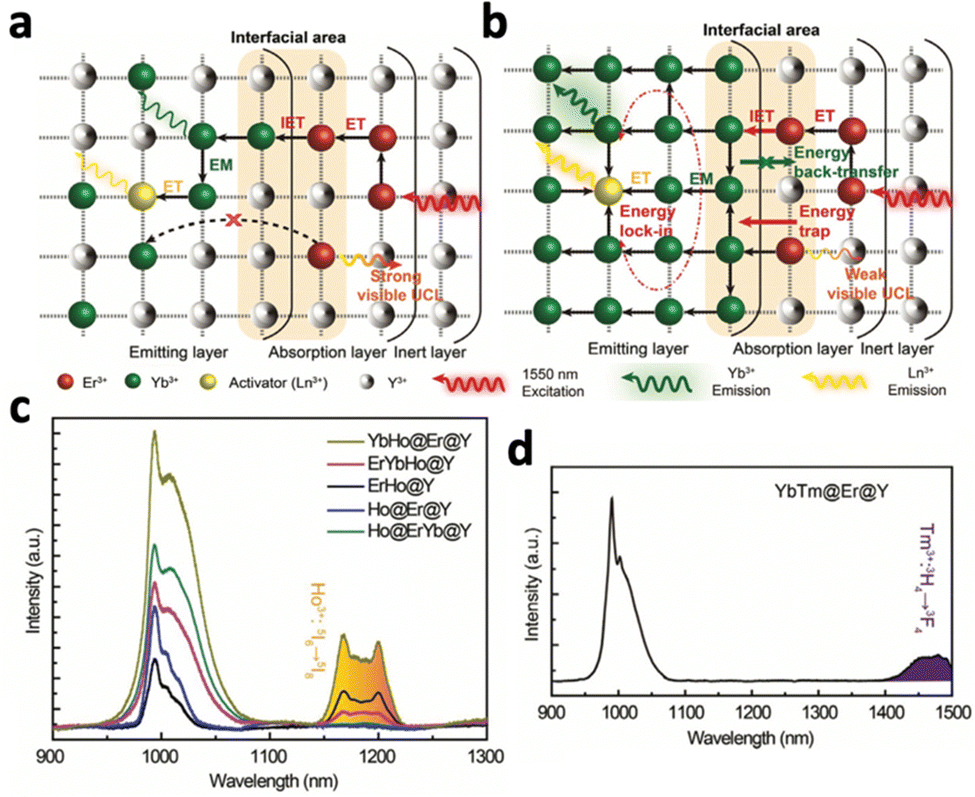 | ||
| Fig. 10 Schematic illustration of energy transfer in the Er3+-sensitized nanomaterial under 1550 nm excitation. (a) Common strategy of low Yb3+ doping and relying on Er3+ to relay energy, which results in low ET efficiency. (b) Proposed design strategy using the locally enriched Yb3+ embedded in the emitting layer relays the excitation energy from the Er3+-sensitized absorption layer to Ln3+. NIR upconversion emission spectra of (c) Yb,Ho@Er@Y, Er,Ho@Y, Er,Yb,Ho@Y, Ho@Er@Y, Ho@Er,Yb@Y and (d) YbTm@Er@Y. Reproduced with permission from ref. 79. Copyright 2022, Royal Society of Chemistry. | ||
It was reported that the hexagonal phase of NaYF4 favours upconversion luminescence due to a significant electron cloud distortion of the cations.76 Aiming to stabilize the hexagonal phase of NaLnF4 nanocrystals, Gd3+ was doped into different systems like NaYF4:Yb3+,Er3+ and NaYbF4:Tm3+.61,80 Higher Gd3+ doping resulted in a higher population of the hexagonal phase, corresponding to a 40-fold stronger upconversion intensity for NaYbF4:Tm3+. This raised interest in the complete substitution of Y3+ ions in NaYF4 with Gd3+ ions to improve upconversion luminescence, producing NaGdF4:Yb3+,Er3+ and NaGdF4:Yb3+,Tm3+.74,81 In addition, high upconversion efficiency was observed in NaGdF4-based nanosystems, since the lowest excited level (6P7/2) of Gd3+ is in the ultraviolet region. This energy level is significantly higher than the excited levels of most sensitizers and activators like Yb3+ and Er3+.81 Therefore, energy loss through energy transfer from Yb3+ and Er3+ to the 4f levels of Gd3+ is avoided.
Apart from NaLnF4, certain other lanthanide-based hosts possessing low phonon energy could also be doped with a sensitizer and activator to exhibit upconversion luminescence. Examples include KYb3F10:Yb3+,Tm3+, Y2O3:Yb3+,Tm3+, Y2O3:Yb3+,Er3+ or Gd2O3:Yb3+,Er3+ and GdOF:Yb3+,Er3+, which possess high stability in the hexagonal phase.82–86 Certain non-lanthanide-based hosts could also emit upconversion luminescence following lanthanide doping, including ZnGa2O4:Yb3+,Tm3+,Eu3+-doped [Eu(THA)3(phen)] (HTHA = 4,4,4-trifluoro-1-(9-hexylcarbazole-3-yl)-1,3-butanedione, phen = 1,10-phenanthroline) and SrF2:Ho3+.87,88 Similar to Er3+, Ho3+ could also act as both the sensitizer and activator. Under 1156 nm excitation, Ho3+ exhibited upconversion luminescence at 554 nm, 653 nm, 755 nm, and 900 nm due to the 5S2,5F4–5I8, 5F5–5I8, 5S2,5F4–5I7 and 5I5–5I8 transitions.87
By bridging Yb3+, Er3+ and Y3+ with the organic linker pyrazine, an upconverting organic–inorganic hybrid was constructed.89 Possessing high surface area and porosity, the upconversion luminescence produced by organic–inorganic hybrids interact more strongly and in closer proximity with biomolecules for therapeutic applications. The nanohybrid absorbed NIR photons of 974 nm with the sensitiser Yb3+, which transferred the energy to the activator Er3+, resulting in efficient emission of red and green upconversion luminescence. The hybrid also enabled the creation of Yb3+–Er3+ pairs, facilitating efficient energy transfer while shielding the energy transfer and relaxation processes from the vibrations of the ligands.
3.2 Downconversion luminescence
Downconversion luminescence refers to conventional fluorescence, where the emission wavelength is longer than the excitation wavelength due to Stoke's shift. Downconversion luminescence can be useful in monitoring deep-tissue therapy as it can potentially produce emissions in the NIR-II region that possess deep biological penetration. In this section, we will discuss the nanomaterials that exhibit downconversion luminescence, especially in the NIR-II region, following NIR activation.However, certain limitations still exist for organic-based nanomaterials that hinder their application in deep tissue light-mediated therapy – such as the broad emission peaks, short luminescence lifetime, susceptibility to photobleaching and the lack of control and tunability over excitation and emission wavelength.
Although NaLnF4 is well-known for its upconversion properties, it can also exhibit downconversion luminescence with similar lanthanide dopants. Not only could Er3+ and Ho3+ be activated by NIR-II luminescence at 1550 nm and 1150 nm respectively (mentioned in the upconversion section), they could also emit NIR-II downconversion luminescence at the same wavelengths.92 By doping different activators like Ho3+ (20%), Pr3+ (3%), Tm3+ (4%) and Er3+ (10%) into NaYbF4, the nanosystem produced emissions in the NIR-II region–1155 nm (Ho3+), 1310 nm (Pr3+), 1475 nm (Tm3+), and 1525 nm (Er3+) following 980 nm excitation.93 The emission peaks were assigned to the transitions 5I6–5I8, 1G4–3H5, 3H4–3F4, and 4I13/2–4I15/2 for Ho3+, Pr3+, Tm3+, and Er3+, respectively.
It has been reported that NIR light at around 808 nm exhibits less attenuation in biological tissues compared to the traditional 980 nm excitation wavelength of Yb3+ ions.94 Therefore, Li et al. co-doped Nd3+ with Yb3+ to form CaF2:Yb3+,Nd3+, where Nd3+ was excited by 808 nm photons to the 4F5/2 energy level.95 Nd3+ then transferred the energy to the 2F5/2 level of Yb3+, leading to the emission of 980 nm photons through 2F5/2–2F7/2 transition. Apart from relaying energy to Yb3+, Nd3+ itself could emit downconversion luminescence in the NIR-I/II regions. Following 808 nm excitation, Nd3+ emitted at 903 nm, 1062 nm, and 1336 nm corresponding to the 4F3/2–4I9/2, 4F3/2–4I11/2, and 4F3/2–4I13/2 transitions.96 Similarly, Tm3+ in LiTmF4 could be excited by either 800 nm or 1208 nm radiation, leading to the emission of NIR-IIc photons (1600–2100 nm) due to the 3F4–3H6 transition of Tm3+, allowing even deeper tissue penetration and a higher SBR in in vivo imaging.8 Apart from Nd3+ and Tm3+, Ni2+ could also be excited by 808 nm. Under 808 nm excitation, ZnGa2O4:Ni2+,Er3+ experienced the population of the 3T2(3F) level in Ni2+.97 A portion of the energy relaxes radiatively from 3T2(3F) to 3A2(3F), emitting 1280 nm luminescence, while another portion of the energy is transferred to the 4I13/2 level of Er3+, which then relaxes and emits light at 1550 nm due to the 4I13/2–4I15/2 radiative transition.
3.3 Persistent luminescent nanoparticles
Persistent luminescence nanoparticles (PLNPs) are luminescent nanoparticles that retain the excitation energy within the material through trapping mechanisms, allowing them to slowly emit photons over extended durations even after the excitation source has been removed.104 Following excitation, electrons are tunnelled to traps and are slowly released to emit luminescence (Fig. 8d). This unique phenomenon does not require a continuous excitation source for light emission, resulting in enhanced signal-to-noise imaging and deep tissue therapy without the need for continuous excitation sources, eliminating side effects like light scattering and tissue damage.However, most PLNPs could only be activated by high-energy excitation sources like UV/X-rays to pump the excitons into the deep traps (Fig. 11a).14,105,106 Although certain PLNPs could be re-activated by NIR light after UV excitation, the intensity and afterglow time were significantly reduced.105,107 To engineer PLNPs that can be directly activated by NIR light, Han's group developed CaSnO3:Bi2+, which exhibited persistent luminescence at 810 nm following 700 nm NIR excitation.108 This nanophosphor operates through an “upconversion-like” carriers’ transition process, as the NIR light first excites the deep trap due to its low energy, and slowly transitions to the shallow traps that have higher energy (Fig. 11b). The emission band at 810 nm was assigned to the 2P3/2(1)–2P1/2 transition of Bi2+ (Fig. 11b). Compared with the traditional PLNP ZnGa2O4:Cr3+ excited with NIR light, CaSnO3:Bi2+ exhibited 10-fold stronger persistent luminescence (Fig. 11c).
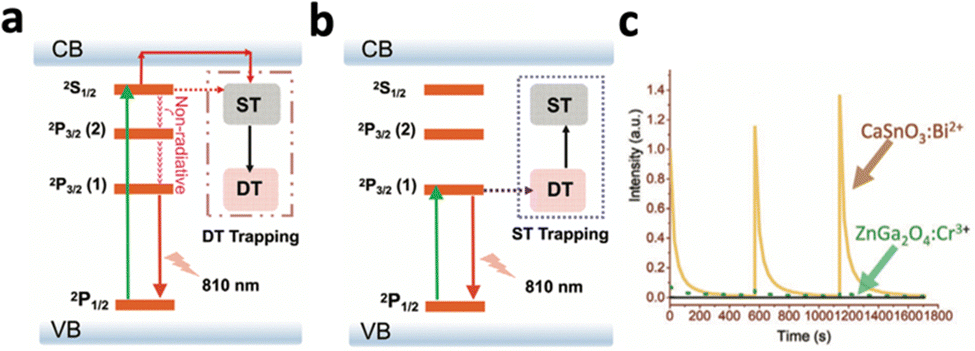 | ||
| Fig. 11 Schematic illustration of the energy transfer mechanism during (a) high-energy (UV) photon-excited afterglow and (b) low-energy (NIR) photon-excited afterglow. (c) Afterglow decay curves of CaSnO3:Bi2+ and ZnGa2O4:Cr3+. Reproduced with permission from ref. 108. Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Efforts have also been made to synthesize upconverting persistent luminescent nanomaterials. One method is to physically combine upconversion nanomaterials with persistent luminescence nanomaterials, such that the emission spectra of the UCNP overlaps with the excitation spectra of PLNP to generate persistent luminescence.109,110 In 2017, Li's group produced a nanohybrid consisting of upconversion nanoparticles (NaYbF4:Tm@NaYF4) and persistent nanoparticles (Zn1.1Ga1.8Ge0.1O4:0.5% Cr).109 Upon 980 nm laser irradiation, the NaYbF4:Tm@NaYF4 UCNP emitted visible light at 345, 360, 452 and 475 nm, activating the Zn1.1Ga1.8Ge0.1O4:0.5% Cr PLNP to give persistent luminescence at 700 nm. Although this nanohybrid allowed deep NIR tissue bioimaging with long afterglow time, the energy transfer efficiency of the system is questionable. Shortly after, Liu et al. directly doped lanthanide ions into a PLNP host crystal, producing Zn3Ga2GeO8:Yb3+,Er3+,Cr3+ that exhibited upconverted persistent luminescence.111 Under 980 nm excitation, Yb3+ underwent excitation to the 2F5/2 level and transferred the energy to the adjacent Er3+, resulting in the population of its excitation state levels at 2H11/2, 4S3/2 and 4F9/2. Er3+ then relayed the energy to the traps located in the crystal lattice. After the cessation of excitation light, the energy was transferred to Cr3+, emitting light at 700 nm via2E–4A2 transition. This nanocrystal exhibited long afterglow time, with detectable luminescence 10 hours after intravenous injection in mice.
The afterglow time of inorganic persistent luminescent nanoparticles mainly depends on the trap density of the nanomaterial. On the other hand, organic-based nanomaterials usually rely on sustained biochemical reactions for prolonged afterglow emissions. For example, many organic-based PLNPs produce dioxetane as an intermediate, which degrades continuously to emit visible photons.53 Hence, many organic afterglow luminescent nanosystems incorporate phenylenevinylene (PPV), which produces singlet oxygen under NIR excitation. This oxidizes the double bonds in the structure of PPV, producing unstable dioxetane intermediates that contribute to afterglow luminescence.112 Efforts have been made to achieve in vivo delivery of PPV through encapsulation with polymers for deep tissue tumour imaging.113,114 Pu et al. synthesized NIR-emitting F12+-ANP, which consists of PPV, EMF12+, and the photosensitiser NIR775 encapsulated in PEG (Fig. 12a).115 Initially, F12+-ANP exhibited suppressed afterglow due to the quenching effect of F12+ species. However, upon interaction with the enriched H2S in the tumour area, F12+-ANP is reduced to F2-ANP, resulting in the recovery of 1O2 production and fluorescence at 580 nm from the produced dioxetane. This resulted in the excitation of NIR775 and afterglow luminescence at 780 nm (Fig. 12b). F12+-ANP added with NaHS exhibited fluorescence at 580 nm and 780 nm while F12+-ANP alone did not exhibit any fluorescence (Fig. 12c). The nanoprobe also showed high afterglow intensity in the presence of H2S with a long half-life of 6.6 minutes (Fig. 12d), while showing an SBR of 71.9 in vivo 12 hours post-injection.
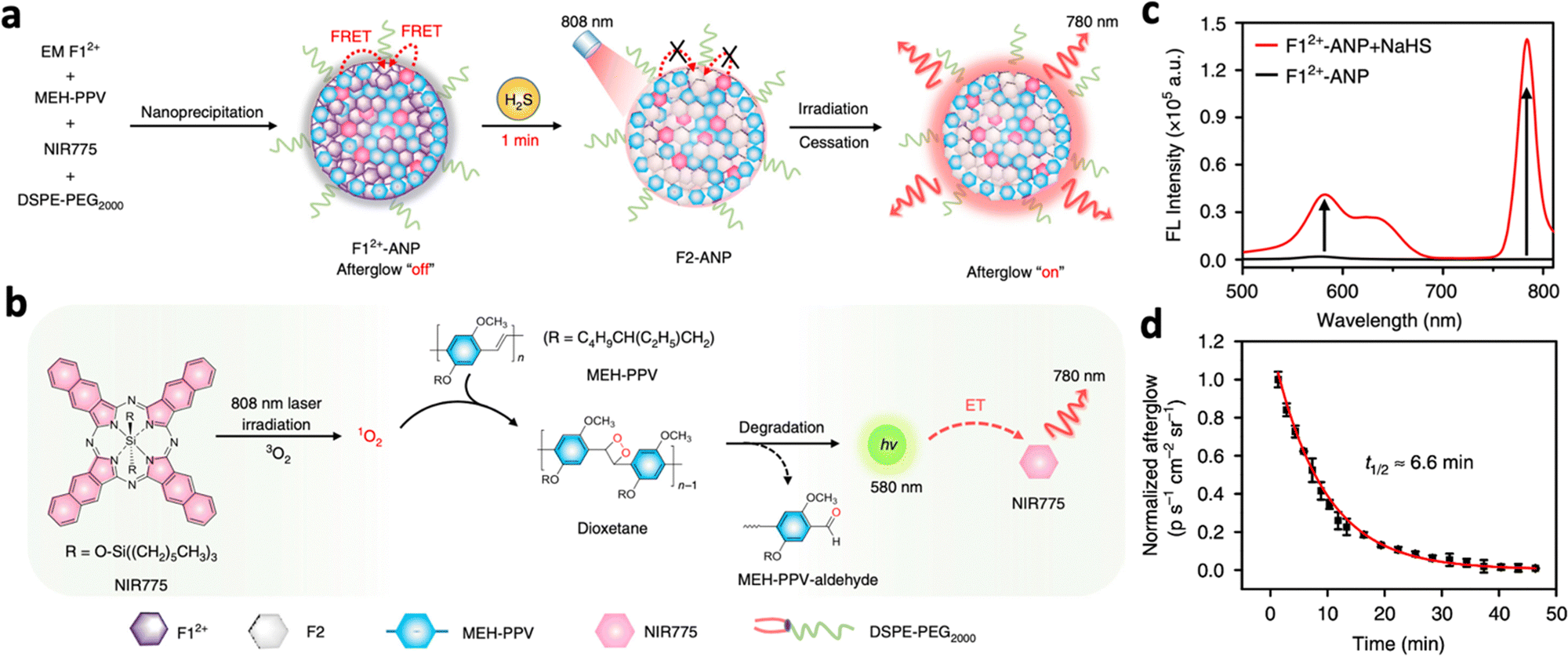 | ||
| Fig. 12 (a) Schematic illustration of the proposed mechanism of H2S-activated NIR afterglow luminescence at 780 nm following irradiation with an 808 nm laser. (b) Schematic illustration of the photoreaction processes to produce NIR afterglow luminescence within activated F12+-ANP (i.e., F2-ANP). (c) Fluorescence spectra of F12+-ANP in the presence or absence of NaHS (excitation: 808 nm). (d) Afterglow decay of F12+-ANP in the presence of H2S monitored at 780 nm. Reproduced with permission from ref. 115. Copyright 2020, Nature Publishing Group. | ||
3.4 Enhancing NIR-excited luminescence
Apart from reducing surface quenching and improving luminescence intensity, heterogenous passive shell coatings could provide additional benefits, such as introducing imaging modalities or improved biocompatibility. For example, coating Gd3+-containing shells, i.e., NaGdF4, could enable T1-weighted MRI for image guided therapy.118,119 In addition, coating the magnetic FexOy shell on NaYF4:Yb3+,Tm3+ enabled T2-enhanced magnetic resonance imaging (MRI) of the lymphatic node.120 In addition, the emission intensity at 475 nm was increased by 2.5-fold simultaneously due to the reduction in surface quenching. Fe3O4 was also coated on UCNPs to enable magnet-guided photodynamic therapy towards the tumour site.6 Apart from coating iron oxides, passive silica coating was also studied extensively since it provides a hydrophilic surface on the UCNPs/DCNPs, which is more suitable for biological applications.121 In addition, silica coating exhibits low cytotoxicity and provides a chemically active surface that can be easily modified to introduce diverse functional groups, catering to the conjugation of biological molecules or functional nanoparticles. In 2008, Zhao et al. reported SiO2-coated Y2O3:Eu3+ nanoparticles, which exhibited 4.13 times luminescence enhancement compared to the uncoated nanoparticles.122 More recently, SiO2 coating on ZnGa2O4:Yb3+,Tm3+ successfully enhanced upconversion luminescence intensity by 12 times at 700 nm and 830 nm following 980 nm excitation.88
Having more than 1 sensitizer located in different layers of active shell coatings allows the nanosystem to be excited by multiple wavelengths, i.e., orthogonal excitation. Our group previously reported NaErF4:Yb/Tm@NaYF4:Yb@NaNdF4:Yb, which could be excited by 808 nm and 980 nm due to the presence of both Yb3+ and Nd3+.127 Under 980 nm excitation, the Yb3+ sensitizers present in the core (NaErF4:Yb/Tm) and two subshells (NaYF4:Yb and NaNdF4:Yb) absorbed the excitation light and transferred it to the Er3+ activators in the core (Fig. 13a). The elevated concentration of Er3+ in the excited state increased the likelihood of cross-relaxation between the 4I11/2 state and 4F7/2 state, promoting the population of the 4F9/2 state and enhanced the 650 nm red upconversion emission (Fig. 13b). When excited with 808 nm light, the Nd3+ ions served as sensitizers and transferred the excitation energy to Yb3+ ions and subsequently to the Er3+ activators in the core (Fig. 13c). However, since the Nd3+ ions are only localized in the outer shell, the absorption of 808 nm light is significantly lower compared to 980 nm light. Moreover, the longer energy migration distance between Nd3+ and Er3+ ions limited energy transfer to Er3+. Consequently, the cross-relaxation effect was less prominent, resulting in green luminescence dominated by the 2H11/2, 4S3/2–4I15/2 transition of Er3+ (Fig. 13d).
 | ||
| Fig. 13 Schematic illustration of energy migration pathways of NaErF4:Yb/Tm@NaYF4:Yb@NaNdF4:Yb under (a) 980 and (b) 808 nm excitations. Proposed upconversion mechanism for (c) red emission generated under 980 nm laser excitation and (d) green emission under 808 nm laser excitation. Reproduced with permission from ref. 127. Copyright 2019, American Chemical Society. | ||
Substituting NaYF4:Yb3+/Tm3+ with Ca2+ caused a 121-fold improvement in upconversion luminescence intensity.129 This is because the substitution of Ca2+ ions with Y3+ in the lattice inhibits the migration and rearrangement of dislocations, thereby suppressing the formation of small-angle grain boundaries. This enhances the optical homogeneity of the crystal and improves upconversion luminescence. Doping Y3+ into CaF2:Nd3+ was also shown to improve downconversion luminescence by 2.38-fold, since it could bind with Nd3+ and prevent the formation of aggregated inactive Nd3+–Nd3+ clusters.136 Doping Li+ or Na+ caused crystal shrinkage which also enhanced luminescence enhancement.128,134 For example, Li+ easily substituted Gd3+ ions in GdF3:Yb3+,Tm3+, creating defects 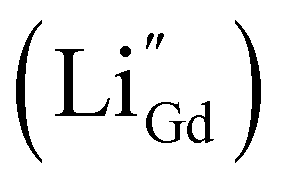 that can be electrically neutralized by F− vacancies
that can be electrically neutralized by F− vacancies 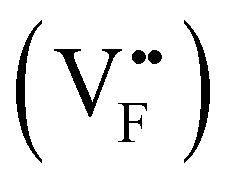 . This substitution and neutralization induced the contraction of the crystal cell of GdF3, reducing symmetry around Tm3+ and increasing the probability of radiative transitions. As a result, under 980 nm excitation, the emission intensity at 800 nm and 474 nm increased 2.2 and 5.2 times, respectively, under 3% of Li+ doping. Ferroelectric polarization of LiNbO3:Yb3+,Tm3+ also enhanced upconversion and downconversion luminescence by 2.6 and 3.2 times through deliberately causing crystal shrinkage.137 Upon application of an increasing electric field to the LiNbO3:Yb3+,Tm3+, the lattice was found to shrink by 0.03 Å.
. This substitution and neutralization induced the contraction of the crystal cell of GdF3, reducing symmetry around Tm3+ and increasing the probability of radiative transitions. As a result, under 980 nm excitation, the emission intensity at 800 nm and 474 nm increased 2.2 and 5.2 times, respectively, under 3% of Li+ doping. Ferroelectric polarization of LiNbO3:Yb3+,Tm3+ also enhanced upconversion and downconversion luminescence by 2.6 and 3.2 times through deliberately causing crystal shrinkage.137 Upon application of an increasing electric field to the LiNbO3:Yb3+,Tm3+, the lattice was found to shrink by 0.03 Å.
Doping certain lanthanide ions could also improve luminescence intensity via energy transfer processes. Doping NaYF4:Yb3+,Tb3+ with Mn2+ ions could increase the upconversion emission of Tb3+ ions by over 30 times due to the more efficient energy transfer between the sensitiser and activator.75 Energy is transferred from excited Yb3+ ions to Mn2+ ions and then to the 5D4 state of Tb3+ ions, which resulted in stronger luminescence following relaxation. In another study, the downconversion luminescence of NaYbF4:Er3+,Nd3+ was enhanced through the suppression of upconversion luminescence by Ce3+ doping.138 Initially, Nd3+ ions absorbed the excitation energy at 808 nm and transmitted it to Yb3+ ions to excite Er3+, resulting in downconversion luminescence at 1530 nm by 4I13/2–4I15/2 (Fig. 14a). Concurrently, a competing process occurs where the 4I11/2 level of Er3+ is excited to its higher 2H11/2 and 4S3/2 levels for upconversion emission. The similar energy gap between 2F5/2–2F7/2 of Ce3+ and 4I11/2–4I13/2 of Er3+ allowed Ce3+ to absorb energy from Er3+, causing them to relax from 4I11/2 to 4I13/2 (Fig. 14c and d). The emission peak at 1530 nm for the nanoparticles doped with 5% Ce3+ was 10.4 times higher than that of nanoparticles without Ce3+ (Fig. 14b).
 | ||
| Fig. 14 (a) Schematic illustration of the energy transfer path from Nd3+ to inner Er3+ for luminescence without (a) and with (c) Ce3+-ion doping. (b) The downconverting NIR fluorescence spectra of NaYbF4:2% Er,y% Ce@NaYF4:10% Yb@NaYF4:50% Nd (y = 0, 2, 5, and 10). (d) Detailed illustration of the energy transfer from Nd3+ to Er3+, emitting NIR-II luminescence at 1525 nm under 808 nm excitation. Reproduced with permission from ref. 138. Copyright 2020, Royal Society of Chemistry. | ||
3.5 NIR-activated light-mediated therapy
NIR light possesses sufficient tissue penetration depth to carry out light-mediated therapy in deep tissue. However, certain NIR wavelengths possess deeper tissue penetration than the other wavelengths, such as NIR light located at around 800 nm or NIR-II light. Therefore, an increasing number of studies have focused on engineering 808 nm or NIR-II-excitable upconversion or downconversion nanomaterials for therapy and imaging respectively. In addition, NIR-IIb and NIR-IIc photons, or magnetic resonance imaging (MRI) are mostly used for imaging-guidance of deep tissue therapy due to their ability to produce high resolution images in deep tissues. Orthogonal excitations were also applied in various therapies since different modalities of the system, i.e., therapy and imaging, could be activated with different excitation wavelength. Finally, better targeting strategies were developed such that the nanosystem is only “activated” in the tumour, minimizing damage to normal cells.In this section, we will cover the major directions of NIR-activated light-based therapy and their challenges.
The initial application of UCNPs for photodynamic therapy was limited by the loading rate of photosensitizers onto the UCNP.150 In addition, the desorption and leakage of PSs from the nanoplatform is also a big concern, since most rely on the electrostatic or hydrophobic interaction between the UCNP and photosensitizers.151 This issue was solved by Zhang's group in 2012 as they covalently linked photosensitizers with the UCNP NaYF4:Yb3+,Er3+, with 100 photosensitizing molecules covalently bonded to every 20 nm UCNP.152
To achieve deep-tissue therapy, it is crucial to engineer nanosystems that could be excited by highly penetrative NIR-II photons. Zeng et al. recently reported NaLuF4:40% Mn20% Er@NaLuF4@SiO2.7 Upon 1532 irradiation, Er3+ was excited to 2H11/2, emitting red light following 4F9/2 to 4I15/2 transition, which activated the photosensitizer ZnPc to produce singlet oxygen. The red upconversion luminescence was also utilised for imaging guidance in vivo following both oral administration and subcutaneous injection. To enhance imaging depth and resolution, NIR-II photons at 1500 nm were produced from NaYF4:Yb3+,Er3+ through downconversion luminescence following 980 nm activation.153 The imaging modality successfully produced angiography of the hindlimb blood vessels and visualized the liver of a mice following a 2 mg injection with a low NIR power density of 0.5 W cm−2. To enhance image resolution, MRI was used to image Gd3+-containing UCNPs due to the T1-weighted MRI properties of Gd3+.154 For example, Wu et al. produced NaGdF4:Yb/Tm@SiO2@TiO2 nanocomposites, which displayed a high r1 relaxivity value of 4.53 mM−1 s−1.74 Despite having higher resolution, the high cost and complexity and MRI has to be taken under consideration.
Orthogonal excitation allows the UCNPs to be excited by 2 different wavelengths, producing 2 different emissions wavelengths – one for PDT and one for imaging/diagnosis.127,155 For example, GdOF:Yb3+,Er3+,Eu3+ coupled with the photosensitizer DHA could carry out PDT and produce NIR-II imaging with orthogonal excitation.156 Under 980 nm NIR irradiation, the Er3+ ions were excited and produced red emissions at 550 nm and 650 nm, which excited DHA to produce singlet oxygen, inducing tumour ablation in vivo after 14 days of treatment. Following 808 nm laser irradiation, Er3+ produced downconverting NIR-II emissions at 1530 nm, which was observed clearly at the tumour site following intravenous injection of the UCNP in mice.
To enhance tumour cells uptake of the nanoparticles, targeting ligands like folate have been conjugated on UCNPs.157,158 Instead of using targeting ligands, magnetic field was shown to increase the cell uptake of transferrin-coated NaYF4:Gd3+,Yb3+,Er3+ loaded with the photosensitizer PpIX on MDA-MB-231 and HeLa cells in vitro.159In vivo injection of UCNPs coated with Fe3O4 while placing external magnets near the tumour area also enhanced the accumulation of UCNPs in tumour cells and increased PDT efficacy.6
Persistent luminescence nanoparticles (PLNPs) do not require constant irradiation for continuous emission, which reduces irradiation time and minimizes the side effects of NIR irradiation on tissue. The physical combination of the UCNP NaYF4:Yb3+,Tm3+ and PLNP SrAl2O4:2% Eu2+,4% Dy3+ resulted in the emission of persistent luminescence at 520 nm following 980 nm excitation.110 After 5 minutes of 980 nm charging, the nanophosphors emitted light for 30 minutes, resulting in the excitation of photosensitizers for PDT (Fig. 15a). A high degree of singlet oxygen generation resulted in 60% reduction in cell viability of HT29 cells after 4 cycles of irradiation (Fig. 15b). To combat the hypoxia environment in the tumour, the nanosystem encapsulated CaO2, which reacted with water to generate oxygen. Higher oxygen content and oxygenated hemoglobin of the tumours was reported in vitro, which resulted in increased generation of singlet oxygen and reduction in tumour volume (Fig. 15c).
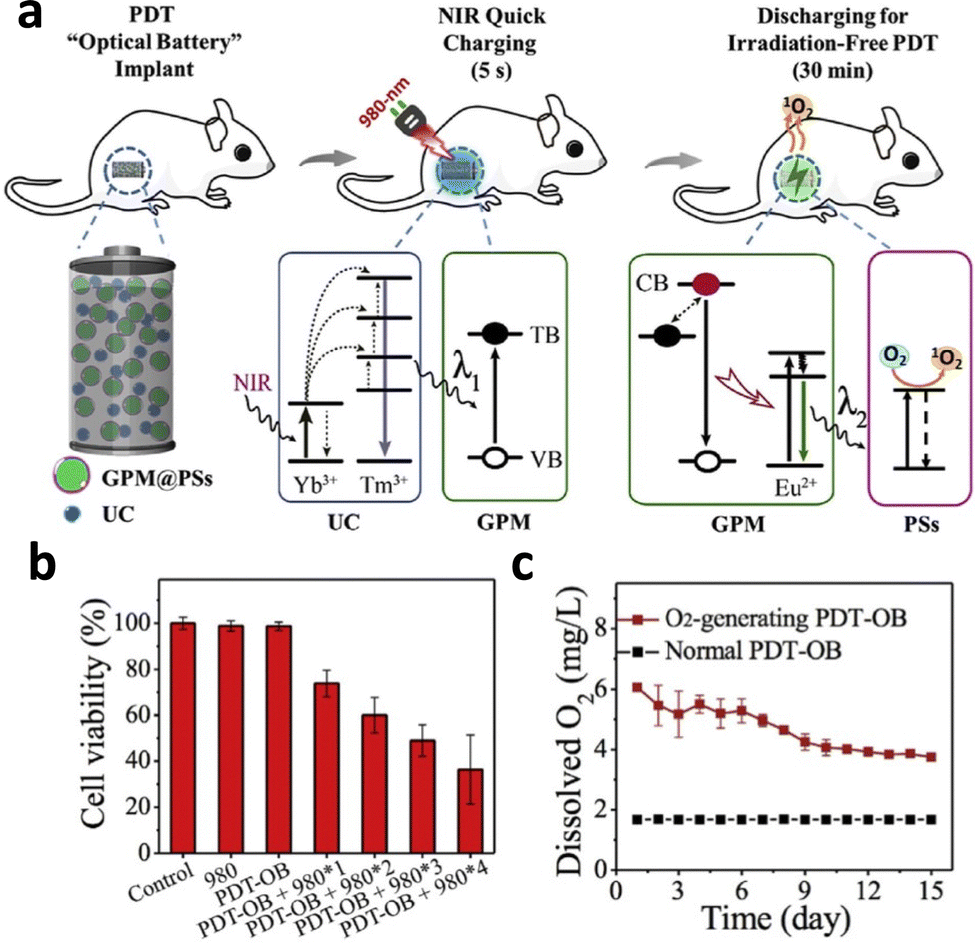 | ||
| Fig. 15 (a) Schematic illustration of the NIR-excited PDT with persistent luminescence (GPM: green persistent luminescence materials; PSs: photosensitizers; UC: upconversion materials; TB: trapping band; and VB: valence band). (b) Viability of HT29 cells treated with different numbers of 980 nm NIR recharging cycles. (c) In vitro comparison of O2 generation ability between CaO2-containing implants and CaO2-free implants. Reproduced with permission from ref. 110. Copyright 2018, Elsevier. | ||
Gd3+-Doped upconverting/downconverting nanoparticles exhibit T2-enhanced MRI modalities that could be used as an image-guidance for PTT.86,160 Fe3O4, having high T2 relaxation time, exhibits MRI modalities while being a photothermal agent itself. A hollow carbon sphere containing Fe3O4 and NaGdF4:Yb3+,Er3+@NaGdF4 UCNPs exhibited a r2 value of 845.13 mM−1 s−1, indicating their effectiveness as a T2 contrast agent.56 Under external magnetic field, the nanoparticles exhibited enhanced accumulation at the tumour site. 980 nm laser irradiation on the nanosystem resulted in visible upconversion emission that excited Fe3O4. Tumour temperature increased and a complete elimination of tumour was observed following treatment. To minimise energy loss during transfer between UCNPs and the photothermal agent, Hao et al. developed carbon dots that produce NIR-II emission in the range of 900–1200 nm under 808 nm excitation while acting as a photothermal agent.65 The CDs showed a high quantum yield (QY) of approximately 0.4% and a photothermal conversion efficiency of 30.6%, which resulted in the near-disappearance of tumour after 6 days of 10-minute 808 nm irradiation daily.
Orthogonal excitation/emission was also applied in PTT to trigger therapy and imaging separately. Chen et al. produced prussian blue (PB)-coated NaErF4@NaYF4@NaNdF4 that could emit different downconversion luminescence under 808 and 980 nm excitation (Fig. 16a).65 Under 808 nm excitation, Nd3+ was excited to produce 1064 nm emission, which activated PB to generate heat (Fig. 16c). The PEGylated nanosystem at 600 μg mL−1 raised the tumour temperature above 42 °C for 10 minutes, significantly more than the water and the nanosystem without PB (Fig. 16d). In vivo studies confirmed the prominent tumour cell ablation and significant reduction in tumour volume compared to other the control groups (Fig. 16e). On the other hand, 980 nm excitation caused Er3+ to produce emission at 1525 nm, which provided a clear, high-resolution and high-contrast image of miniature blood vessels, brain tissue, and internal organs in in vivo imaging (Fig. 16b and f). Even with a 2-fold higher laser power and 80-fold longer integration time, 1064 nm luminescence exhibited lower resolution and could only capture an unclear image (Fig. 16g). This further highlights the importance of using NIR-IIb or NIR-IIc luminescence for imaging-guided therapy.
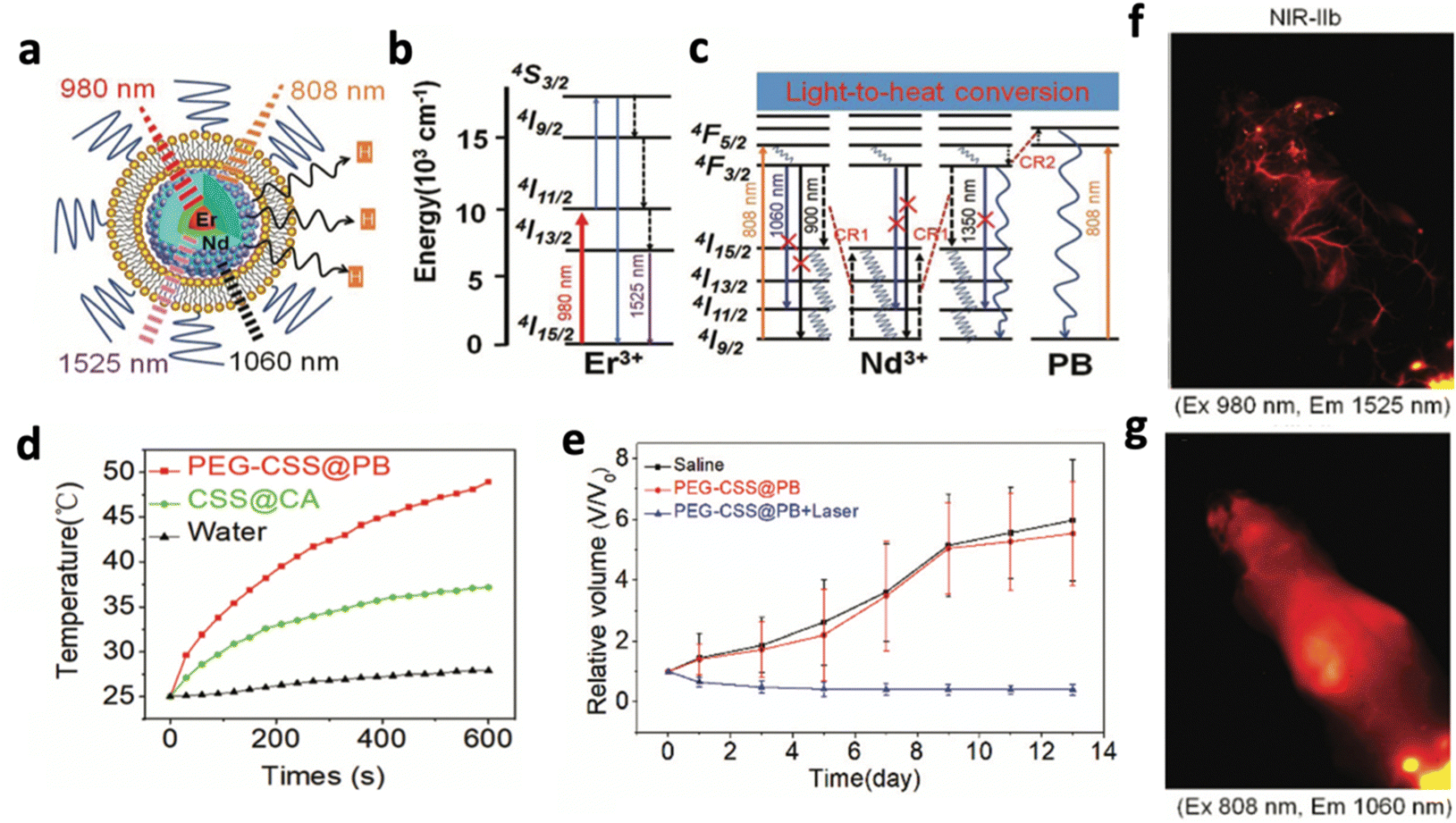 | ||
| Fig. 16 Schematic illustrations of (a) the design of NaErF4@NaYF4@NaNdF4 exhibiting NIR-II luminescence under orthogonal excitation, (b) energy transition diagrams of the 1525 nm luminescence of Er3+ under 980 nm excitation and (c) energy transition diagrams of the 1060 nm luminescence of Nd3+ under 808 nm excitation, with cross-relaxation pathways between Nd3+ ions and Nd3+ ions with PB. (d) Comparison of temperature increases of the PEG-CSS@PB nanocomposite, CSS@CA nanocomposite and water, excited by a 808 nm laser (1 W cm−2). (e) Comparison of tumour volume following different groups of treatment. In vivo NIR II luminescence imaging of the mouse with a tail vein injection of PEG-CSS@PB, acquired with (f) 1525 nm and (g) 1064 nm luminescence. Reproduced with permission from ref. 65. Copyright 2019, Royal Society of Chemistry. | ||
Trans platinum complex trans,trans,trans-[Pt(N3)2-(NH3)(py)(O2CCH2CH2COOH)2] (DPP) could be activated by UV/blue light to yield toxic platinum complexes, which could achieve cancer chemotherapy.161,162 Therefore, DPP was conjugated onto the surface of NaYF4:Yb3+,Tm3+@NaGdF:Yb3+ UCNPs, where a 980 nm laser activated the UCNPs to produce UV emissions, triggering the generation of toxic platinum complexes and caused significant tumour inhibition in vivo.119 Apart from directly generating toxic substances, UCNPs could trigger drug release from nanocomposites with the use of photothermal effect.163 For example, Yang et al. assembled Gd2O3:Yb3+,Er3+ in mesoporous silica loaded with gold nanocrystals and the target drug doxorubicin (Dox).164 Upon 980 nm excitation, the UCNPs emitted green light, which overlapped with the surface plasmon resonance (SPR) band of gold nanoparticles. This caused the gold nanocrystals to generate heat and release the loaded Dox, while the elevated tissue temperature enhanced the cellular uptake of Dox.
UCNPs were also applied to provide imaging guidance for drug delivery, but most of them do not provide imaging specifically for the site of drug release.67,163 To enhance the specificity of imaging, a nanosystem consisting of UCNPs coated with dye-doped and drug-loaded macroporous silica shells protected by hyaluronic acid (HA) was developed.165 While intact, the Ho3+-containing UCNPs produced 660 nm upconversion under 980 nm excitation, which excited the doped Cy5.5 dye to emit at 710 nm. Following specific degradation by hyaluronidase in tumour cells, the nanosystem disintegrated and released the drug load while restoring luminescence at 660 nm. Intratumoural injection of this nanosystem displayed no signal at 660 nm for the first 48 hours but started showing increasing intensity after 48 hours, which indicated a specific and progressive drug release. To enhance the resolution and imaging depth, a NIR-II emitting Nd3+-MOF loaded with chloroquine (CQ) and coated with HA was developed.166 Following 808 nm excitation, Nd-MOF crystals exhibited emissions at 1064 and 1337 nm in the NIR-II region, which peaked 12 hours after intravenous injection in vivo.
Photothermal therapy and chemotherapy could be combined by making use of drug delivery systems that release drugs upon an increase in temperature. For example, NaGdF4:Yb/Er@NaGdF4 UCNPs were functionalised with mesoporous silica shells, and loaded with doxorubicin (DOX) and gold nanoparticles.167 Upon 980 nm laser irradiation, the green emission from UCNPs excited gold nanoparticles due to SPR band overlap, causing a rise in temperature to 70.7 °C after 7 min of the irradiation while simultaneously releasing the drug. The presence of gold nanoparticles caused an increase in DOX release efficiency from 38.6% to 78.9%. To provide an imaging modality for this combinational therapy, Nd3+-based downconversion nanoparticles were conjugated with CuS and Dox. 808 nm irradiation not only excited CuS to generate heat but also excited Nd3+ to emit NIR-II fluorescence at 1064 nm. Following 8 minutes of irradiation, the nanosystem exhibited a 5.1 °C temperature rise, releasing Dox and reducing the cell viability of 4T1 cells from 90% to 40% in vitro. The axillary lymph nodes in breast tumour exhibited clear fluorescence 1 hour post-injection in vivo and remained evident for 4 hours with high contrast.
By loading drugs together with UCNPs and photosensitizers, PDT efficacy could be enhanced without the need to increase oxygen level in tumour tissue.168,169 Similar to photothermal–chemotherapy, it is important to incorporate drug release systems that are also triggered by NIR irradiation, to minimize side effects on healthy cells. Gong et al. coloaded NaYF4:Yb3+,Tm3+,Er3+ UCNPs with the photosensitiser Rose Bengal (RB) and hydrophobic drug AB3 inside the polymer PNBMA.170 The red upconversion at 650 nm induced by Er3+ activated RB to generate singlet oxygen, while the UV upconversion by Tm3+ triggered the PNBMA to undergo a hydrophobic-to-hydrophilic transition, releasing the hydrophobic drug AB3. The targeted combinational therapy displayed remarkable antitumour effects, outperforming the individual treatments of chemotherapy or PDT.
By combining photothermal therapy and photodynamic therapy, the heat generated in PTT improved blood flow to the tumour area, enhancing the intratumoural oxygen level and promoting PDT.171 Therefore, photothermal agents with higher surface area to bind with UCNPs and photosensitizers have been explored. For example, MoS2 nanosheets could be excited directly by an 808 nm laser (1 W cm−2) to produce heat that could reach 55.3 °C after 60 min of irradiation.172 Nanographene oxide (NGO) was also shown to increase temperature to 65 °C within 2 minutes of 808 nm irradiation.125 MoS2 and NGO both exhibit a large surface area and ability to covalently graft with UCNPs as well as photosensitisers like Ce6 or ZnPc.172,173 To simplify the synthesis process, photothermal agents that could generate ROS under NIR irradiation have been studied. For example, FePc can emit ROS and exhibits a photothermal conversion efficiency of 42.5% under 730 nm irradiation.174 In addition, the photothermal agent Cu2−xS nanodots caused a 52 °C temperature increase under 1064 nm irradiation, while generating hydroxyl radicals through a Fenton-like reaction with H2O2.175
3.6 Strengths and weaknesses of the NIR-mediated therapy
NIR-activated therapy has several strengths that contribute to its growing popularity in biomedical applications. One of the main advantages is the higher tissue penetration capability of NIR light compared to conventional UV/visible sources. Certain NIR wavelengths fall within the “optical window” of biological tissues, allowing them to penetrate deeper than UV/visible light into biological tissues for light-mediated therapy. Another advantage of NIR-activated therapy is that NIR provides the versatility to tune the wavelength for specific purposes. For example, 808 nm excitation wavelength could be used instead of 980 nm for deeper tissue penetration; NIR-IIb or NIR-IIc emission wavelength could be used for high-resolution deep-tissue imaging. In addition, orthogonal excitation in several nanosystems allowed them to produce different emission wavelengths that perform different functions, such as therapy or imaging. The orthogonal emissions at 2 different wavelengths could also be monitored simultaneously to track 2 processes together. Additionally, NIR-activated therapy benefits from the widespread availability of equipment. NIR light sources and detectors are readily found in many research laboratories and clinical settings. This accessibility makes it easier for researchers and clinicians to implement NIR-activated therapy in their studies and medical practices. In contrast to chemiluminescence-activated therapy, NIR-activated therapy offers the advantage of external control over the activation and deactivation of luminescence. This capability allows for the precise modulation of the luminescent signal. Furthermore, NIR-activated luminescence typically exhibits stronger intensity than chemiluminescence-based approaches, enabling more effective therapeutic outcomes.However, NIR-activated therapy has certain limitations and disadvantages that need to be considered. NIR light can generate heat when absorbed by tissues, which may lead to thermal damage in surrounding healthy tissues. Careful control over the NIR light dosage and monitoring of tissue temperature are essential to mitigate the risk of overheating and ensure the safety of the therapy. Another limitation is the depth of tissue penetration. While NIR light offers higher tissue penetration than UV and visible light, its penetration depth is still limited compared to other excitation sources like X-rays and ultrasound. This constraint restricts the treatment of deep-seated tissues and tumours.
4. X-ray-excited luminescence
X-rays, having higher tissue penetration than most of the other excitation modalities, could activate many nanosystems to carry out light-mediated therapeutics in deep tissue. The generation of X-ray-excited luminescence is usually dependent on the direct ejection of electrons. High energy X-ray photons eject electrons in atomic orbitals that results in the creation of holes, where subsequent filling of these holes by electrons from higher orbitals results in X-ray fluorescence.176 However, the efficiency of this process is heavily dependent on the electronic structure of the atom, since competing processes may occur, such as Compton scattering, Rayleigh scattering or emission of Auger electrons.1774.1 X-ray-excitable luminescent nanomaterials
X-ray-responsive nanomaterials typically comprise elements with high atomic numbers, characterized by significant X-ray attenuation coefficients. These nanomaterials can efficiently absorb X-rays and generate luminescence. Hence, most X-ray excitable luminescent nanomaterials consist of lanthanides and heavy metals due to their high atomic number. In addition, certain semiconductors and organic nanomaterials exhibit X-ray excitable luminescence due to the direct band-to-band excitation by X-rays. However, they are less studied due to their lower X-ray absorption coefficients.Different from NIR-excited luminescence, most X-ray-excited luminescent nanomaterials do not require a sensitiser, since X-rays have enough energy to cause a direct band-to-band excitation (Fig. 17a and b). In contrast, certain sensitisers, such as Yb3+, may even quench X-ray luminescence through cross-relaxation and charge trapping processes.178 Band-to-band excitation by X-rays allows many nanomaterials to emit in the visible range or even in the deep UV range with strong intensity, with or without the presence of an activator. This would be beneficial for activating light-sensitive molecules like photosensitisers, or cause direct UV damage to the target cells.
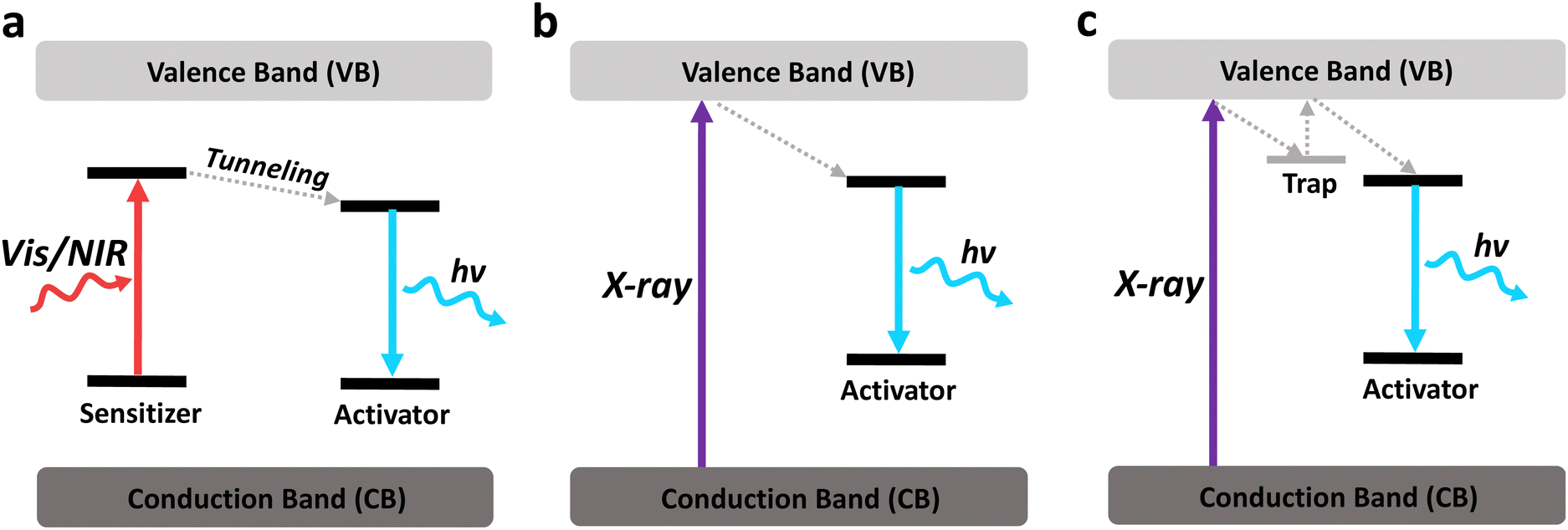 | ||
| Fig. 17 Schematic illustrations of the typical mechanisms of (a) downconversion luminescence upon excitation with visible or NIR light, (b) X-ray-excited luminescence and (c) X-ray-excited persistent luminescence. | ||
Activator ions that have energy levels located near the 5d orbital are often chosen to accept energy from the excited host crystal to emit light of different wavelengths. Eu3+, for example, gives red luminescence at 592 nm (5D0–7F1), 618 nm (5D0–7F2) and 698 nm (5D0–7F4).184 Sm3+ gives red luminescence at 554 nm (4G5/2–6H9/2), 596 nm (4G5/2–6H7/2), 646 nm (4G5/2–6H5/2) and 708 nm (4G5/2–6H3/2).185 Tb3+ gives green emission at 490 nm (5D4–7F6), 545 nm (5D4–7F5), 585 nm (5D4–7F4) and 621 nm (5D3–7F6).186 Gd3+ is often co-doped with Tb3+ to facilitate the energy transfer from 5d electrons to Tb3+ ions, since 6IJ and 6PJ energy levels of Gd3+ are located close to the 5D4 level of Tb3+.183
Due to the high energy of X-rays, they can excite many lanthanide ions inducing emission of light in the UV range. For example, Pr3+ is capable of emitting light in the UVC spectrum at 235, 245, 263, and 274 nm, attributed to the characteristic inter-configurational 4f15d1–4f2 transition in Pr3+.187 Ce3+ ions can also emit UV at 360 nm due the transition from the lowest level of the 5d configuration to the 2F5/2 and 2F7/2 levels.188 Tm3+ ions are also known for their UV emissions at 353 and 368 nm due to the 3P0–3F4 and 1D2–3H6 transitions, accompanied by NIR emission at 807 nm due to the 3H4–3H6 transition of Tm3+ ions.189,190 Not only can UV emissions activate most of the photosensitive molecules for deep tissue therapy, but they can also directly cause DNA damage in the targeted tissue while minimizing the damage to surrounding tissues due to their low biological penetration.187
However, different from most lanthanide-based nanomaterials, metal-based compounds can emit X-ray-excited luminescence even without the addition of activator ions. Although offering more simplicity in design, metal-based nanomaterials usually produce broader emissions with less tunability in emission wavelength. For example, GSH-Au NPs were first shown to exhibit X-ray-excited luminescence at 645 nm and 800 nm in 2013 by Chen et al., as X-ray photons above the L3 absorption edge of Au (∼12 keV) have sufficient energy to knock out the inner-shell electrons.197 In order to alter emission wavelengths of Au NPs, changes in thir surface modifications have to be made. For example, bovine serum albumin (BSA)-directed Au nanoclusters produced an emission at 667 nm while lysozyme-coated Au nanoclusters produced an emission at 421 nm.198 This is due to the varying degrees of inelastic scattering between the electrons in the organic coating and the ejected photoelectrons, resulting in different levels of hole–electron generation. In addition, the conjugation of activator-based organic compound europium bromoacetate (EuBA) onto Au nanorods resulted in an emission shift to around 630 nm.199 Similarly, copper–cysteamine nanoparticles also exhibited 645 nm emission following X-ray excitation.200 Other than gold and copper, molybdenum (Mo6) complexes also have a high atomic number and have demonstrated emissions at 690 nm with ethylene oxide surface modification following X-ray irradiation.201
Other than metal nanoparticles, metal oxides were also found to exhibit X-ray excited luminescence due to their large band gap and the presence of bulk and surface defect sites.202 For example, Al2O3 nanotubes, TiO2 NPs and ZnO NPs exhibited X-ray excited luminescence due to their oxygen vacancies.203–205 ZnO gives an emission at 382 nm and 510 nm following X-ray excitation, where the former is due to band-gap emission (3.24 eV) and the latter is due to oxygen vacancies.205 Similarly, X-ray excitation of ZnS NPs resulted in UV emission at 330 nm.206 Combining several heavy metals together in a nanocomposite could also result in efficient X-ray absorption. Shan et al. reported CsZrCl6, which gave an emission peak at 464 nm, mainly due to the recombination of triplet states.207 The accumulation of triplet states within the nanocomposite could also boost its ability to generate ROS, which could be applied in many ROS-based therapies.
Zinc-based nanosystems are favourable for X-ray excited afterglow luminescence due to their high X-ray absorption coefficient. For example, ZnS:Cu,Co-A was reported to emit 578 nm under X-ray irradiation, but afterglow luminescence time was just 10 minutes long.209 To improve the X-ray excited afterglow of the nanosystem, other Zn-based nanosystems were developed, namely, zinc gallate (ZnGa2O4), zinc silicate (ZnSi2O4), or variations of zinc gallogermanate.210 X-ray-activated ZnGa2O4:Cr3+ was first discovered in 2017 by Hao's group.211 They reported ZnGa2O4:Cr3+, which exhibited 6 hours of persistent luminescence at 700 nm after the cessation of the soft X-ray excitation source with low excitation power (45 kVp, 0.5 mA), due to the characteristic electron transition of Cr3+ from 2E to 4A2 (Fig. 18a and b). Although this nanosystem was originally discovered for UV-excitation, X-ray excitation increased the penetration depth from 3 mm to 20 mm (Fig. 18c). More importantly, the PLNPs could be recharged with the same X-ray dosage without a decrease in luminescence intensity. Later on, work has beencarried out to further optimise this nanosystem. For example, tungsten W(VI) was doped to improve X-ray absorption, which resulted in a 1.3-fold increase in luminescence intensity due to the additional electrons ejected.106 The afterglow lifetime was also increased due to the readjustment of trap depth and density by W(VI). Mn2+ was also doped into ZnGa2O4 to shift the emission to from 700 nm to 530 nm, due to the transition of Mn2+ from 4T1 to 6A1.212
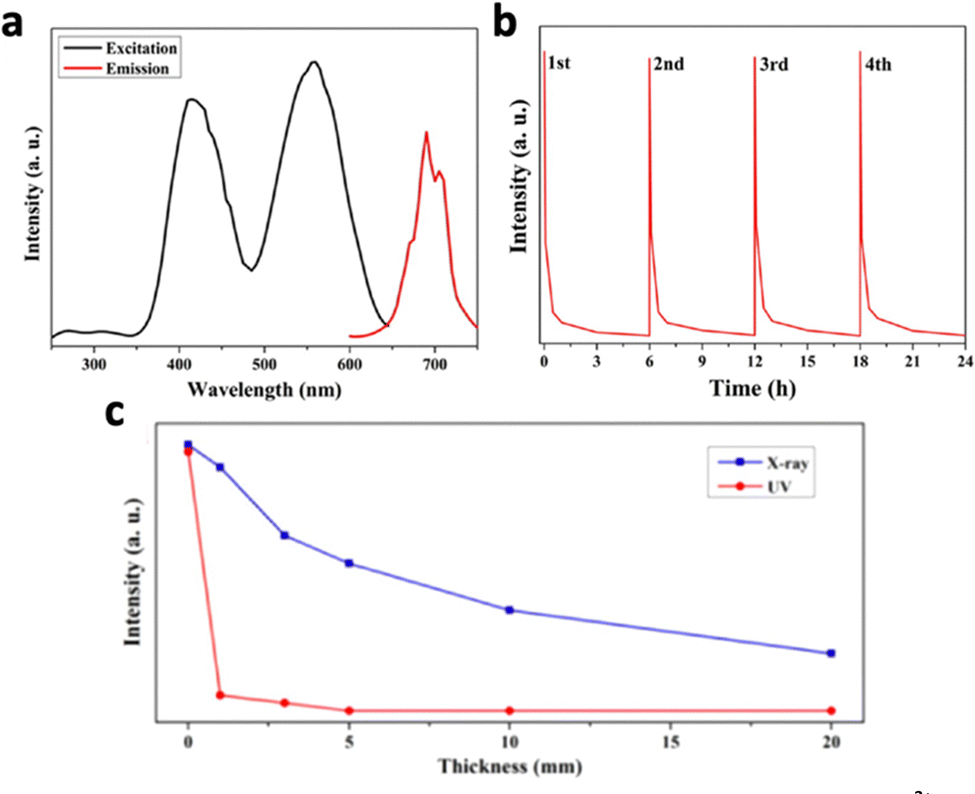 | ||
| Fig. 18 (a) Excitation spectrum (black) and emission spectrum (red) of ZnGa2O4:Cr3+. (b) 4 Cycles of persistent luminescence decay curves by ZnGa2O4:Cr3+ following cessation of X-ray irradiation (5 min, 45 kVp). (c) Luminescence intensity of ZnGa2O4:Cr3+ excited by X-ray (blue) and 365 nm UV (red) through pork tissues with different thicknesses (0, 1, 3, 5, 10, and 20 mm). Reproduced with permission from ref. 211. Copyright 2017, American Chemical Society. | ||
Zinc gallogermanate, having different variations, is synthesized by doping germanium(IV) into zinc gallate. Zn3Ga2Ge2O10:0.5% Cr3+,0.1% Mn2+ was produced to generate red luminescence at 698 nm and green luminescence at 532 nm due to the transitions of Cr3+ and Mn2+.213 This nanosystem is particularly useful due to its ability to generate visible and NIR afterglow emissions, which could be used to activate photosensitisers and carry out deep tissue imaging guidance simultaneously. To enhance the resolution of imaging guidance, Liu et al. developed Zn2Ga3Ge0.75O8:Cr3+,Nd3+ that exhibited NIR-I and NIR-II afterglow following X-ray irradiation.214 Not only did the nanoparticle exhibit emissions at 696 nm due to Cr3+, the presence of Nd3+ resulted in emissions at 895, 1067 and 1340 nm, originating from the 4F3/2–4I9/2, 4F3/2–4I11/2, and 4F3/2–4I13/2 transitions of Nd3+. Following cessation of X-ray irradiation, the 700 nm afterglow peak of Cr3+ (2E–4A2) partly overlaps with the absorption peaks of Nd3+ at 745 nm (4I9/2–4F7/2 + 4S3/2) and 805 nm (4I9/2–4F5/2 + 2H9/2). The energy transfer from Cr3+ to Nd3+ enabled afterglow luminescence at 696 nm and 1067 nm for 800 and 10 min, respectively. Besides Nd3+, Yb3+ also serves as a good candidate for NIR-II emission in the 950–1150 nm range, attributed to its phonon-assisted transition of from the 2F5/2 state to the 2F7/2 state.215
On the other hand, NaLnF4-based PLNPs like NaYF4:Ln3+ or NaGdF4:Ln3+ have also shown high X-ray absorption and afterglow luminescence, due to the presence of high-Z elements.216,217 In addition, the presence of Na+ and F− in the crystal lattice induces the formation of vacancies and Frenkel defects, which facilitates the development of trapping sites for the trapping of excited electron–hole pairs.218 The electron–hole pairs are subsequently captured by different activators for afterglow emission. For example, a multi-layered NaYF4 doped with different activator ions (Er3+, Nd3+ and Ho3+) exhibited emissions peaking at 1064 nm, 1180 nm and 1525 nm.219 These peaks correspond to the 4F3/2–4I11/2 transition of Nd3+, 5I6–5I8 transition of Ho3+ and 4I13/2–4I15/2 transition of Er3+, respectively. The afterglow by Er3+ at 1064 nm remained for over 72 hours following the cessation of X-ray irradiation.
Liu et al. discovered that NaLuF4 has better X-ray absorption compared to NaYF4 or NaGdF4.208 NaLuF4 has an atomic number Zmax = 71 and X-ray absorption coefficient of Kα = 63.31 keV, surpassing that of NaYF4 (Zmax = 39, Kα = 17.05 keV) or NaGdF4 (Zmax = 64, Kα = 50.24 keV). NaLuF4:Tb3+ displayed strong afterglow emission peaks at 584 nm (5D4–7F4), 546 nm (5D4–7F5) and 489 nm (5D4–7F6) according to the optical transitions of Tb3+.208 Notably, the post-excitation afterglow intensity of NaLuF4:15% Tb3+ nanocrystals is 3 times stronger than that of NaYF4:15% Tb3+ nanocrystals. The afterglow duration of NaLuF4:15% Tb3+ was reported to be 30 days, which is much longer than the 15 day afterglow duration of ZnGa2O4:Cr3+, and many other commonly studied X-ray-activated PLNPs like SrAl2O4:Eu2+,Dy3+ and ZnS:Cu2+/CO2+. Other than Tb3+ ions, other activators like Sm3+, Pr3+ and Dy3+ could also accept energy from NaLuF4 to emit persistent luminescence.218 This is because the 3P1 level of Pr3+, 4F9/2 level of Dy3+ and 4G5/2 level of Sm3+ are all located in close proximity to the traps in NaLuF4.
4.2 Enhancing X-ray-excited luminescence
Other than doping elements directly into the host crystals, placing the element in close proximity to the crystal could also enhance luminescence intensity. For example, it was found that the molybdenum cluster compound (n-Bu4N)2[Mo6I8(OOC-1-adamantane)6] embedded in a polystyrene (PS) matrix caused an increase in radioluminescence intensity.229 This is because the polystyrene matrix can be excited by X-rays, transferring the energy efficiently to the cluster. In addition, Gali et al. grew ZnGa2O4:Cr3+ (ZGO) on top of SiC nanoparticles, which acted as an additional X-ray absorber to transfer the energy to the ZnGa2O4:Cr3+ for a 14-fold radioluminescence enhancement.230
Despite the outstanding luminescence enhancement, the usage of high temperature treatments poses hazards and increases production costs. One method for reducing surface quenching in X-ray-excited luminescence from nanomaterials is to simply increase the particle size. For example, Nd3+ was doped into Zn2Ga3Ge0.75O8:Cr3+, which resulted in an increase in the nanoparticle size from 45.4 nm to 86.2 nm and a subsequent 2-fold enhancement in luminescence intensity located at 696 nm.214 Instead of increasing the particle size, gold nanoclusters (2.5 nm) was assembled and aggregated into aggregation-induced emission (AIE) clustoluminogens with the help of poly(allyl-amine hydrochloride) (PAH).233 The clustoluminogens exhibited a size of 65.6 nm and a 5.2-fold enhancement in luminescence intensity at 570 nm.
Since increasing particle size might limit therapeutic applications due to the difficulties in penetrating biological barriers, designing core–shell structured nanophosphors with a passive shell coating would be a better strategy. It was realised that the NaYF4 coating on NaLuF4:Tb3+ caused a 1.5-fold enhancement in luminescence intensity and a 6.5-fold increase in afterglow luminescence intensity.208 In addition, NaYF4:Er3+@NaYF4 with a 35 nm core and 7 nm shell led to a remarkable amplification of approximately 25-fold in luminescence intensity, compared with NaYF4:Er3+ alone (22 nm).219 Developing core–shell structured nanophosphors could also enable the simultaneous doping of different activator ions other than Er3+ in different layers, separated by inert layers of NaYF4. This prevents the cross-relaxation and concentration quenching when 2 or more activators are doped in the same crystal lattice.
Apart from engineering hexagonal nanophosphors, enhancing the crystallinity of the host crystal can also improve the luminescence intensity. For example, Li+ ions were doped into Y2O3:Yb3+,Er3+ to enhance the crystallinity of the crystal lattice, which showed enhanced X-ray-activated luminescence.178 Depositing nano-sized TiO2 with CH3NH3PbI3 also showed an 2-fold enhancement in luminescence intensity at 790 nm along with an increase in luminescence lifetime.234 The ordered nanoporous architecture of TiO2 nanotube arrays confines the three-dimensional [PbI6]4− octahedral lattice, effectively curtailing ion migration and minimizing octahedral aggregation. This confinement mechanism significantly enhances the stability and crystallinity of the CH3NH3PbI3 organometal halide perovskite, thus advancing its luminescence performance.
Recently, Han et al. reported that Li+ and Yb3+ co-doping in Zn2SIO4:Mn2+,Yb3+ could enhance luminescence by shifting the crystal to the favourable phase.210 A coexistence of α phase (410) and β phases (023) is present in Zn2SiO4, where the nanocrystals exhibit the most optimal afterglow time and luminescence intensity with an α/β ratio of 2.76 (Fig. 19c). However, Zn2SiO4:Mn2+ exhibited an α/β ratio of 0.42 (Fig. 19b). Introducing Yb3+ and Li+ ions into the crystal caused a preferential substitution of higher-valence Zn2+ ions within the ZnO6 octahedra of the β-phase structure, due to the higher valence of Yb3+ and Li+. This substitution tendency takes place before Yb3+ ions replace the lower-valence Zn2+ ions within the ZnO4 tetrahedra of the α-phase structure and accelerates the creation of the β crystal phase. As a result, Zn2SiO4:Mn2+,Yb3+,Li+ exhibited an α/β ratio of 3.57, accompanied by a 9-fold enhancement in X-ray excited luminescence intensity compared to Zn2SiO4:Mn2+ (Fig. 19a and b). The band gap of Zn2SIO4:Mn2+,Yb3+,Li+ was measured to be 4.51 eV, higher than the 4.02 eV band gap of Zn2SIO4:Mn2+. This led to the formation of deeper traps and increased trap density, contributing to a stronger and longer afterglow luminescence.
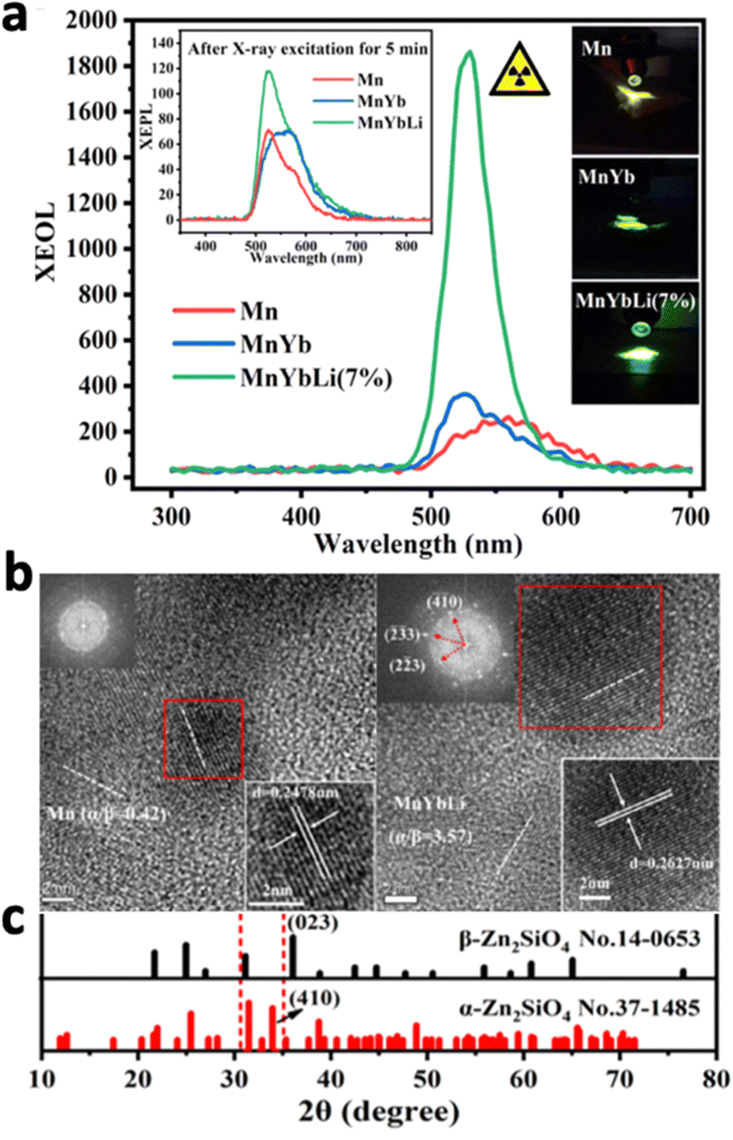 | ||
| Fig. 19 (a) X-ray excited luminescence spectrum and afterglow spectrum (inlet) of Mn, MnYb, and MnYbLi (7%) samples after X-ray excitation for 5 min. (b) High resolution TEM images of ZnSi2O4:Mn (α/β = 0.42, left) and ZnSi2O4:Mn,Yb,Li (α/β = 3.57, right) samples. (c) XRD patterns of α-Zn2SIO4 and β-Zn2SIO4. Reproduced with permission from ref. 210. Copyright 2023, American Chemical Society. | ||
4.3 X-ray-activated light-mediated therapy
To apply X-ray-excited luminescent nanomaterials to deep tissue therapy, merely inducing luminescence in nanomaterials is not enough. Since X-rays are highly ionising, it is important to reduce the dose of X-ray excitation while maintaining effectiveness for therapy. Therefore, several nanomaterials were proposed which only required a low X-ray dosage (as low as 0.09 Gy) for effective therapy. More importantly, the emergence of long afterglow luminescent nanomaterials could further reduce the exposure time to X-rays.Many light-based therapy rely on the generation of ROS to kill the target cells. Therefore, methods have been developed to increase the oxygen content in tumour to enhance ROS generation, or even deplete molecules from the ROS defence system of the target cells, such as glutathione (GSH). In addition, combined therapies have also been shown to boost overall therapeutic efficacy. Furthermore, X-ray-excited luminescence usually falls into the UV/visible spectrum, activating many of the light-sensitive molecules for photodynamic therapy or drug release. In this section, we will discuss the applications of X-ray-excited luminescence in photodynamic therapy, radiodynamic therapy, gas therapy and chemotherapy.
The efficacy of the X-ray-activated photodynamic therapy depends heavily on the efficiency of the photosensitizer to generate ROS. Following excitation by the luminescent nanomaterials, electron–hole pairs are generated in the photosensitizer. These holes exhibit a propensity to react with water molecules, resulting in the formation of hydroxyl radicals (˙OH). However, a portion of the electron–hole pairs would recombine, resulting in the limited production of ˙OH and reduced effectiveness of the therapy. Therefore, Bu et al. conjugated the cisplatin prodrug Pt(IV) with LiLuF4:Ce3+ and the photosensitizer Ag3PO4, where Pt(IV) acted as an electron acceptor.237 This facilitated the separation of holes from electrons, thereby fostering an increased yield of ˙OH. Upon X-ray irradiation, LiLuF4:Ce3+ produced emissions at 305 and 325 nm due to the 5d to 4f transition of Ce3+, which overlapped with the 295 nm excitation peak of Ag3PO4, causing the enhanced production of hydroxyl radicals. Furthermore, cisplatin is produced as Pt(IV) accepts electrons, directly attacking the DNA of the target cells.239 The additional therapeutic effect from this nanosystem resulted in almost 100% HeLa cells eradication at 6 Gy while the control group without Pt(IV) only caused 90% cell eradication. More importantly, the cell killing effect of this nanosystem was retained under hypoxia conditions, where the other control groups without Pt(IV) exhibited a significant reduction in cell eradication.
Not only did this study improve the efficiency in ROS generation from photosensitizers, but it also suggested that combining chemotherapeutic drugs with PDT could enhance therapeutic efficacy in a hypoxic tumour microenvironment. Therefore, attempts have been made to co-deliver chemotherapeutic drugs, such as 5-FU, together with the scintillating nanoparticle to boost the therapeutic efficacy.240 Li et al. co-loaded CaF2:Ce3+,Tb3+, Rose Bengal and sunitinib inside polyamidoamine (PAMAM) dendrimers, where sunitinib (SU) inhibits multiple receptor tyrosine kinases (RTK), suppressing angiogenesis in tumour.192In vitro studies on 4T1 cells confirmed an additional 20% tumour cell eradication in the presence of sunitinib, where the combined nanosystem achieved over 80% of cell eradication. Even at a lower radiation dose of 0.5 Gy, this approach yielded substantial tumour regression after 4 days of treatment.
In order to reduce side effects on other healthy tissues, efforts have been made to reduce the X-ray dosage and develop X-ray activated PLNPs that shorten X-ray irradiation time. Yang et al. produced tungsten(W)-doped ZnGa2O4:Cr (ZGO:Cr/W) coupled with the photosensitizer ZnPcS4, enabling photodynamic therapy with a low dose of X-rays (0.18 Gy).106 The presence of Cr3+ caused the generation of NIR luminescence peaking at 696 nm, which activated ZnPcS4 for the production of singlet oxygen. Following 2 minutes of X-ray irradiation, ZGO:Cr/W–ZnPcS4 exhibited increased singlet oxygen production, which persisted for 40 minutes more after the switching off of X-ray excitation due to the afterglow effect. In vitro PDT effects on cells demonstrated only 25% cell viability after 3 2-minute 0.09 Gy X-ray irradiation cycles.
Despite the effectiveness of the treatment, there is no imaging modality to guide the PDT process, since most of the NIR luminescence is quenched by the photosensitizer. This caused the development of Zn3Ga2Ge2O10:Cr3+,Mn2+ (ZGGCM), which could emit NIR (698 nm) and green (532 nm) afterglow from Cr3+ and Mn2+ ions, respectively.213 The green luminescence is used to activate Rose Bengal, while the NIR luminescence is used as an imaging modality to monitor the therapy. Intravenous administration of a pre-X-ray-excited ZGGCM solution to normal mice resulted in strong afterglow signals in the liver and lungs that lasted for at least 9 minutes. Apart from imaging with NIR luminescence, Gd3+-containing nanomaterials could also be imaged through computed tomography (CT) or magnetic resonance imaging (MRI) due to the strong X-ray absorption and long relaxation time of Gd3+.183
It has been recently reported that NaLuF4:Tb3+ conjugated with Rose Bengal (RB) and the ligand targeting amyloid-β (ScNPs@RB/Ab) could carry out PDT against Alzheimer's disease.14 Following intravenous injection of the nanosystem in vivo, the targeting amyloid-β caused a 6.6-fold enhancement in blood–brain barrier penetration efficiency compared to ScNPs@RB. 0.12 Gy X-rays successfully penetrated the scalp and excited ScNPs@RB/Ab in the brain, outperforming other excitation sources like NIR and green light (Fig. 20a and b). NaLuF4:Tb3+ emitted green luminescence, which excited RB to produce singlet oxygen (1O2) for oxygenation and suppression amyloid-β self-assembly (Fig. 20c). As a result, neurotoxic aggregated amyloid-β plaques could not be formed. After 18 days of treatment, spatial memory and learning of the mice were evaluated using the Morris water maze test. The group injected with ScNPs@RB/Ab and subjected to X-ray irradiation displayed improved cognitive function, with shorter escape latencies, increased time in the target quadrant, and more platform crossings (Fig. 20d–f). Moreover, Aβ plaque load in the cortex and hippocampus was significantly reduced, accompanied by a substantial decrease in soluble and insoluble Aβ42 levels (Fig. 20g–j). On the other hand, both ScNPs@RB/Ab and X-rays alone showed limited effects on cognitive function and Aβ deposition.
 | ||
| Fig. 20 (a) Transmittance of X-ray NIR and green light through the scalp, cranium and different thicknesses of brain tissue. (b) Schematic illustration for the X-ray activated photodynamic therapy for Alzheimer's disease by inhibiting Aβ aggregation in the brain. (c) Spectral overlap between X-ray activated luminescence from NaLuF4:Tb3+ and the absorption spectrum of Rose Bengal. In vivo comparison of (d) escape latency, (e) time in target quadrants and (f) times across the platform of Alzheimer's disease mice model in the Morris water maze test. Quantification of Aβ plaques in the (g) cortex and (h) hippocampus, as well as (i) soluble and (j) insoluble Aβ42 levels in the brain after different treatments. Reproduced with permission from ref. 14. Copyright 2023, Elsevier. | ||
Since these nanosystems could produce ROS without the quenching of their luminescence, their luminescence could be used for other purposes. For example, their luminescence could be used to activate a photosensitizer for additional generation of ROS. Au NPs were able to generate hydroxyl radicals under X-ray excitation while producing luminescence at 570 nm, to excite the conjugated Rose Bengal.233 The combined effect from both PDT and RT showed a better therapeutic effect than any of the treatments alone. Other than generating additional ROS, the luminescence generated could also be applied for imaging purposes in NIR-I and NIR-II regions.201,242 Yang et al. produced NIR-II-emitting Nd3+-doped black phosphorus quantum dots that produced ROS following X-ray irradiation.242 Nd3+ ions, having high X-ray attenuation, absorbed X-ray and relayed the energy to BP QDs to generate ROS (Fig. 21a). Concurrently, part of the excited Nd3+ relaxed and emitted NIR-II luminescence peaking at 1050 nm (Fig. 21b). In vivo studies revealed the strong NIR-II FL signals precisely located in the brain glioblastoma (GBM) site, peaking at 12 hours post-injection (Fig. 21c). This resulted in a reduction of HIF-1α and CD31 expression in the GBM region, accompanied by the suppression of intracranial GBM growth.
 | ||
| Fig. 21 (a) Schematic illustration of energy transfer from Nd3+ ions to BP QDs for ROS generation following X-ray irradiation. (b) NIR-II emission spectrum of Nd3+-doped BP QDs. (c) Real-time NIR-II luminescence imaging of mice intravenously injected with Nd3+-doped BP QDs. Reproduced with permission from ref. 242. Copyright 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Radiodynamic therapy or photodynamic therapy rely on ROS generation and are often limited by the hypoxic tumour microenvironment. Without the use of ROS, LuPO4:Pr3+ produced UV light following X-ray irradiation to kill tumour cells through oxygen-independent processes.187 LuPO4:Pr3+ converted X-rays into UVC radiation through a unique 4f15d1-4f transition in Pr3+, resulting in a localized emission within the 220–285 nm range. UVC radiation induces a significant DNA damage primarily affecting cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6-4PPs), leading to cell cycle arrest and inactivation. Due to the strong absorption of UVC photons within a few micrometers, neighboring cells near the scintillating particles are impacted while the normal surrounding tissue is spared. Remarkably, at a concentration of 2.5 mg mL−1 LuPO4:Pr3+, almost 90% of fibroblast (HFF1) cells were eradicated under 2 Gy X-ray irradiation.
Apart from utilising NO, Yang et al. designed Au-TiO2 coated with PLNP ZnS:Cu,Co-A (Au-TiO2@ZnS:Cu,Co-A) for the generation of H2 in tumour cells, which exhibited anti-cancer and anti-inflammatory properties.209 Under X-ray irradiation, electrons at the Au–TiO2 NR heterojunction were transferred to Au NRs to undergo a reduction reaction with water to produce H2. Following the cessation of X-ray irradiation, ZnS:Cu,Co-A produced afterglow luminescence at 578 nm. This enabled Au NRs to inject hot electrons into the conduction band of TiO2 to catalyze H2 generation, which is observed even 10 minutes after cessation of X-ray irradiation. Concurrently, the holes generated are captured by sacrificial agents or OH− ions, yielding reactive oxygen species. Au–TiO2@ZnS showed concentration- and time-dependent cytotoxicity on MC38 cancer cells in vitro, as well as a 90.9% tumour supression rate in vivo.
Instead of solely relying on ROS generation, Hao et al. developed a CO-based gas therapy that could also activate anti-tumour immunity.13 They combined NaLuF4:Gd3+,Tb3+@NaLuF4 with PhotoCORM (ScNPs-PhotoCORM), where green emission from Tb3+ activated PhotoCORM to generate CO and ROS up to 8 cm deep in biological tissue following X-ray irradiation. Simultaneously, CO reversed the deep tissue immunosuppressive TME and activated adaptive anti-tumour immunity (Fig. 22a). Tumour-bearing mice treated with ScNPs-PhotoCORM exhibited elevated levels of pro-inflammatory IL-6 and TNF-α, while exhibiting lower levels of anti-inflammatory cytokine IL-10 (Fig. 22b–d). Furthermore, the mice exhibited enhanced levels of interferon-γ (IFN-γ) and CD8, confirming the activation of in vivo adaptive anti-tumour immunity (Fig. 22e and f). Both the primary and distant tumours exhibited significant suppression in tumour growths, confirming the successful activation of deep tissue anti-tumour immunity response and TME reversal (Fig. 22g and h). To further enhance the efficacy of CO-based gas therapy, LiLuF4:Ce3+ and UV-photosensitive Mn2(CO)10 were combined.246 UV emission from LiLuF4:Ce3+ following X-ray irradiation resulted in CO and MnO2 release from Mn2(CO)10. MnO2 generated hydroxyl radicals through Fenton-like activity, while depleting glutathione (GSH) to impair the cellular antioxidant defence system. In vivo studies with the nanosystem showed superior tumour growth control through DNA damage and inhibiting glycolysis.
 | ||
| Fig. 22 (a) Schematic illustration of CO-based gas therapy which involves ROS generation and immune activation to a pro-inflammatory state. (b) Comparison of in vivo mice tumour slices of different treatment groups stained with (b) IL-10, (c) TNF-α, (d) IL-6, (e) IFN-γ and (f) CD8. Comparison of (g) primary and (h) distant tumour growth in 4T1 tumour-bearing mice with different treatments. Reproduced with permission from ref. 13. Copyright 2021, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4.4 Strengths and weaknesses of X-ray-activated light-mediated therapy
X-ray activated luminescence deep-tissue therapy holds both promising strengths and weaknesses as a therapeutic approach. One of its primary strengths lies in its ability to penetrate deep tissues in the body, which enables the treatment in deep tissue which is challenging to access using UV, NIR or visible light. For instance, X-rays can penetrate bones, enabling light-based therapeutics in the brain through the cranium. Additionally, the use of high-energy X-ray excitation allows direct band-to-band excitation of nanomaterials, which causes highly efficient generation of UV or visible light. The vast production of UV/visible light enabled the activation of light-sensitive molecules, such as photosensitisers, photocleavable linkers which operate mostly in the UV/visible range. In addition, the highly efficient UV production could also cause direct cell damage to the target cells while minimizing healthy cell damage due to the low penetration of UV light. Compared to NIR-induced upconversion luminescence which requires the energy accumulation of several photons, X-ray-excited UV/visible emissions are much more efficient. The high luminescence efficiency also results in brighter luminescence and longer afterglow in various nanosystems.However, there are weaknesses that accompany the application of X-ray-excited luminescence in deep tissue therapy. One significant concern is the potential for radiation-induced damage to healthy tissue. The radioactive nature of X-rays inadvertently damages the DNA of healthy cells which could cause adverse side effects and complications. In addition, since X-rays favour the generation of UV/visible light, NIR emissions from X-ray-excited nanomaterials are rarely seen, which limits their potential applications to photothermal therapy, or NIR-II deep tissue imaging and monitoring. Furthermore, X-ray-generating equipment are scarce and often costly, which limits the accessibility and availability of X-ray activated luminescence therapy.
5. Ultrasound-excited luminescence
Ultrasound, possessing deep tissue penetration and being relatively safe, has been used extensively as an imaging modality since the 1960s. Out of all the excitation methods listed before, nanomaterials that exhibit ultrasound-excited luminescence are rarer and much less studied. However, ultrasound-activated light therapy remains as one of the most promising therapeutic modality.In this section, we will discuss 2 ways by which ultrasound can generate light through nanomaterial-mediated processes – nanomaterial-mediated sonoluminescence and ultrasound-excited mechano-luminescence, as well as their application in deep tissue therapy.
5.1 Nanomaterial-mediated sonoluminescence
Sonoluminescence, the emission of light following acoustic cavitation of microbubbles, was discovered over 2 decades ago.11 Following ultrasound irradiation, acoustic cavitation occurs in the liquid medium, causing the generation of microbubbles that undergo rapid compression and expansion, and finally collapse (Fig. 23a). The collapse of microbubbles usually results in a temperature change in the gas over thousands of degrees in a very short time resulting in gas ionization and light emission, usually in the UV range.248 Certain nanomaterials can also accept energy from sonoluminescence to emit at a different wavelength (Fig. 23a). | ||
| Fig. 23 Schematic illustration of the mechanism of (a) nanomaterial-mediated sonoluminescence and (b) ultrasound-excited mechano-luminescence. | ||
However, sonoluminescence is weak and lacks tunability in emission wavelength. Therefore, nanomaterials were used in several studies to improve the intensity and modify the emission wavelength of sonoluminescence, fostering their application in deep tissue therapy. For example, carbon nanodots were shown to alter the original blue sonoluminescence into orange.249 The original blue sonoluminescence results from the interaction between hydroxyl free radicals (˙OH) in water and the collapsing bubbles. Following the addition of carbon nanodots, they captured ˙OH and formed C- and O-based functional groups like –COOH bonds and CO molecules. This interaction suppressed ˙OH radicals and resulted in a weaker UV/blue emission. On the other hand, –COOH or C-based molecules like CO were excited by the collapse of bubbles and emitted at 610 nm following radiative recombination. Nanomaterials can also enhance the intensity of sonoluminescence. A nanoconjugate composed of protoporphyrin IX and gold nanoparticles (Au–PpIX) was able to enhance sonoluminescence at 350–650 nm by over 4 times.250 This is because gold nanoparticles functioned as cavitation nuclei, increasing the microbubble formation. In addition, PpIX was activated by ultrasound to generate free radicals which resulted in greater sonoluminescence. Similarly, biochar-supported ZnO (ZnO-BC) nanorods also enhanced sonoluminescence by reducing the energy threshold required for bubble generation, increasing the number of nucleation sites and microbubbles that led to stronger sonoluminescence.251
One of the most prominent applications of nanomaterial-mediated sonoluminescence is sonodynamic therapy, where the generation of sonoluminescence causes the excitation of photosensitisers and generation of reactive oxygen species (ROS). Grebinyk et al. reported that the combination of ultrasound irradiation and 20 μM C60 fullerene reduced the viability of HeLa cells.252 C60 fullerene exhibited a broad-band absorption from 320 to 580 nm, which overlapped with the UV-peaked broad emission spectrum of sonoluminescence. 60 s of 1 MHz ultrasound irradiation caused ROS generation and reduction in cell viability of HeLa cells to 59%, while ultrasound or C60 alone had negligible effects on cell viability. To reduce the distance between the site of sonoluminescence and photosensitizers, Wang et al. synthesised microbubbles with Rose Bengal as the wall (RB-MB), where the ultrasound-induced collapse of microbubbles generated sonoluminescence to directly excite Rose Bengal.253 Under 1 MHz ultrasound irradiation, the RB-MBs exhibited enhanced ROS production compared to other control groups. In addition, RB-MBs exhibited 76.5% tumour inhibition in the HT-29 tumour mice model as a result of a more efficient energy transfer, whereas the combination of Rose Bengal NPs and ultrasound alone exhibited 49.2% tumour inhibition. Sonodynamic therapy can also be enhanced through increasing the efficiency of ROS generation of photosensitizers. Au144 clusters were deposited on TiO2, acting as an electron acceptor that prevented the rapid electron–hole recombination of TiO2.254 Hence, ˙OH production was enhanced by 2 fold. The efficient electron-trapping was enabled due to the slightly lower energy level of the LUMO in Au144 compared to the excited state of TiO2.
To reduce ultrasound irradiation time and enable low-background imaging/monitoring, sonodynamic therapy can also be coupled with ultrasound-induced afterglow. Recently, Pu's group developed an organic nanosystem (NCBS/DPAs SNAP) that was able to produce singlet oxygen and ultrasound-induced afterglow through dioxetane-mediated emission.255 Under ultrasound irradiation, sonoluminescence activated the photosensitizer silicon 2,3-naphthalocyanine bis(trihexylsilyloxide) (NCBS) to produce singlet oxygen, which converted the sonoafterglow substrate dicyanomethylene-4H-benzothiopyran-phenoxyl-adamantylidene (DPAs) into active dioxetane substrates. NCBS absorbed the energy from dioxetane and emitted afterglow luminescence at 780 nm (Fig. 24a). Following 5 minutes of 1 MHz ultrasound irradiation, the nanosystem induced 90% cell death in 4T1 cancer cells. To enhance the therapeutic specificity of the nanosystem, DPAs were silenced with ONOO− responsive moieties (Pro-DPAs). This nanosystem was inactive in healthy tissues populated with M0 and M2 macrophages, while exhibiting a 3.5 fold increase in ROS generation and luminescence in a M1 macrophage-populated tumour microenvironment due to the overproduction of ONOO−. An SBR of around 90 was also observed in a living mouse under 1.8 cm tissue depth, which is 4.0 and 47.4 times higher than that for photoafterglow and fluorescence, respectively. This nanosystem serves as an excellent candidate for tumour-specific sonodynamic therapy with deep tissue imaging capabilities.
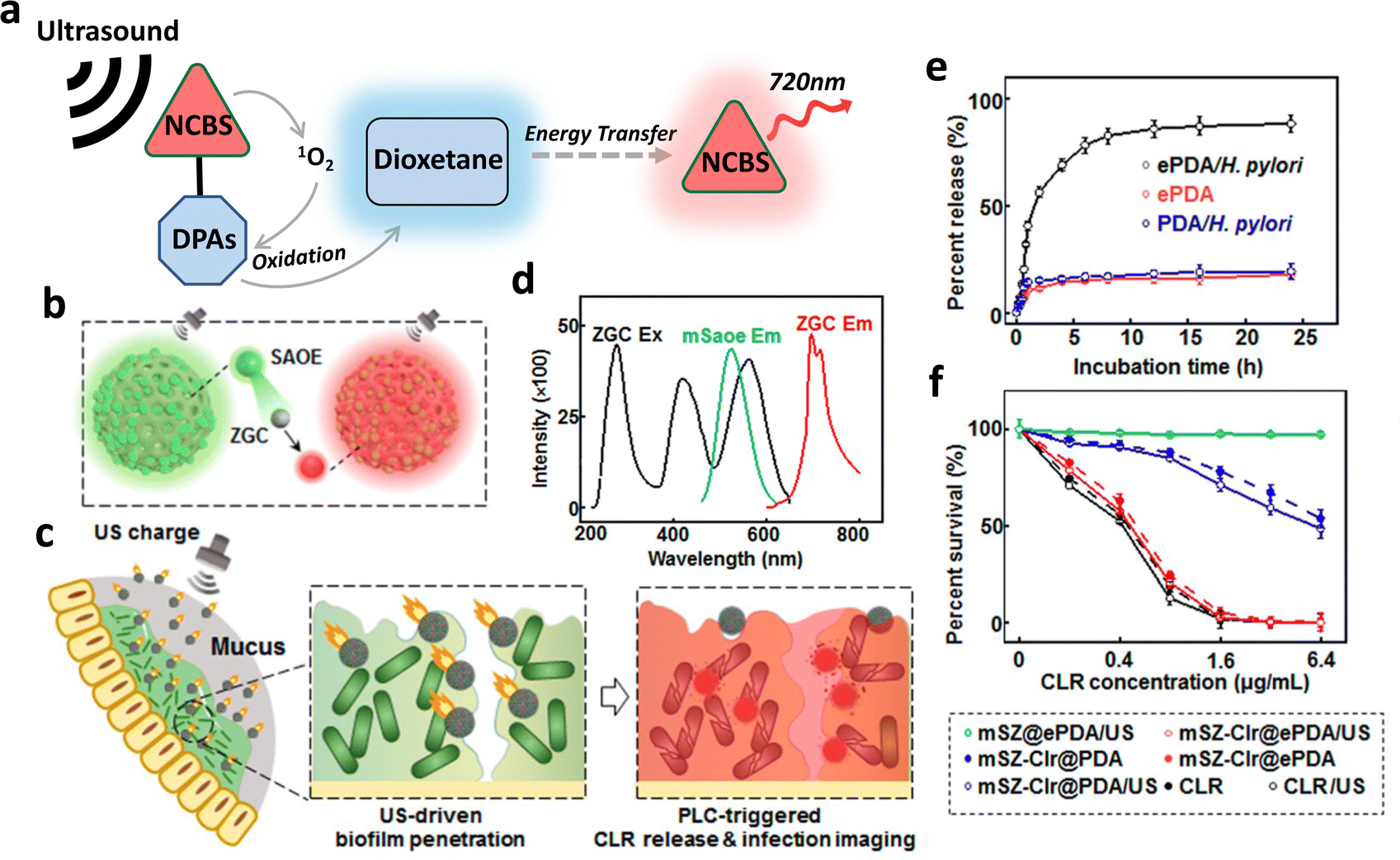 | ||
| Fig. 24 (a) Schematic illustration of the mechanism of nanomaterial-mediated sonoluminescence afterglow through generation of singlet oxygen and dioxetane intermediates. (b) Schematic illustration of the mechanism of ultrasound-excited mechanoluminescence through energy transfer between SAOE and ZGC, emitting red afterglow luminescence. (c) Schematic illustration of PLC-triggered drug release and ultrasound-activated NIR afterglow for H. pylori treatment and imaging. (d) Excitation and emission spectra of ZGC and the emission spectrum of SAOE. (e) Percentage release of CLR from the ePDA-coated or non-coated nanosystem alone or incubated with H. pylori. (f) Survival rate of H. pylori following treatment with different groups at different CLR concentration. Reproduced with permission from ref. 256. Copyright 2022, American Chemical Society. | ||
5.2 Ultrasound-excited mechanoluminescence
Although nanomaterials can enhance sonoluminescence or alter its emission wavelength, there are still insufficient strategies to control sonoluminescence, as its occurrence and intensity depends on many different parameters of the liquid medium.257On the other hand, ultrasound-activated mechanoluminescence depends mainly on the characteristics of the nanoparticle, which could be more easily controlled. Mechanoluminescence generally refers to the emission of light under external mechanical stress or deformation of the crystal.12 In the nanomaterial world, it was realised that ultrasound could induce mechanical stress or deformation in nanomaterials, enabling mechanoluminescence through different mechanisms (Fig. 23b). For example, ultrasound could release electrons trapped inside Sr2MgSi2O7:Eu2+,Dy3+ (SMSO), resulting in emission at 470 nm. When SMSO is exposed to 10 seconds of 365 nm UV irradiation, an electron from Eu2+ is excited to the conduction band, which is then trapped by 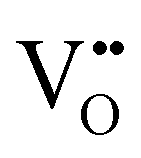 and
and 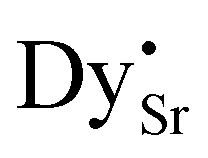 defects.258 Upon 1.5 MHz ultrasound irradiation, the trapped electron is released back to the ground state of Eu2+, causing luminescence peaking at 470 nm due to the 4f65d1 to 4f7 transition of Eu2+. By incorporating it with caesium lead halide quantum dots inside a polydimethylsiloxane (PDMS) pixel array, the emission wavelength can even be tuned to 515 nm (green) or 640 nm (red).
defects.258 Upon 1.5 MHz ultrasound irradiation, the trapped electron is released back to the ground state of Eu2+, causing luminescence peaking at 470 nm due to the 4f65d1 to 4f7 transition of Eu2+. By incorporating it with caesium lead halide quantum dots inside a polydimethylsiloxane (PDMS) pixel array, the emission wavelength can even be tuned to 515 nm (green) or 640 nm (red).
Ultrasound irradiation (1 MHz) can also induce mechanoluminescence in SrAl2O4:Eu2+ (SAOE) at 525 nm without the need for UV pre-irradiation. When combined with persistent luminescent ZnGa2O4:Cr3+ (ZGC) inside mesoporous silica nanoparticles (mSZ), the mechanoluminescence at 525 nm from SAOE excited ZGC, emitting afterglow at 715 nm (Fig. 24b and d).256 The nanosystem was loaded with the drug clarithromycin (CLR) and coated with quenching polydopamine layers (ePDA). The presence of phospholipase C from H. pylori degraded the ePDA layers and released CLR while restoring the NIR luminescence (Fig. 24c). 82.4% of CLR was released following 25 hours of incubation with H. pylori, whereas only 15.8% of CLR release was observed in PBS (Fig. 24e). In addition, afterglow was only detected in H. pylori mice, and the intensity increased with increasing H. pylori concentrations. Incubating this nanosystem with H. pylori reduced cell viability to 15.3% at 6.4 μg mL−1 (Fig. 24f). The identical nanosystem was also applied for a photothermal/NO-based combined therapy.259 The NIR afterglow produced under ultrasound irradiation activated PDA layers to liberate NO and induces a photothermal effect. The liberation of NO and the temperature gradient on the NPs caused them to self-propel, enhancing endocytosis into tumour cells by 2 fold. The NPs also exhibited sustained NO release for 2 hours, accompanied by an 11 °C temperature increase which persisted for 30 minutes after the cessation of ultrasound activation. This combined therapy completely halted the tumour growth after 27 days of treatment, while exhibiting the highest apoptosis rate (95.2%) and the lowest proliferation rate (8.3%) compared to other treatments.
5.3 Strengths and weaknesses of ultrasound-activated luminescence-based therapy
The utilization of ultrasound-activated phototherapy for the treatment of deep tissues presents both noteworthy advantages and limitations. Among its advantages, ultrasound possesses a superior tissue penetration depth compared to traditional UV/vis light and near-infrared (NIR) light, rendering it suitable for deep tissue applications. Importantly, ultrasound is regarded as a safer modality than X-rays as it does not involve ionizing radiation, reducing the risk of harming healthy cells. Additionally, the equipment required for ultrasound generation is simpler and more cost-effective compared to X-ray generating equipment.However, it is important to note that ultrasound does not possess enough penetration depth compared to X-rays, especially through bone, where it experiences severe scattering. This limits the application of ultrasound-activated light therapy in areas shielded by bone, such as the brain. Despite being the most studied form of ultrasound-activated luminescence, sonoluminescence is still an unpredictable and random process. The difficulty in controlling the intensity, location and occurrence of such process makes it challenging for application in therapeutic purposes. Although ultrasound-excited mechanoluminescent nanoparticles can solve such problems, currently there are limited reports on nanomaterials that produce strong and consistent luminescence under ultrasound activation. This greatly impedes the application of such a mechanism in deep tissue therapy. Finally, although ultrasound does not cause radiation damage, it might induce unwanted cavitation in other healthy cells, which can cause unwanted tissue heating.
To widely apply ultrasound-excited luminescence in deep tissue therapy in the future, it is essential to produce low-dose ultrasound-activatable luminescent nanomaterials that rely on a controllable mechanism to produce luminescence.
6. Conclusions and outlook
Deep tissue light-mediated therapy is promising due to the precise control of therapy and its effectiveness. Much work has been performed to overcome the bottleneck of traditional light-based therapy which involves using low-penetrating UV or visible light to activate therapy. Studies on chemiluminescence-, NIR-, X-ray- and ultrasound-activated light therapy have proved sufficient specificity and therapeutic efficacy, despite having individual advantages and drawbacks. Chemiluminescence allows autofluorescence-free luminescence generation, but it lacks external control over luminescence and therapy activation. NIR enables tuneable excitation and emission wavelength that could be applied for different theragnostic purposes, i.e., orthogonal excitation/emission, but its penetration depth is limited. X-rays possess the deepest tissue penetration depth and generate strong luminescence, but they cause radiation damage to surrounding healthy cells. Ultrasound possesses sufficient tissue penetration combined with limited side effects on healthy tissue, but the mode of luminescence generation is not very well controlled and limited reports on ultrasound-activatable nanoparticles are available.Other than carrying out the therapy alone, image-guided therapy has been studied more recently. This is because it provides a modality to monitor the therapy which results in greater precision and efficiency, with the help of CT, MRI, or luminescence in the NIR-IIb/NIR-IIc spectrum. Efforts have also been made to develop “activatable” nanosystems that only become activated at target sites, exhibiting strong specificity when combined with external light triggering.
However, challenges still exist in deep tissue therapy with the mentioned 4 excitation modes. First of all, external irradiation using NIR, X-rays or ultrasound frequently results in unwanted irradiation on the healthy tissue, which may lead to unwanted side effects. NIR causes tissue heating and X-ray causes radiation damage while ultrasound causes unwanted cavitation that also leads to tissue heating. Therefore, since it is hard to eliminate such side effects, it is important to engineer nanosystems that can be activated by low dose of NIR, X-ray and ultrasound. Second, certain activation modes still do not yield luminescence with high intensity or efficacy, which includes chemiluminescence, sonoluminescence or certain NIR-activated luminescence. Efforts would be required to develop more controllable, efficient and brighter nanosystems following such activation. Finally, there has also been an emergence of combined therapy, such as the combination of chemotherapy and photodynamic therapy, resulting in a better therapeutic effect. In these systems, it is important to ensure that both therapies are activated by light, or activated by the specific local environment of the target cells to ensure specificity of the treatment.
Author contributions
Chung Yin Tsang: conceptualisation, writing – original draft, and writing – review and editing; Yong Zhang: conceptualisation, writing – review and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support received from the National Medical Research Council of Singapore (NMRC, MOH-000640, and MOH-001114-00), and the City University of Hong Kong (project number 9380160).Notes and references
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed
.
- B. Simmons, R. Griffith, L. Falto-Aizpurua and K. Nouri, J. Eur. Acad. Dermatol. Venereol., 2015, 29, 1275–1279 CrossRef CAS PubMed
- N. Fomina, J. Sankaranarayanan and A. Almutairi, Adv. Drug Delivery Rev., 2012, 64, 1005–1020 CrossRef CAS PubMed
- B. C. Wilson and G. Adam, Med. Phys., 1983, 10, 824–830 CrossRef CAS PubMed
- D. J. Burgess, Nat. Rev. Cancer, 2012, 12, 737 Search PubMed
- Y. Liu, Y. Liang, P. Lei, Z. Zhang and Y. Chen, Adv. Sci., 2023, 10, e2203669 CrossRef PubMed
- S. Bi, Z. Deng, J. Huang, X. Wen and S. Zeng, Adv. Mater., 2023, 35, e2207038 CrossRef PubMed
- Y. Chang, H. Chen, X. Xie, Y. Wan, Q. Li, F. Wu, R. Yang, W. Wang and X. Kong, Nat. Commun., 2023, 14, 1079 CrossRef CAS PubMed
- S. Diao, J. L. Blackburn, G. Hong, A. L. Antaris, J. Chang, J. Z. Wu, B. Zhang, K. Cheng, C. J. Kuo and H. Dai, Angew. Chem., 2015, 127, 14971–14975 CrossRef
- X. Qian, Y. Zheng and Y. Chen, Adv. Mater., 2016, 28, 8097–8129 CrossRef CAS PubMed
- M. P. Brenner, S. Hilgenfeldt and D. Lohse, Rev. Mod. Phys., 2002, 74, 425 CrossRef CAS
- A. Feng and P. F. Smet, Materials, 2018, 11, 484 CrossRef PubMed
- Y. Li, M. Jiang, Z. Deng, S. Zeng and J. Hao, Adv. Sci., 2021, 8, e2004391 CrossRef PubMed
- M. Ma, J. Wang, H. Jiang, Q. Chen, Y. Xiao, H. Yang and L. Lin, Acta Biomater., 2023, 155, 635–643 CrossRef CAS PubMed
- Z.-F. Zhang, H. Cui, C.-Z. Lai and L.-J. Liu, Anal. Chem., 2005, 77, 3324–3329 CrossRef CAS PubMed
- M. Vacher, I. Fdez. Galván, B.-W. Ding, S. Schramm, R. Berraud-Pache, P. Naumov, N. Ferre, Y.-J. Liu, I. Navizet and D. Roca-Sanjuan, Chem. Rev., 2018, 118, 6927–6974 CrossRef CAS PubMed
- A. García-Campaña and W. Baeyens, Analusis, 2000, 28, 686–698 CrossRef
- H. An, C. Guo, D. Li, R. Liu, X. Xu, J. Guo, J. Ding, J. Li, W. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 17230–17243 CrossRef CAS PubMed
- Z. Chen, Y. Tan, K. Xu, L. Zhang, B. Qiu, L. Guo, Z. Lin and G. Chen, Biosens. Bioelectron., 2016, 75, 8–14 CrossRef CAS PubMed
- M. Wu, L. Wu, J. Li, D. Zhang, S. Lan, X. Zhang, X. Lin, G. Liu, X. Liu and J. Liu, Theranostics, 2019, 9, 20–33 CrossRef CAS PubMed
- Z. Zhou, X. Liu, L. Yue and I. Willner, ACS Nano, 2018, 12, 10725–10735 CrossRef CAS PubMed
- H. Liu, Y. Su, D. Deng, H. Song and Y. Lv, Anal. Chem., 2019, 91, 9174–9180 CrossRef CAS PubMed
- S. Y. Yin, W. Liu, K. Zhang and J. Li, ACS Appl. Bio Mater., 2021, 4, 3490–3498 CrossRef CAS PubMed
- L. Cai, L. Deng, X. Huang and J. Ren, Anal. Chem., 2018, 90, 6929–6935 CrossRef CAS PubMed
- Z. Gu, A. Fu, L. Ye, K. Kuerban, Y. Wang and Z. Cao, ACS Sens., 2019, 4, 2922–2929 CrossRef CAS PubMed
- Y. Hou, R. Han, Y. Sun, C. Luo and X. Wang, Anal. Chim. Acta, 2022, 1195, 339386 CrossRef CAS PubMed
- L. Han, Y. Li and A. Fan, Luminescence, 2018, 33, 751–758 CrossRef CAS PubMed
- Y. Wang, M. Wang, L. Han, Y. Zhao and A. Fan, Talanta, 2018, 182, 523–528 CrossRef CAS PubMed
- Z. Abolghasemi-Fakhri, M. Amjadi and J. L. Manzoori, Spectrochim. Acta, Part A, 2019, 216, 85–90 CrossRef CAS PubMed
- Y. Tian, Y. Zhang, X. Lu, D. Xiao and C. Zhou, J. Mater. Chem. B, 2023, 11, 2200–2206 RSC
- H. Chen, F. Gao, R. He and D. Cui, J. Colloid Interface Sci., 2007, 315, 158–163 CrossRef CAS PubMed
- F. Zheng, W. Ke, Y. Zhao and C. Xu, Electrophoresis, 2019, 40, 2218–2226 CrossRef CAS PubMed
- Y. Qi, B. Li and F. Xiu, Spectrochim. Acta, Part A, 2014, 128, 76–81 CrossRef CAS PubMed
- Y. Wu, J. Wang and H. Cui, Anal. Bioanal. Chem., 2022, 414, 367–375 CrossRef CAS PubMed
- W. H. Chen, M. Vázquez-González, A. Kozell, A. Cecconello and I. Willner, Small, 2018, 14, 1703149 CrossRef PubMed
- L. He, Z. W. Jiang, W. Li, C. M. Li, C. Z. Huang and Y. F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 28868–28876 CrossRef CAS PubMed
- Z. Wang, F. Liu and C. Lu, Biosens. Bioelectron., 2014, 60, 237–243 CrossRef CAS PubMed
- S. Bi, B. Xiu, J. Ye and Y. Dong, ACS Appl. Mater. Interfaces, 2015, 7, 23310–23319 CrossRef CAS PubMed
- Y. Sun, L. Shi, Q. Wang, L. Mi and T. Li, Anal. Chem., 2019, 91, 3652–3658 CrossRef CAS PubMed
- J. Hassanzadeh and A. Khataee, Talanta, 2018, 178, 992–1000 CrossRef CAS PubMed
- M. Zhou, Q. Chen, A. Wang, J. Li and Y. Ma, Luminescence, 2019, 34, 673–679 CrossRef CAS PubMed
- M. Amjadi, J. L. Manzoori, T. Hallaj and M. H. Sorouraddin, Spectrochim. Acta, Part A, 2014, 122, 715–720 CrossRef CAS PubMed
- T. Hallaj, M. Amjadi, J. L. Manzoori and N. Azizi, Luminescence, 2017, 32, 1174–1179 CrossRef CAS PubMed
- E. Delnavaz and M. Amjadi, Mikrochim. Acta, 2021, 188, 278 CrossRef CAS PubMed
- J. Li, Y. Han, X. Li, L. Xiong, L. Wei and X. Cheng, Luminescence, 2021, 36, 79–84 CrossRef CAS PubMed
- Z. Lin, X. Dou, H. Li, Y. Ma and J. M. Lin, Talanta, 2015, 132, 457–462 CrossRef CAS PubMed
- Y. Haghighi Shishavan and M. Amjadi, Luminescence, 2022, 37, 734–741 CrossRef CAS PubMed
- S. Han, Z. Fan, X. Chen, Y. Wu and J. Wang, Spectrochim. Acta, Part A, 2017, 183, 103–108 CrossRef CAS PubMed
- Y. Li, J. Wang, Y. Yang and S. Han, Luminescence, 2020, 35, 773–780 CrossRef CAS PubMed
- M. Amjadi, T. Hallaj and F. Mirbirang, Mikrochim. Acta, 2020, 187, 191 CrossRef CAS PubMed
- Y. H. Seo, A. Singh, H. J. Cho, Y. Kim, J. Heo, C. K. Lim, S. Y. Park, W. D. Jang and S. Kim, Biomaterials, 2016, 84, 111–118 CrossRef CAS PubMed
- H. Chen, H. Li and J. M. Lin, Anal. Chem., 2012, 84, 8871–8879 CrossRef CAS PubMed
- R. Chen, L. Zhang, J. Gao, W. Wu, Y. Hu and X. Jiang, J. Biomed. Biotechnol., 2011, 2011, 679492 Search PubMed
- S. Y. Yin, W. Liu, J. Yang and J. Li, J. Mater. Chem. B, 2021, 9, 5877–5886 RSC
- Y. C. Chen, Y. J. Liu, C. L. Lee, K. Y. Pham, D. Manoharan, S. Thangudu, C. H. Su and C. S. Yeh, Adv. Healthcare Mater., 2022, 11, e2201613 CrossRef PubMed
- J. Wang, C. Yao, B. Shen, X. Zhu, Y. Li, L. Shi, Y. Zhang, J. Liu, Y. Wang and L. Sun, Theranostics, 2019, 9, 608–619 CrossRef CAS PubMed
- Q. Mao, J. Fang, A. Wang, Y. Zhang, C. Cui, S. Ye, Y. Zhao, Y. Feng, J. Li and H. Shi, Angew. Chem., Int. Ed., 2021, 60, 23805–23811 CrossRef CAS PubMed
- Y. Zhou, W. Wu, P. Yang, D. Mao and B. Liu, Biomaterials, 2022, 288, 121693 CrossRef CAS PubMed
- C. Sun, Z. Wang, L. Yue, Q. Huang, S. Lu and R. Wang, J. Mater. Chem. B, 2020, 8, 8878–8883 RSC
-
P. N. Prasad, Introduction to biophotonics, John Wiley & Sons, 2004 Search PubMed
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed
- H. Li, E. Heydari, Y. Li, H. Xu, S. Xu, L. Chen and G. Bai, Nanomaterials, 2023, 13, 219 CrossRef CAS PubMed
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed
- Q. Liu, J. Tian, Y. Tian, Q. Sun, D. Sun, F. Wang, H. Xu, G. Ying, J. Wang, A. K. Yetisen and N. Jiang, ACS Nano, 2021, 15, 515–525 CrossRef CAS PubMed
- X. Wang, H. Li, F. Li, X. Han and G. Chen, Nanoscale, 2019, 11, 22079–22088 RSC
- H. M. Gong, S. Xiao, X. R. Su, J. B. Han and Q. Q. Wang, Opt. Express, 2007, 15, 13924–13929 CrossRef CAS PubMed
- Z. Cheng, R. Chai, P. Ma, Y. Dai, X. Kang, H. Lian, Z. Hou, C. Li and J. Lin, Langmuir, 2013, 29, 9573–9580 CrossRef CAS PubMed
- R. Tian, W. Sun, M. Li, S. Long, M. Li, J. Fan, L. Guo and X. Peng, Chem. Sci., 2019, 10, 10106–10112 RSC
- J. Park, A. Estrada, K. Sharp, K. Sang, J. A. Schwartz, D. K. Smith, C. Coleman, J. D. Payne, B. A. Korgel, A. K. Dunn and J. W. Tunnell, Opt. Express, 2008, 16, 1590–1599 CrossRef CAS PubMed
- N. Akizuki, S. Aota, S. Mouri, K. Matsuda and Y. Miyauchi, Nat. Commun., 2015, 6, 8920 CrossRef CAS PubMed
- S. Míguez-Lago, I. F. A. Mariz, M. A. Medel, J. M. Cuerva, E. Maçôas, C. M. Cruz and A. G. Campaña, Chem. Sci., 2022, 13, 10267–10272 RSC
- Y. Zhang, T. T. Shen, H. L. Zhang, A. M. Kirillov, H. J. Cai, J. Wu, W. S. Liu and Y. Tang, Chem. Commun., 2016, 52, 4880–4883 RSC
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC
- L. Zhang, L. Zeng, Y. Pan, S. Luo, W. Ren, A. Gong, X. Ma, H. Liang, G. Lu and A. Wu, Biomaterials, 2015, 44, 82–90 CrossRef CAS PubMed
- K. Prorok, M. Olk, M. Skowicki, A. Kowalczyk, A. Kotulska, T. Lipiński and A. Bednarkiewicz, Nanoscale Adv., 2019, 1, 3463–3473 RSC
- G. S. Yi and G. M. Chow, Adv. Funct. Mater., 2006, 16, 2324–2329 CrossRef CAS
- C. Mi, J. Zhou, F. Wang and D. Jin, Nanoscale, 2019, 11, 12547–12552 RSC
- Q. Feng, W. Zheng, J. Pu, Q. Chen and W. Shao, Front. Chem., 2021, 9, 690833 CrossRef CAS PubMed
- H. Wang, Y. Xu, T. Pang, B. Chen, F. Xin, M. Xing, M. Tian, Y. Fu, X. Luo and Y. Tian, Nanoscale, 2022, 14, 962–968 RSC
- J. A. Damasco, G. Chen, W. Shao, H. Ågren, H. Huang, W. Song, J. F. Lovell and P. N. Prasad, ACS Appl. Mater. Interfaces, 2014, 6, 13884–13893 CrossRef CAS PubMed
- X. Wang, K. Liu, G. Yang, L. Cheng, L. He, Y. Liu, Y. Li, L. Guo and Z. Liu, Nanoscale, 2014, 6, 9198–9205 RSC
- J. Li, Y. Long, Q. Zhao, S. Zheng, Z. Fang and B. O. Guan, Nanomaterials, 2021, 11, 1033 CrossRef CAS PubMed
- Q. Y. Meng, B. J. Chen, S. C. Lü, J. T. Sun and X. R. Qu, Guangpuxue Yu Guangpu Fenxi, 2010, 30, 1224–1228 CAS
- H. Niioka, S. Fukushima, M. Ichimiya, M. Ashida, J. Miyake, T. Araki and M. Hashimoto, Microscopy, 2014, 63(Suppl 1), i29 CrossRef PubMed
- J. Liu, L. Huang, X. Tian, X. Chen, Y. Shao, F. Xie, D. Chen and L. Li, Int. J. Nanomed., 2017, 12, 1–14 CrossRef PubMed
- C. Wang, L. Xu, J. Xu, D. Yang, B. Liu, S. Gai, F. He and P. Yang, Dalton Trans., 2017, 46, 12147–12157 RSC
- S. Ryszczyńska and T. Grzyb, Methods Appl. Fluoresc., 2022, 10, 024001 CrossRef PubMed
- Y. Li, R. Wang, W. Zheng and Y. Li, Inorg. Chem., 2019, 58, 8230–8236 CrossRef CAS PubMed
- Z. Giedraityte, M. Tuomisto, M. Lastusaari and M. Karppinen, ACS Appl. Mater. Interfaces, 2018, 10, 8845–8852 CrossRef CAS PubMed
- Y. Li, G. Bai, S. Zeng and J. Hao, ACS Appl. Mater. Interfaces, 2019, 11, 4737–4744 CrossRef CAS PubMed
- F. Huang, Y. Li, J. Liu, J. Zhang, X. Wang, B. Li, H. Chang, Y. Miao and Y. Sun, ACS Appl. Bio Mater., 2021, 4, 5695–5706 CrossRef CAS PubMed
- S. Sekiyama, M. Umezawa, S. Kuraoka, T. Ube, M. Kamimura and K. Soga, Sci. Rep., 2018, 8, 16979 CrossRef PubMed
- Z. Zhang, Y. Yang, M. Zhao, L. Lu, F. Zhang and Y. Fan, ACS Appl. Bio Mater., 2022, 5, 2935–2942 CrossRef CAS PubMed
- D. E. Hudson, D. O. Hudson, J. M. Wininger and B. D. Richardson, Photomed. Laser Surg., 2013, 31, 163–168 CrossRef PubMed
- C. Cao, M. Xue, X. Zhu, P. Yang, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 18540–18548 CrossRef CAS PubMed
- D. Zhao, X. Han, S. Wang, J. Liu, Y. Lu and C. Li, Chemistry, 2020, 26, 3145–3151 CrossRef CAS PubMed
- M. Lesniak, M. Kochanowicz, A. Baranowska, P. Golonko, M. Kuwik, J. Zmojda, P. Miluski, J. Dorosz, W. A. Pisarski, J. Pisarska and D. Dorosz, Nanomaterials, 2021, 11, 2115 CrossRef CAS PubMed
- S. Li, Q. Ma, C. Wang, K. Yang, Z. Hong, Q. Chen, J. Song, X. Song and H. Yang, Anal. Chem., 2022, 94, 2641–2647 CrossRef CAS PubMed
- F. Bertorelle, K. D. Wegner, M. Perić Bakulić, H. Fakhouri, C. Comby-Zerbino, A. Sagar, P. Bernadó, U. Resch-Genger, V. Bonačić-Koutecký, X. Le Guével and R. Antoine, Chemistry, 2022, 28, e202200570 CrossRef CAS PubMed
- W. Zhang, S. Chen, P. Sun, S. Ye, Q. Fan, J. Song, P. Zeng, J. Qu and W. Y. Wong, Adv. Healthcare Mater., 2022, 11, e2200467 CrossRef PubMed
- S. Mateos, J. Lifante, C. Li, E. C. Ximendes, T. Muñoz-Ortiz, J. Yao, M. de la Fuente-Fernández, L. García Villalón, Á. M. Granado, I. Zabala Gutierrez, J. Rubio-Retama, D. Jaque, D. H. Ortgies and N. Fernández, Small, 2020, 16, e1907171 CrossRef PubMed
- O. Yarema, M. Yarema and V. Wood, Chem. Mater., 2018, 30, 1446–1461 CrossRef CAS
- J. Li, T. Guan, D. Tu, W. Lian, P. Zhang, S. Han, F. Wen and X. Chen, Chem. Commun., 2022, 58, 2204–2207 RSC
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. Bos, P. Dorenbos, M. Bessodes and D. Gourier, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed
- J. L. Li, J. P. Shi, C. C. Wang, P. H. Li, Z. F. Yu and H. W. Zhang, Nanoscale, 2017, 9, 8631–8638 RSC
- L. Song, P. P. Li, W. Yang, X. H. Lin, H. Liang, X. F. Chen, G. Liu, J. Li and H. H. Yang, Adv. Funct. Mater., 2018, 28, 1707496 CrossRef
- X. Zhao, K. C. Zhao, L. J. Chen, Y. S. Liu, J. L. Liu and X. P. Yan, Chem. Sci., 2020, 12, 442–452 RSC
- X. Chen, Y. Li, K. Huang, L. Huang, X. Tian, H. Dong, R. Kang, Y. Hu, J. Nie, J. Qiu and G. Han, Adv. Mater., 2021, 33, e2008722 CrossRef PubMed
- X. Qiu, X. Zhu, M. Xu, W. Yuan, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 32583–32590 CrossRef CAS PubMed
- L. Hu, P. Wang, M. Zhao, L. Liu, L. Zhou, B. Li, F. H. Albaqami, A. M. El-Toni, X. Li, Y. Xie, X. Sun and F. Zhang, Biomaterials, 2018, 163, 154–162 CrossRef CAS PubMed
- Z. Xue, X. Li, Y. Li, M. Jiang, G. Ren, H. Liu, S. Zeng and J. Hao, Nanoscale, 2017, 9, 7276–7283 RSC
- R. D. Scurlock, B. Wang, P. R. Ogilby, J. R. Sheats and R. L. Clough, J. Am. Chem. Soc., 1995, 117, 10194–10202 CrossRef CAS
- C. Xie, Y. Lyu, X. Zhen, Q. Miao and K. Pu, ACS Appl. Bio Mater., 2018, 1, 1147–1153 CrossRef CAS PubMed
- C. Xie, X. Zhen, Q. Miao, Y. Lyu and K. Pu, Adv. Mater., 2018, 30, e1801331 CrossRef PubMed
- L. Wu, Y. Ishigaki, Y. Hu, K. Sugimoto, W. Zeng, T. Harimoto, Y. Sun, J. He, T. Suzuki, X. Jiang, H. Y. Chen and D. Ye, Nat. Commun., 2020, 11, 446 CrossRef CAS PubMed
- G.-S. Yi and G.-M. Chow, Chem. Mater., 2007, 19, 341–343 CrossRef CAS
- L. Wu, M. Jia, D. Li and G. Chen, Nano Lett., 2023, 23, 2862–2869 CrossRef CAS PubMed
- T. Wang, M. Yang, J. Huang, Y. Zhao, H. Wang, S. Leng, J. Chen, G. Sun and J. Liu, Sci. Bull., 2017, 62, 903–912 CrossRef CAS PubMed
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS PubMed
- A. Xia, Y. Gao, J. Zhou, C. Li, T. Yang, D. Wu, L. Wu and F. Li, Biomaterials, 2011, 32, 7200–7208 CrossRef CAS PubMed
- N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2011, 44, 925–935 CrossRef CAS PubMed
- Q. Lü, A. Li, F. Guo, L. Sun and L. Zhao, Nanotechnology, 2008, 19, 205704 CrossRef PubMed
- S. Dong, J. Xu, T. Jia, M. Xu, C. Zhong, G. Yang, J. Li, D. Yang, F. He, S. Gai, P. Yang and J. Lin, Chem. Sci., 2019, 10, 4259–4271 RSC
- X. Chen, Y. Tang, A. Liu, Y. Zhu, D. Gao, Y. Yang, J. Sun, H. Fan and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 14378–14388 CrossRef CAS PubMed
- A. Gulzar, J. Xu, D. Yang, L. Xu, F. He, S. Gai and P. Yang, Dalton Trans., 2018, 47, 3931–3939 RSC
- W. Wang, M. Zhao, L. Wang and H. Chen, Mikrochim. Acta, 2019, 186, 630 CrossRef PubMed
- M. Tang, X. Zhu, Y. Zhang, Z. Zhang, Z. Zhang, Q. Mei, J. Zhang, M. Wu, J. Liu and Y. Zhang, ACS Nano, 2019, 13, 10405–10418 CrossRef CAS PubMed
- X. Chuai, Z. Liu, Y. Liu, C. He and W. Qin, J. Nanosci. Nanotechnol., 2014, 14, 3687–3689 CrossRef CAS PubMed
- J. Zhao, Y. Hu, S. W. Lin, U. Resch-Genger, R. Zhang, J. Wen, X. Kong, A. Qin and J. Ou, J. Mater. Chem. B, 2020, 8, 6481–6489 RSC
- J. Yin, H. Zheng, W. Zhang, L. Shen, R. Lai, L. Tian, F. Zhao and Y. Shao, Opt. Express, 2022, 30, 32459–32473 CrossRef CAS PubMed
- Y. Li, F. Li, Y. Huang, H. Wu, J. Wang, J. Yang, Q. Xiao and H. Lin, RSC Adv., 2019, 9, 18070–18075 RSC
- Y. Zhong, Z. Ma, F. Wang, X. Wang, Y. Yang, Y. Liu, X. Zhao, J. Li, H. Du, M. Zhang, Q. Cui, S. Zhu, Q. Sun, H. Wan, Y. Tian, Q. Liu, W. Wang, K. C. Garcia and H. Dai, Nat. Biotechnol., 2019, 37, 1322–1331 CrossRef CAS PubMed
- D. Kang, H. J. Ahn, J. Lee, S. K. Kim, J. Pyun, C. S. Song, S. J. Kim and J. Lee, Biosens. Bioelectron., 2021, 190, 113369 CrossRef CAS PubMed
- D. Karthickraja, G. A. Kumar, D. K. Sardar, S. Karthi, G. C. Dannangoda, K. S. Martirosyan, M. Prasath, M. Gowri and E. K. Girija, Mater. Sci. Eng., C, 2021, 125, 112095 CrossRef CAS PubMed
- T. Song, M. Zhang, Y. Liu, J. Yang, Z. Gong, H. Yan, H. Zhu, D. Yan, C. Liu and C. Xu, RSC Adv., 2018, 8, 10954–10963 RSC
- Z. F. Yu, J. P. Shi, J. L. Li, P. H. Li and H. W. Zhang, J. Mater. Chem. B, 2018, 6, 1238–1243 RSC
- E. Pan, G. Bai, J. Zhou, L. Lei and S. Xu, Nanoscale, 2019, 11, 11642–11648 RSC
- C. Cao, N. Wu, W. Yuan, Y. Gu, J. Ke, W. Feng and F. Li, Nanoscale, 2020, 12, 8248–8254 RSC
- S. J. Kwon, G. Y. Lee, K. Jung, H. S. Jang, J. S. Park, H. Ju, I. K. Han and H. Ko, Adv. Mater., 2016, 28, 7899–7909 CrossRef CAS PubMed
- L. M. Wiesholler, C. Genslein, A. Schroter and T. Hirsch, Anal. Chem., 2018, 90, 14247–14254 CrossRef CAS PubMed
- D. Lu, S. K. Cho, S. Ahn, L. Brun, C. J. Summers and W. Park, ACS Nano, 2014, 8, 7780–7792 CrossRef CAS PubMed
- Y. Ji, W. Xu, N. Ding, H. Yang, H. Song, Q. Liu, H. Ågren, J. Widengren and H. Liu, Light: Sci. Appl., 2020, 9, 184 CrossRef CAS PubMed
- Y. Zhang, J. Wang, F. Nan and Q. Q. Wang, RSC Adv., 2018, 8, 20056–20060 RSC
- Z. Zhang, Y. Liu, Y. Fang, B. Cao, J. Huang, K. Liu and B. Dong, Adv. Sci., 2018, 5, 1800748 CrossRef PubMed
- W. Zhang, T. Chen, L. Su, X. Ge, X. Chen, J. Song and H. Yang, Anal. Chem., 2020, 92, 6094–6102 CrossRef CAS PubMed
- J. Huang, X. Zhang, S. Li, F. Qu, B. Huang, R. Cui, Y. Liu, W. Hu, X. Yang and Y. Zhang, Anal. Chem., 2023, 95, 3761–3768 CrossRef CAS PubMed
- A. R. Hong, J. S. Han, G. Kang, H. Ko and H. S. Jang, Materials, 2020, 13, 5338 CrossRef CAS PubMed
- T. Yu, D. M. Wei, Z. Li, L. J. Pan, Z. L. Zhang, Z. Q. Tian and Z. Liu, Chem. Commun., 2020, 56, 1976–1979 RSC
- Q. Wang, T. Liang, J. Wu, Z. Li and Z. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 29303–29312 CrossRef CAS PubMed
- D. K. Chatterjee and Z. Yong, 2008.
- J. Shan, S. J. Budijono, G. Hu, N. Yao, Y. Kang, Y. Ju and R. K. Prud'homme, Adv. Funct. Mater., 2011, 21, 2488–2495 CrossRef CAS
- K. Liu, X. Liu, Q. Zeng, Y. Zhang, L. Tu, T. Liu, X. Kong, Y. Wang, F. Cao, S. A. Lambrechts, M. C. Aalders and H. Zhang, ACS Nano, 2012, 6, 4054–4062 CrossRef CAS PubMed
- K. Tezuka, M. Umezawa, T. I. Liu, K. Nomura, K. Okubo, H. C. Chiu, M. Kamimura and K. Soga, ACS Appl. Bio Mater., 2021, 4, 4462–4469 CrossRef CAS PubMed
- S. Jin, L. Zhou, Z. Gu, G. Tian, L. Yan, W. Ren, W. Yin, X. Liu, X. Zhang, Z. Hu and Y. Zhao, Nanoscale, 2013, 5, 11910–11918 RSC
- H. Zhao, Y. Li, X. Zhang, K. Wu, J. Lv, C. Chen, H. Liu, Z. Shi, H. Ju and Y. Liu, Biomaterials, 2022, 291, 121873 CrossRef CAS PubMed
- L. Cai, Z. Wang, B. Lin, K. Liu, Y. Wang, Y. Yuan, X. Tao and R. Lv, Nanoscale Adv., 2022, 4, 2224–2232 RSC
- F. Ai, Q. Ju, X. Zhang, X. Chen, F. Wang and G. Zhu, Sci. Rep., 2015, 5, 10785 CrossRef CAS PubMed
- J. Choi and S. Y. Kim, J. Biomater. Appl., 2022, 37, 646–658 CrossRef CAS PubMed
- D. Wang, L. Zhu, Y. Pu, J. X. Wang, J. F. Chen and L. Dai, Nanoscale, 2017, 9, 11214–11221 RSC
- R. Lv, P. Yang, G. Chen, S. Gai, J. Xu and P. N. Prasad, Sci. Rep., 2017, 7, 13562 CrossRef PubMed
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed
- G. Kuang, H. Lu, S. He, H. Xiong, J. Yu, Q. Zhang and Y. Huang, Adv. Healthcare Mater., 2021, 10, e2100938 CrossRef PubMed
- Y. Yu, Y. Huang, W. Feng, M. Yang, B. Shao, J. Li and F. Ye, RSC Adv., 2021, 11, 29065–29072 RSC
- N. Niu, F. He, P. Ma, S. Gai, G. Yang, F. Qu, Y. Wang, J. Xu and P. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 3250–3262 CrossRef CAS PubMed
- F. Zheng, C. Wang, T. Meng, Y. Zhang, P. Zhang, Q. Shen, Y. Zhang, J. Zhang, J. Li, Q. Min, J. Chen and J. J. Zhu, ACS Nano, 2019, 13, 12577–12590 CrossRef CAS PubMed
- R. Cui, W. Sun, M. Liu, J. Shi and Z. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 59164–59173 CrossRef CAS PubMed
- H. Cai, T. Shen, A. M. Kirillov, Y. Zhang, C. Shan, X. Li, W. Liu and Y. Tang, Inorg. Chem., 2017, 56, 5295–5304 CrossRef CAS PubMed
- Y. Cao, K. Wang, P. Zhu, X. Zou, G. Ma, W. Zhang, D. Wang, J. Wan, Y. Ma, X. Sun and J. Dong, Colloids Surf., B, 2022, 213, 112393 CrossRef CAS PubMed
- Z. Wei, X. Liu, D. Niu, L. Qin and Y. Li, ACS Appl. Bio Mater., 2020, 3, 4655–4664 CrossRef CAS PubMed
- G. Chen, R. Jaskula-Sztul, C. R. Esquibel, I. Lou, Q. Zheng, A. Dammalapati, A. Harrison, K. W. Eliceiri, W. Tang, H. Chen and S. Gong, Adv. Funct. Mater., 2017, 27 Search PubMed
- Y. Liu, Y. Liu, W. Bu, C. Cheng, C. Zuo, Q. Xiao, Y. Sun, D. Ni, C. Zhang and J. Liu, Angew. Chem., 2015, 127, 8223–8227 CrossRef
- J. Han, H. Xia, Y. Wu, S. N. Kong, A. Deivasigamani, R. Xu, K. M. Hui and Y. Kang, Nanoscale, 2016, 8, 7861–7865 RSC
- J. Xu, A. Gulzar, Y. Liu, H. Bi, S. Gai, B. Liu, D. Yang, F. He and P. Yang, Small, 2017, 13, 1701841 CrossRef PubMed
- J. Liu, J. Zhang, F. Huang, Y. Deng, B. Li, R. Ouyang, Y. Miao, Y. Sun and Y. Li, Acta Biomater., 2020, 113, 570–583 CrossRef CAS PubMed
- K. Du, S. Zhao, J. Feng, X. Gao, K. Liu, X. Wang, M. Zhang, Y. Li, Y. Lu and H. Zhang, J. Mater. Chem. B, 2021, 9, 7216–7228 RSC
- X. Chen, J. Song, X. Chen and H. Yang, Chem. Soc. Rev., 2019, 48, 3073–3101 RSC
- G. Song, L. Cheng, Y. Chao, K. Yang and Z. Liu, Adv. Mater., 2017, 29, 1700996 CrossRef PubMed
- I. N. Stanton, J. A. Ayres and M. J. Therien, Dalton Trans., 2012, 41, 11576–11578 RSC
- D. Maiti, H. Yu, B. S. Kim, M. Naito, S. Yamashita, H. J. Kim and K. Miyata, ACS Appl. Bio Mater., 2022, 5, 5477–5486 CrossRef CAS PubMed
- B. P. Quigley, C. D. Smith, S. H. Cheng, J. S. Souris, C. A. Pelizzari, C. T. Chen, L. W. Lo, C. S. Reft, R. D. Wiersma and P. J. La Riviere, Med. Phys., 2017, 44, 5367–5377 CrossRef CAS PubMed
- M. C. Micheletto, J. Guidelli É and A. J. Costa-Filho, ACS Appl. Mater. Interfaces, 2021, 13, 2289–2302 CrossRef CAS PubMed
- G. R. Waetzig, G. A. Horrocks, J. W. Jude, L. Zuin and S. Banerjee, Nanoscale, 2016, 8, 979–986 RSC
- F. Ahmad, X. Wang, Z. Jiang, X. Yu, X. Liu, R. Mao, X. Chen and W. Li, ACS Nano, 2019, 13, 10419–10433 CrossRef CAS PubMed
- L. Sudheendra, G. K. Das, C. Li, D. Stark, J. Cena, S. Cherry and I. M. Kennedy, Chem. Mater., 2014, 26, 1881–1888 CrossRef CAS PubMed
- D. Kirsanova, V. Polyakov, V. Butova, P. Zolotukhin, A. Belanova, Z. Gadzhimagomedova, M. Soldatov, I. Pankin and A. Soldatov, Nanomaterials, 2021, 11, 3212 CrossRef CAS PubMed
- X. Zhong, X. Wang, G. Zhan, Y. Tang, Y. Yao, Z. Dong, L. Hou, H. Zhao, S. Zeng, J. Hu, L. Cheng and X. Yang, Nano Lett., 2019, 19, 8234–8244 CrossRef CAS PubMed
- M. Müller, Y. Wang, M. R. Squillante, K. D. Held, R. R. Anderson and M. Purschke, Radiother. Oncol., 2018, 129, 589–594 CrossRef PubMed
- D. Manoharan, L. C. Chang, L. C. Wang, Y. S. Shan, F. C. Lin, L. C. Wu, H. S. Sheu, W. P. Su and C. S. Yeh, ACS Nano, 2021, 15, 9084–9100 CrossRef CAS PubMed
- Q. Zhang, B. Yan, F. Lei and H. H. Chen, Nanoscale, 2012, 4, 7646–7648 RSC
- D. Avram, B. Cojocaru, A. Urda, I. Tiseanu, M. Florea and C. Tiseanu, Phys. Chem. Chem. Phys., 2015, 17, 30988–30992 RSC
- M. V. Rezende, P. J. Montes, A. B. Andrade, Z. S. Macedo and M. E. Valerio, Phys. Chem. Chem. Phys., 2016, 18, 17646–17654 RSC
- Z. Jiang, L. He, X. Yu, Z. Yang, W. Wu, X. Wang, R. Mao, D. Cui, X. Chen and W. Li, ACS Nano, 2021, 15, 11112–11125 CrossRef CAS PubMed
- L. Q. Guan, S. Shi, X. W. Niu, S. C. Guo, J. Zhao, T. M. Ji, H. Dong, F. Y. Jia, J. W. Xiao, L. D. Sun and C. H. Yan, Adv. Sci., 2022, 9, e2201354 CrossRef PubMed
- H. Chen, G. D. Wang, Y. J. Chuang, Z. Zhen, X. Chen, P. Biddinger, Z. Hao, F. Liu, B. Shen, Z. Pan and J. Xie, Nano Lett., 2015, 15, 2249–2256 CrossRef CAS PubMed
- B. Cline and J. Xie, Methods Mol. Biol., 2022, 2394, 811–822 CrossRef CAS PubMed
- L. Zhang, X. Wang, X. Wang, X. Wang, Y. Luo, C. Tan, L. Jiang, Y. Wang and W. Liu, Inorg. Chem., 2023, 62, 6421–6427 CrossRef CAS PubMed
- C. Yang, L. Ma, J. Maley, R. Sammynaiken, R. Feng, G. Xiang and W. Chen, J. Biomed. Nanotechnol., 2013, 9, 1827–1836 CrossRef CAS PubMed
- Y. Osakada, G. Pratx, C. Sun, M. Sakamoto, M. Ahmad, O. Volotskova, Q. Ong, T. Teranishi, Y. Harada, L. Xing and B. Cui, Chem. Commun., 2014, 50, 3549–3551 RSC
- L. Luo, W. Sun, Y. Feng, R. Qin, J. Zhang, D. Ding, T. Shi, X. Liu, X. Chen and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 12591–12599 CrossRef CAS PubMed
- S. Shrestha, J. Wu, B. Sah, A. Vanasse, L. N. Cooper, L. Ma, G. Li, H. Zheng, W. Chen and M. P. Antosh, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16823–16828 CrossRef CAS PubMed
- K. Kirakci, T. N. Pozmogova, A. Y. Protasevich, G. D. Vavilov, D. V. Stass, M. A. Shestopalov and K. Lang, Biomater. Sci., 2021, 9, 2893–2902 RSC
- K. Vanheusden, C. Seager, W. T. Warren, D. Tallant and J. Voigt, Appl. Phys. Lett., 1996, 68, 403–405 CrossRef CAS
- Z. Wang, C. Li, L. Liu and T. K. Sham, J. Chem. Phys., 2013, 138, 084706 CrossRef PubMed
- A. Vejdani Noghreiyan, M. R. Sazegar, S. A. Mousavi Shaegh and A. Sazgarnia, Photodiagn. Photodyn. Ther., 2020, 30, 101770 CrossRef CAS PubMed
- L. Armelao, F. Heigl, S. Brunet, R. Sammynaiken, T. Regier, R. I. Blyth, L. Zuin, R. Sankari, J. Vogt and T. K. Sham, ChemPhysChem, 2010, 11, 3625–3631 CrossRef CAS PubMed
- H. Deng, L. Lin, S. Wang, G. Yu, Z. Zhou, Y. Liu, G. Niu, J. Song and X. Chen, Adv. Mater., 2019, 31, e1903443 CrossRef
- F. Zhang, Y. Zhou, Z. Chen, M. Wang, Z. Ma, X. Chen, M. Jia, D. Wu, J. Xiao, X. Li, Y. Zhang, Z. Shi and C. Shan, Adv. Mater., 2022, 34, e2204801 CrossRef
- X. Ou, X. Qin, B. Huang, J. Zan, Q. Wu, Z. Hong, L. Xie, H. Bian, Z. Yi, X. Chen, Y. Wu, X. Song, J. Li, Q. Chen, H. Yang and X. Liu, Nature, 2021, 590, 410–415 CrossRef CAS PubMed
- Y. Wu, L. Su, M. Yuan, T. Chen, J. Ye, Y. Jiang, J. Song and H. Yang, Angew. Chem., Int. Ed., 2021, 60, 12868–12875 CrossRef CAS PubMed
- X. Jiang, X. Gao, L. Li, P. Zhou, S. Wang, T. Liu, J. Zhou, H. Zhang, K. Huang, Y. Li, M. Wang, Z. Jin, E. Xie, W. Liu and G. Han, ACS Appl. Mater. Interfaces, 2023, 15, 21228–21238 CrossRef CAS PubMed
- Z. Xue, X. Li, Y. Li, M. Jiang, H. Liu, S. Zeng and J. Hao, ACS Appl. Mater. Interfaces, 2017, 9, 22132–22142 CrossRef CAS PubMed
- Z. Xue, M. Jiang, H. Liu, S. Zeng and J. Hao, Biomaterials, 2020, 263, 120384 CrossRef CAS PubMed
- H. Jiang, R. Wang, Q. Zhang, L. Song, X. Sun, J. Shi and Y. Zhang, Nanoscale, 2022, 14, 15451–15461 RSC
- R. Jiang, J. Yang, Y. Meng, D. Yan, C. Liu, C. Xu and Y. Liu, Dalton Trans., 2020, 49, 6074–6083 RSC
- S. Zheng, J. Shi, X. Fu, C. Wang, X. Sun, C. Chen, Y. Zhuang, X. Zou, Y. Li and H. Zhang, Nanoscale, 2020, 12, 14037–14046 RSC
- Y. Zhuang, D. Chen, W. Chen, W. Zhang, X. Su, R. Deng, Z. An, H. Chen and R. J. Xie, Light: Sci. Appl., 2021, 10, 132 CrossRef CAS PubMed
- J. Ma, W. Zhu, L. Lei, D. Deng, Y. Hua, Y. M. Yang, S. Xu and P. N. Prasad, ACS Appl. Mater. Interfaces, 2021, 13, 44596–44603 CrossRef CAS PubMed
- L. Lei, Y. Wang, W. Xu, R. Ye, Y. Hua, D. Deng, L. Chen, P. N. Prasad and S. Xu, Nat. Commun., 2022, 13, 5739 CrossRef CAS PubMed
- P. Pei, Y. Chen, C. Sun, Y. Fan, Y. Yang, X. Liu, L. Lu, M. Zhao, H. Zhang, D. Zhao, X. Liu and F. Zhang, Nat. Nanotechnol., 2021, 16, 1011–1018 CrossRef CAS PubMed
- P. S. Kim, P. Zhang and T. K. Sham, Langmuir, 2004, 20, 4690–4695 CrossRef CAS PubMed
- L. Siller, S. Krishnamurthy, L. Kjeldgaard, B. R. Horrocks, Y. Chao, A. Houlton, A. K. Chakraborty and M. R. Hunt, J. Phys.: Condens. Matter, 2009, 21, 095005 CrossRef CAS PubMed
- W. Sun, T. Shi, L. Luo, X. Chen, P. Lv, Y. Lv, Y. Zhuang, J. Zhu, G. Liu, X. Chen and H. Chen, Adv. Mater., 2019, 31, e1808024 CrossRef PubMed
- L. Liu, T. K. Sham, W. Han, C. Zhi and Y. Bando, ACS Nano, 2011, 5, 631–639 CrossRef CAS PubMed
- S. G. Ryan, M. N. Butler, S. S. Adeyemi, T. Kalber, P. S. Patrick, M. Zaw Thin, I. F. Harrison, D. J. Stuckey, M. Pule and M. F. Lythgoe, Sci. Rep., 2019, 9, 19223 CrossRef CAS
-
R. Leo William, Techniques for Nuclear and Particle Physics Experiments: a How-to Approach, Springer, 1994 Search PubMed
- Y. Osakada, G. Pratx, L. Hanson, P. E. Solomon, L. Xing and B. Cui, Chem. Commun., 2013, 49, 4319–4321 RSC
- D. Zhang, H. Zhang, X. Zhang, T. K. Sham, Y. Hu and X. Sun, Phys. Chem. Chem. Phys., 2016, 18, 6406–6410 RSC
- H. Chen, T. Moore, B. Qi, D. C. Colvin, E. K. Jelen, D. A. Hitchcock, J. He, O. T. Mefford, J. C. Gore, F. Alexis and J. N. Anker, ACS Nano, 2013, 7, 1178–1187 CrossRef CAS PubMed
- K. Kirakci, P. Kubát, K. Fejfarová, J. Martinčík, M. Nikl and K. Lang, Inorg. Chem., 2016, 55, 803–809 CrossRef CAS PubMed
- D. Beke, M. V. Nardi, G. Bortel, M. Timpel, Z. Czigány, L. Pasquali, A. Chiappini, G. Bais, M. Rudolf, D. Zalka, F. Bigi, F. Rossi, L. Bencs, A. Pekker, B. G. Márkus, G. Salviati, S. E. Saddow, K. Kamarás, F. Simon and A. Gali, Chem. Mater., 2021, 33, 2457–2465 CrossRef CAS PubMed
- M. Xin and W. H. Cao, Guangpuxue Yu Guangpu Fenxi, 2009, 29, 2272–2275 CAS
- H. Chen, F. Wang, T. Moore, B. Qi, D. Sulejmanovic, S. J. Hwu, O. T. Mefford, F. Alexis and J. N. Anker, J. Mater. Chem. B, 2017, 5, 5412–5424 RSC
- W. Sun, L. Luo, Y. Feng, Y. Cai, Y. Zhuang, R. J. Xie, X. Chen and H. Chen, Angew. Chem., Int. Ed., 2020, 59, 9914–9921 CrossRef CAS PubMed
- H. Li, Z. Chen, Z. Sang, X. Zhang and Y. Wang, RSC Adv., 2020, 10, 43773–43782 RSC
- S. Clement, W. Deng, E. Camilleri, B. C. Wilson and E. M. Goldys, Sci. Rep., 2016, 6, 19954 CrossRef CAS PubMed
- H. Chen, X. Sun, G. D. Wang, K. Nagata, Z. Hao, A. Wang, Z. Li, J. Xie and B. Shen, Mater. Horiz., 2017, 4, 1092–1101 RSC
- H. Wang, B. Lv, Z. Tang, M. Zhang, W. Ge, Y. Liu, X. He, K. Zhao, X. Zheng, M. He and W. Bu, Nano Lett., 2018, 18, 5768–5774 CrossRef CAS PubMed
- M. Isikawa and E. Guidelli, ACS Appl. Mater. Interfaces, 2022, 14, 324–336 CrossRef CAS PubMed
- J. Liu, H. Wang, X. Yi, Y. Chao, Y. Geng, L. Xu, K. Yang and Z. Liu, Adv. Funct. Mater., 2017, 27, 1703832 CrossRef
- R. Sang, F. Deng, A. Engel, E. Goldys and W. Deng, Biomed. Pharmacother., 2022, 155, 113837 CrossRef CAS PubMed
- Y. C. Chuang, C. H. Chu, S. H. Cheng, L. D. Liao, T. S. Chu, N. T. Chen, A. Paldino, Y. Hsia, C. T. Chen and L. W. Lo, Theranostics, 2020, 10, 6758–6773 CrossRef CAS PubMed
- Z. Li, C. Zhao, Q. Fu, J. Ye, L. Su, X. Ge, L. Chen, J. Song and H. Yang, Small, 2022, 18, e2105160 CrossRef PubMed
- W. Fan, B. C. Yung and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 8383–8394 CrossRef CAS PubMed
- A. W. Carpenter and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC
- F. Zhang, S. Liu, N. Zhang, Y. Kuang, W. Li, S. Gai, F. He, A. Gulzar and P. Yang, Nanoscale, 2020, 12, 19293–19307 RSC
- Z. Du, X. Wang, X. Zhang, Z. Gu, X. Fu, S. Gan, T. Fu, S. Xie and W. Tan, Angew. Chem., Int. Ed., 2023, 62, e202302525 CrossRef CAS PubMed
- K. Sarkar, S. E. Torregrossa-Allen, B. D. Elzey, S. Narayanan, M. P. Langer, G. A. Durm and Y. Y. Won, Mol. Pharm., 2022, 19, 2776–2794 CrossRef CAS PubMed
- M. Ashokkumar, J. Lee, S. Kentish and F. Grieser, Ultrason. Sonochem., 2007, 14, 470–475 CrossRef CAS
- D. Song, W. Xu, M. Luo, M. Zhang, H. Wen, X. Cheng, X. Luo and Z. Wang, Nanoscale, 2021, 13, 14130–14138 RSC
- A. Sazgarnia, A. Shanei, H. Eshghi, M. Hassanzadeh-Khayyat, H. Esmaily and M. M. Shanei, Ultrasonics, 2013, 53, 29–35 CrossRef CAS PubMed
- P. Gholami, L. Dinpazhoh, A. Khataee and Y. Orooji, Ultrason. Sonochem., 2019, 55, 44–56 CrossRef CAS PubMed
- A. Radivoievych, B. Kolp, S. Grebinyk, S. Prylutska, U. Ritter, O. Zolk, J. Glökler, M. Frohme and A. Grebinyk, Int. J. Mol. Sci., 2023, 24, 1020 CrossRef CAS PubMed
- R. Hou, X. Liang, X. Li, X. Zhang, X. Ma and F. Wang, Biomater. Sci., 2020, 8, 2526–2536 RSC
- K. Kawamura, A. Ikeda, A. Inui, K. Yamamoto and H. Kawasaki, J. Chem. Phys., 2021, 155, 124702 CrossRef CAS PubMed
- C. Xu, J. Huang, Y. Jiang, S. He, C. Zhang and K. Pu, Nat. Biomed. Eng., 2023, 7, 298–312 CrossRef CAS PubMed
- D. Zhou, Z. Zhang, B. Qiu, D. Zhang, S. Xie, K. Huang and X. Li, ACS Appl. Mater. Interfaces, 2022, 14, 26418–26430 CrossRef CAS
- B. P. Barber, C. Wu, R. Löfstedt, P. H. Roberts and S. J. Putterman, Phys. Rev. Lett., 1994, 72, 1380 CrossRef CAS PubMed
- F. Yang, H. Cui, X. Wu, S.-J. Kim and G. Hong, Nanoscale, 2023, 15, 1629–1636 RSC
- Z. Zhang, H. Yan, W. Cao, S. Xie, P. Ran, K. Wei and X. Li, ACS Nano, 2023, 17, 16089–16106 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |