
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancements in catalysis for plastic resource utilization
Yao
Chen
,
Lele
Bai
,
Dening
Peng
,
Xinru
Wang
,
Meijun
Wu
and
Zhenfeng
Bian
 *
*
MOE Key Laboratory of Resource Chemistry and Shanghai Key Laboratory of Rare Earth Functional Materials, Shanghai Normal University, Shanghai 200234, China. E-mail: bianzhenfeng@shnu.edu.cn
First published on 25th July 2023
Abstract
The widespread production and utilization of plastic products have become ingrained in our society, resulting in a staggering amount of plastic waste, severe environmental challenges, and resource depletion. To address this issue sustainably, catalytic technology has emerged as a promising approach to break down non-biodegradable polymers into tiny organic molecules for secondary applications. Among these methods, photochemical processes utilizing light as an energy source offer controlled degradation of plastics under mild and environmentally friendly reaction conditions. Considering the pressing issue of plastic pollution, this paper presents a comprehensive overview of novel techniques for recycling waste plastics, delving into the underlying scientific principles of catalytic degradation. Moreover, we propose directions for feasibility studies on managing plastic pollution and advancing the development of plastic products.
Environmental significanceThe escalating growth of the plastics industry and widespread plastic consumption have resulted in a grave global environmental challenge: plastic pollution. Conventional methods like landfilling or incineration have proven ineffective in addressing this issue, leading to additional environmental hazards such as landfill degradation and the release of harmful gases. In contrast, plastic resource transformation offers a distinct solution. By breaking down long chains of giant plastic molecules into tiny organic compounds, it presents a two-fold solution: resolving energy concerns caused by dwindling oil supplies while addressing plastic pollution. This dual-purpose approach has captured the attention of numerous researchers, although its implementation poses significant challenges. In recent years, catalytic technology has emerged as a preferred choice for the breakdown and recycling of diverse waste polymers, owing to its remarkable efficiency and selectivity. Notably, the integration of environmentally friendly and sustainable photocatalysis has breathed new life into plastics, offering one of the most promising avenues for achieving high-value utilization of waste plastics. Thus, this study aims to provide an overview of catalytic technologies employed in plastic treatment, along with a comprehensive examination and discussion of catalytic depolymerization mechanisms, as well as the potential for recycling waste plastics. By focusing on these aspects, we strive to contribute to the advancement of sustainable solutions for plastic pollution and foster a better understanding of the immense possibilities catalytic technologies offer in transforming plastic waste into valuable resources. |
1. Introduction
Since the 1950s, humans have produced some 9.2 billion tonnes of plastic, 60% of which has been landfilled, incinerated or dumped directly into rivers, lakes and oceans.1 A report published by the Organisation for Economic Co-operation and Development (OECD) in 2022 states that in the last 20 years, annual global plastic production has soared from 234 million tonnes in 2000 to 460 million tonnes in 2019. Plastic waste has also increased from 156 million tonnes in 2000 to 353 million tonnes in 2019. Global plastic production is expected to reach 1.2 billion tonnes per year by 2060, by which time the amount of plastic waste is also expected to nearly triple.2 While plastics have brought convenience to our lives, they have also inflicted severe harm on the natural environment and human health through plastic pollution.3–5 In the face of global environmental pollution and resource depletion caused by waste plastics, the idea of sustainable development and circular economy has been widely accepted by all countries.6 However, the development of the global recycled plastics industry is relatively slow and varies greatly from region to region due to the bottlenecks in recycling technology and the lack of economic viability of plastics. As a result, the life cycle of plastics has not yet been formed.7Plastics, mainly composed of high molecular weight synthetic resins, undergo processing with cross-linking agents like plasticizers and antioxidants to acquire their chemical resistance and rigidity.8 Consequently, the degradation of plastics in nature alone through light, weathering and microbial bacteria takes a long period of time, while non-degradable plastics such as polyethylene (PE) remain largely unaffected.9 Generally, thermoplastic, homogeneous, and relatively clean waste plastics can be physically recycled, while structurally complex and difficult-to-degrade thermoset waste plastics necessitate chemical interventions.10–13 The utilization of chemical reagents to break down the long chains of plastic macromolecules can lead to a range of high-value chemical intermediates, thereby offering a sustainable approach to treating plastic waste in the long run (Fig. 1).14–16 It is crucial, however, to address the issue of secondary pollution during the chemical degradation and recycling processes.17,18 As a result, traditional methods like open burning, which lead to significant environmental harm, are being phased out.19,20 Emerging, green, efficient, and low-cost methods of plastic recycling and treatment are becoming a vibrant area of research at present.
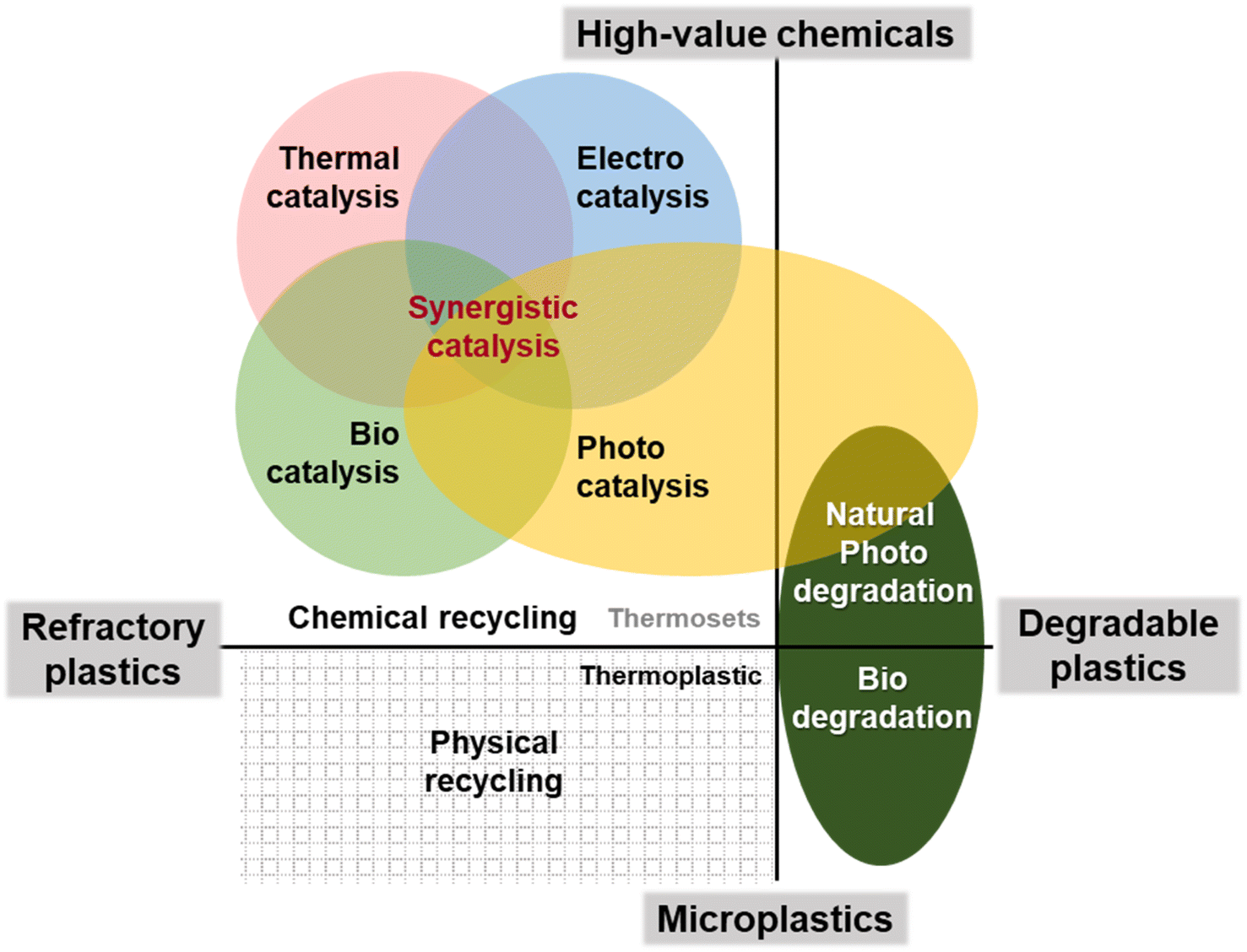 | ||
| Fig. 1 Schematic diagram of the idea to apply catalytic technology in the resource recovery of waste plastics. | ||
Given the pressing need to combat plastic pollution, it is imperative that we pursue sustainable solutions that mitigate environmental impact, foster efficiency, and offer affordability. The exceptional stability of plastics poses a challenge when it comes to their degradation and recycling, as it requires surmounting the energy barrier associated with breaking long polymer chains.21 Chemical oxidation serves as an effective means of degrading plastics through incineration or gasification, yielding heat or energy feedstocks such as syngas.22 Alternatively, chemical cracking transforms waste plastics into fuel oil and chemical feedstocks through thermal or catalytic processes.23 Among these methods, catalytic technology stands out by promoting or accelerating reactions through a reduction in activation energy. This advantageous feature enables rapid reaction rates and high selectivity in product formation.24
Catalytic treatment of waste plastics minimizes the energy requirements for degradation while selectively harnessing the carbon, hydrogen, and oxygen present in the plastics.25,26 This approach not only converts plastics into alternative fuel or small molecule monomers but also yields high-value products and chemical raw materials, effectively turning waste plastics into valuable resources. Various catalytic strategies, utilizing different energy sources such as thermal cracking catalysis, biocatalysis, electrochemical catalysis, and photocatalysis, are demonstrating feasibility and showcasing their respective advantages in waste plastics treatment. For instance, environmentally friendly, energy-driven photocatalytic strategies facilitate the release of hydrogen from plastic decomposition or enable selective depolymerization of plastics into liquid hydrocarbon fuels of appropriate molecular weight through hydrogenolysis. However, catalytic technologies also face challenges in terms of efficiency, necessitating further improvement.27,28 This paper provides a comprehensive overview of catalytic technologies employed in the treatment of plastics, shedding light on the degradation process of plastic macromolecules from both a structural and catalytic mechanism perspective. Additionally, a brief analysis of the issues encountered by catalytic technologies is presented to elucidate more ecologically and naturally beneficial catalytic strategies. Through a deeper understanding of catalytic processes and their potential drawbacks, we aim to contribute to the advancement of efficient and sustainable solutions for plastic treatment, paving the way for a greener future.
2. Molecular structures of hard-to-degrade plastics and their resource recovery technologies
The ease of plastic degradation is heavily influenced by its molecular structure.29 It is well known that plastics are polymers with a large relative molecular mass, exhibit a crucial parameter known as molecular weight. A higher molecular weight signifies a longer molecular chain composed of monomers, resulting in enhanced resistance against corrosion. Variations in functional groups within the plastics molecular structure distinguish different types of polymers, consequently affecting the mechanical, thermal, and chemical resistance of the plastic. Hence, selecting an appropriate degradation method is imperative, considering the plastic's molecular structure. Table 1 presents various common types of challenging-to-degrade plastic structures alongside their corresponding resource recovery technologies.| Plastics | Polymer | Monomer | Molecular weight | Aggregation method | Recovery technology |
|---|---|---|---|---|---|
| Poly(amide) |

|
— | 341.5 | Ring opening polymerization | — |
| Poly(carbonate) |

|
— | 290.3 | Ring opening polymerization | Biocatalysis |
| Poly(ethylene) |

|
CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–100000 000–100000 |
Addition poly-merization | Thermocatalysis |
| Microwave catalysis | |||||
| Poly(ethylene terephthalate) |

|
— | 20000–30000 |
Ring opening polymerization | Thermocatalysis |
| Electrocatalysis | |||||
| Biocatalysis | |||||
| Photocatalysis | |||||
| Poly(lactic acid) |

|
— | 60000 |
Ring opening polymerization | Photocatalysis |
| Poly(propylene) |

|
CH3–CHCH2 |
500–8000 | Addition poly-merization | Photocatalysis |
| Poly(styrene) |

|
C6H5CHCH2 |
80000–150000 |
Addition poly-merization | Photocatalysis |
| Poly(urethane) |
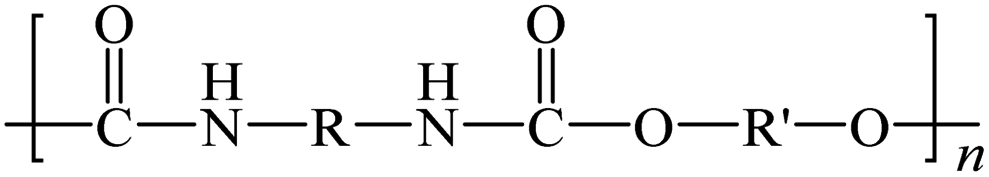
|
— | 80000–150000 |
Ring opening polymerization | Thermocatalysis |
| Poly(methyl methacrylate) |

|
C5H8O2 | 80000–200000 |
Addition poly-merization | — |
| Poly(vinyl chloride) |

|
CH2CHCl |
50000–110000 |
Addition poly-merization | Photocatalysis |
2.1 Structural analysis of hard-to-degrade plastics
To date, there is a wide range of nearly a hundred different plastic materials known. Depending on their behavior in nature, plastics can be categorized into two main groups: degradable plastics and non-degradable plastics.30–33 Degradable plastics can be further classified into several types. First, there are photodegradable plastics that are infused with photosensitizers, enabling them to break down under the influence of light. Second, biodegradable plastics can be decomposed by microorganisms, facilitating their natural degradation over time.34 Lastly, there are water-degradable plastics that are water-soluble, allowing them to dissolve when exposed to water.In contrast, non-degradable plastics mostly possess a highly stable polymer structure, making them resistant to natural degradation. Examples of non-degradable plastics include PE, poly(vinyl chloride) (PVC), poly(styrene) (PS), poly(urethane) (PU), and poly(methyl methacrylate) (PUMA). These plastics are synthesized through addition polymerization or condensation polymerization processes, wherein multiple monomers are linked together to form long-chain molecules, resulting in a three-dimensional network structure.35 Addition polymers, such as PE, form strong carbon–carbon bonds within the polymer, making them chemically inert. Such polymers are also referred to as “full carbon-backbone polymers”. On the other hand, condensation polymers are more susceptible to hydrolysis at high temperatures and in humid environments.36–39 Another significant factor influencing plastic degradation is the organization of their molecular structure. Thermoplastics can exhibit either semi-crystalline or amorphous properties. Semi-crystalline polymers, like PE, poly(acetal), and nylon, undergo a distinct solid-to-liquid transition at specific temperatures.40 Amorphous polymers, such as PS and polycarbonate (PC), do not melt but rather soften when heated above their glass transition temperature.41,42 Additionally, semi-crystalline plastics generally exhibit greater resistance to organic chemicals compared to amorphous plastics. For proper degradation and recycling, plastics require macroscopic fragmentation and intramolecular depolymerization. Precisely breaking the bonds between monomer molecules is the key to depolymerization reactions.
2.2 Waste plastics resource technologies
To protect the environment, direct incineration for energy recovery is not considered feasible. Instead, researchers have turned to thermochemical technologies to indirectly repurpose plastics as an alternative to fossil fuels. These technologies involve converting plastic waste into liquid fuels through processes like pyrolysis and gasification.47,48 However, the efficiency of large-scale thermochemical treatment for plastic waste is still limited.49,50 To enhance the conversion efficiency of waste plastics, Hu et al. introduced chemical cyclic redox technology into the thermochemical treatment process.51 They employed a cyclic process involving Fe and Fe2AlOx to achieve a syngas concentration of 92.84%. Additionally, they designed a two-zone reactor to facilitate thermal cracking and oxidative hydrolysis of plastics, resulting in a carbon conversion rate of 81.03% (Fig. 2a and b). In addition to direct heating methods, the rapid heating capabilities of microwaves have shown promise in transforming waste plastics into energy. Wan Mahari et al. have successfully utilized microwave-induced high-speed heating to obtain high-quality liquid oils and combustible gases from waste plastics within a short timeframe.52 It appears that if the macromolecular chains in waste plastics can be broken down through thermochemical means, further control of the reaction conditions may enable the production of high-value chemicals with different properties.
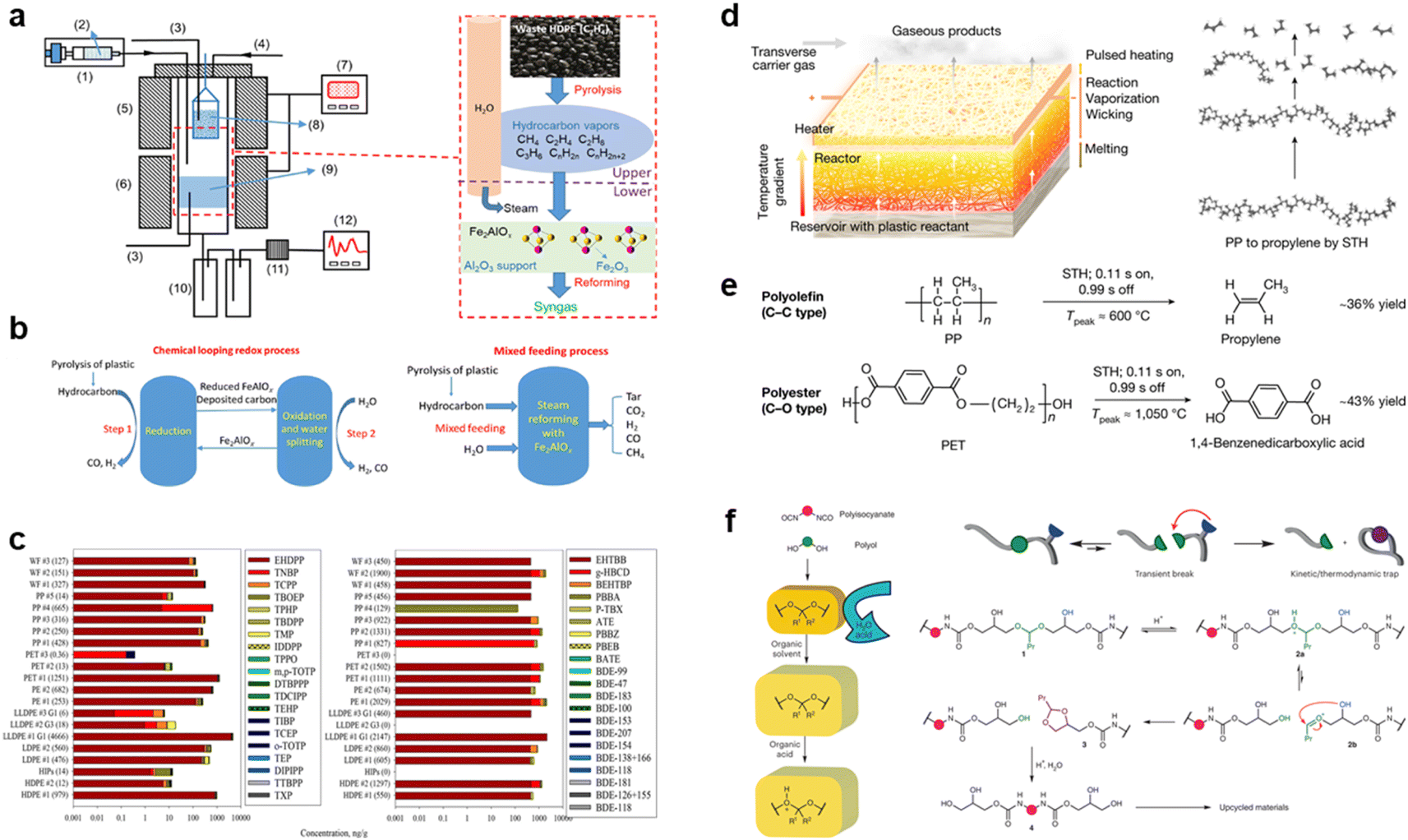 | ||
| Fig. 2 (a) Display of the experimental system and reaction process. (b) Schematic of the chemical looping redox process and mixed feeding process for plastic conversion. Reproduced with permission from ref. 51 Copyright 2022, ACS. (c) Chemical distribution (stacked bar, x-axis) of the targeted organophosphate ester plasticizers and flame retardants and halogenated flame retardants. Reproduced with permission from ref. 53 Copyright 2023, ACS. (d) Schematic demonstrating the STH process in the bilayer configuration and the molecular transformation from polymer to monomer using PP as a model plastic. (e) The depolymerization reactions of PP (as a model polyolefin) and PET (as a model polyester) to their monomers by STH. Reproduced with permission from ref. 60 Copyright 2023, Springer Nature. (f) Polyurethanes and proposed CATCH cleavage. Reproduced with permission from ref. 61 Copyright 2023, Springer Nature. | ||
Thermal cracking is a straightforward process based on free radical reactions. Strong heat is applied to break C–C and C–H bonds, generating free radicals that subsequently combine to form monomers or low molecular compounds.55,56 Recent research has focused on utilizing pyrolysis to convert waste plastics into valuable carbon nanomaterials like graphite or graphene. Smith et al. successfully transformed commercial poly(propylene) (PP) into carbon materials using conventional fused deposition modeling (FDM) with 3D printing.57 Wyss et al. proposed a method for rapid production of 1D graphite materials from waste plastics by optimizing the heat source with flash joule heating (FJH).58 In just a few milliseconds at temperatures above 3100 K, the plastic polymer rapidly reconstitutes into highly ordered sp2 heterogeneous flakes, bypassing the need for lengthy chemical vapor deposition processes. On top of thermal cracking, catalysis can further enhance the efficiency and product selectivity of plastic cracking, as will be highlighted in the next section.59 Catalysis can further enhance the efficiency and selectivity of plastic cracking.
Chemical depolymerization, on the other hand, can break down waste plastic polymers into monomers without the need for a catalyst. Dong and colleagues achieved selective depolymerization of PP and poly(ethylene terephthalate) (PET) into monomers using a far-from-equilibrium thermochemical plastic depolymerization method.60 Their strategy involved employing an electrified space-time heating (STH) unit, which prevents random breaking of C–C and C–H bonds and avoids the formation of by-products (Fig. 2d and e). The choice of reagent in depolymerization reactions significantly affects the treatment of waste plastics. For instance, if organic acids are selected as reagents for breaking long-chain plastics, it is possible to completely depolymerize PU waste's main chain at room temperature using acyclic acetal units of glycerol. This process, referred to as cyclization-triggered chain (CATCH) cleavage by Morado et al., demonstrates promising applications not only for PU but also for other types of waste plastics (Fig. 2f).61
3. Catalytic technology for waste plastics resource recovery
In principle, the desired characteristics of plastic materials for both service and recycling are inherently contradictory. They must possess stability and durability during their useful life, while also undergoing rapid and complete degradation under mild conditions for effective recycling. Consequently, innovative technologies are needed to overcome the kinetic and thermodynamic barriers associated with plastic decomposition and manage the existing pollution caused by non-degradable plastics.62 Catalysis plays a crucial role in this context by reducing the activation energy of reactions and altering the reaction pathway, thereby enhancing reaction rate and selectivity.63 Various energy-driven catalytic processes, such as thermocatalysis, electrocatalysis, biocatalysis, field energy catalysis, and photocatalysis, have shown promising results in transforming waste plastics into valuable resources.64,653.1 Thermocatalysis strategies
The incorporation of catalysts into the thermal cracking process offers significant benefits in terms of alleviating harsh reaction conditions for waste plastic depolymerization. This not only improves the efficiency of the reaction but also enhances product distribution, ultimately increasing its value.66 However, the thermal stability of the catalyst poses a major challenge due to the elevated temperatures involved in waste plastic thermal cracking. Yu et al. employed a calixarene La0.6Ca0.4Co1−xFexO3−δ precatalyst to effectively deconstruct medical waste plastic materials into carbon nanotubes and hydrogen at 850 °C.67 Remarkably, even after 10 consecutive high-temperature cycles, the catalyst maintained the highest yields of carbon nanotubes (245 mg gcat−1) and hydrogen (24.92 mmol gcat−1) for waste plastic treatment. Through varying the reaction temperature and pre-catalyst composition, they identified the Fe/Co ratio in the precatalyst as a key factor in enhancing reactivity and product selectivity. In another study, Dong and colleagues achieved the decomposition of waste plastics and conversion of hydrocarbons using ZSM-5 catalysts at 500 °C.68 over time, the reaction products exhibited exceptional selectivity for short-chain olefins, aromatics, alkanes, hydrogen, coke, and methane (CH4) due to the unique pore structure of ZSM-5 and appropriate reaction conditions (500 °C, 1 atm).Moreover, the use of a precious metal platinum (Pt) catalyst further reduced the cracking temperature of waste plastics.69,70 Li et al. developed a Pt nanoparticle (Pt@S-1) catalyst encapsulated by β-zeolite and silica molecular sieve-1, achieving an impressive 89.5% naphtha yield at 250 °C with 96.8% selectivity for C5–C9 hydrocarbons using a tandem catalytic reaction (Fig. 3a).71 The suitable acidity of the zeolite catalyst and the size effect of the Pt nanoparticles facilitated rapid breaking of C–H/C–C bonds in the waste plastic molecules and effectively controlled the narrow distribution of alkanes. Additionally, Zhang et al. directly converted various grades of waste PE plastics into liquid alkyl aromatics (∼C30) at low temperatures by solidly loading Pt onto γ-alumina (Fig. 3b).72 Notably, they overcame the thermodynamic barrier to obtain long-chain alkane products by modulating the barometric pressure during the reaction. Too low pressure was insufficient for plastic hydrolysis, while excessive pressure led to aromatic hydrogenation. Considering the wide range of plastic types, each waste plastic exhibits distinct thermal decomposition temperatures and chain-breaking mechanisms, leading to the continuous emergence of new thermocatalysts.73–75
 | ||
| Fig. 3 (a) Relationship between isomer/normal (i/n) and Pt/Brønsted acid sites (nPt/na) over different catalysts. Reproduced with permission from ref. 71 Copyright 2023, ACS. (b) Overall PE conversion to alkyl aromatics and alkylnaphtheness, and proposed mechanism of tandem polyethylene hydrogenolysis/aromatization via dehydrocyclization. Reproduced with permission from ref. 72 Copyright 2020, AAAS. (c) Membrane-electrode-assembly (MEA) for EG oxidation and the MEA setup for paired HER(−)//EG oxidation(+). Reproduced with permission from ref. 85 Copyright 2021, Springer Nature. (d) Schematic illustrations of the electrocatalytic upgrading process of triglycerides and waste PET bottles. Reproduced with permission from ref. 87 Copyright 2023, ACS. | ||
3.2 Electrocatalysis strategies
Electrocatalysis is a powerful method of accelerating reactions through charge transfer at the interface of electrodes and electrolytes.76 Since 2021, Zhao's research group has been at the forefront of green electrocatalytic reforming of PET waste plastics to co-produce formic acid and hydrogen. Their initial focus was on converting ethylene glycol (EG) derived from waste plastics into formate using copper-based nanowire catalysts, which exhibited high selectivity.77 Density functional theory (DFT) calculations demonstrated that the CuO catalyst facilitated the breaking of C–C bonds, leading to the production of the glyoxal intermediate. Building upon their success, the group further optimized the electrocatalyst design and synthesized NiCo2O4 electrocatalysts that efficiently hydrolyze PET, resulting in formic acid with a Faraday efficiency of up to 90%.78 To make the process even more sustainable, photovoltaics were employed as an energy source to drive the electrocatalytic cracking of plastics, enhancing the value of formic acid production while generating hydrogen (H2).79 Solar energy not only served as an electricity source but also promoted charge separation on the catalyst, thereby enhancing the reaction efficiency.80 Through the introduction of Fe2O3/Ni(OH)x photoanode catalysts, photoelectrocatalysis (PEC) technology achieved the conversion of PET to formic acid with a Faraday efficiency of approximately 100%.81 Light played a crucial role in exciting electron–hole separation on the anode catalyst, thereby improving the selectivity for degradation products of waste plastics. It is worth noting that photocatalysis itself is also employed in waste plastic degradation management, which will be discussed in detail in the subsequent section.In the electrocatalytic recovery of waste PET, transition metal Co- and Ni-based catalysts have exhibited high selectivity for formate.82–84 Zhou et al. developed nickel (Ni)-modified cobalt phosphide (CoNi0.25P) electrocatalysts as membrane electrodes, achieving selectivity above 80% for formate products (Fig. 3c).85 Behera et al. utilized a one-dimensional (1D) coordination polymer based on cobalt (Co) as an electrocatalyst, resulting in a 100% yield of terephthalic acid (TPA) and 80% selectivity of potassium dicarboxylate (KDF) at a low starting potential of 1.27 V.86 Furthermore, besides formic acid, other valuable compounds can also be obtained from waste PET. Yan et al. successfully cleaved glycerol from PET waste, yielding lactic and glycolic acids by employing gold (Au) loaded on nickel hydroxide (Fig. 3d).87 The selectivity of these products was attributed to the formation of alcohol salts through interactions between adjacent OH groups on the intermediate molecule, resulting in an increased local current density facilitated by Au.
3.3 Biocatalysis strategies
The biocatalytic treatment process for plastics primarily relies on enzymatic catalysis.88 Nature harbors a vast array of active enzymes and microorganisms that possess the capability to break down plastic copolymers, including PET enzyme (PETases), keratinases, and hydrolases.89,90 In the case of PETases, Kalathil et al. discovered that Ideonella sakaiensis (I. sakaiensis), a PETases, can completely mineralize PET into carbon dioxide, water, and adenosine triphosphate (ATP) when exposed to oxygen. Under anaerobic conditions, PET is broken down into acetic acid and ethanol (Fig. 4a).91I. sakaiensis also exhibits the ability to generate electricity from waste plastic, especially when co-cultivated with the electrogenic Geobacter sulfurreducens. Naturally, enzymes are found within organisms as well as in the natural environment. Eiamthong et al. identified highly active MG8 enzymes in the human salivary metagenome for PET hydrolysis, surpassing the degradation efficiency of hydrolases used in both nature and industrial production.92 They genetically recoded the MG8 gene fragment to obtain a covalent binding agent that could facilitate the bio-functionalization of PET. | ||
| Fig. 4 (a) Anaerobic PET conversion by Ideonella sakaiensis. Reproduced with permission from ref. 91 Copyright 2022, Wiley. (b) WT PETase protein structure rendered by the output of MutCompute. (c) Predictions based on both WT PETase and ThermoPETase were ranked by the fold change in the probabilities between the predicted and the WT amino acid. Reproduced with permission from ref. 93 Copyright 2022, Springer Nature. | ||
However, the uncontrolled survival of enzymes in nature and their lack of stability during catalytic reactions with waste plastics pose challenges. Moreover, the diverse range of enzymes in nature makes it difficult to fully comprehend their survival requirements and performance. To address this, researchers conducted a comprehensive analysis of waste plastic hydrolysis enzymes using machine learning algorithms (Fig. 4b and c).93 Their findings revealed that the FAST-PETases effectively enables a closed-loop recycling process for PET. By studying native enzymes, genetic engineering techniques can be employed to develop specific enzyme catalysts for the degradation of waste plastics.94 Zhu et al. developed a BIND-PETases derived from the original enzyme, which exhibits a 30 day survival period at 4 °C and can directly degrade PET microplastics in wastewater without becoming inactivated.95 Alternatively, in combination with inorganic nanomaterial Fe3O4, the enzyme achieves nearly 100% microplastic removal and degradation efficiency.96 Apart from enzymatic catalysis, the use of bio-based materials in catalytic depolymerization has proven effective in breaking down plastic molecules. Saito and his colleagues experimented with vanillin derivatives as bio-based feedstocks, which offer high purity of recovered PC monomers without causing chemical secondary pollution to the environment.97 Overall, biocatalytic degradation of waste plastics is a viable approach, but it necessitates better control and design of enzyme catalysts to enhance its efficacy.98,99
3.4 Other catalysis strategies
Organocatalysis has emerged as an effective method for decomposing waste plastics, serving as a valuable complement to transition metal-based and biocatalysis.100 Given that plastics are inherently organic polymers, organic reagents offer distinct advantages for recycling waste plastics based on the principle of similar solubility. Organocatalytic degradation can be categorized into alcoholysis, phosphate ester, and alcohol–amine reactions, depending on the specific reagents employed. Furthermore, it can be combined with organic polymerization to establish closed-loop recycling pathways for plastics.101,102 In the study by Yang et al., a modular series of dinuclear organoboron catalysts was designed for the synthesis of chemically recyclable poly(cyclopentene)carbonate (PCPC) and its subsequent depolymerization after use.103 The catalytic efficiency of PCPC polymerization achieved remarkable results, with a metal-free organocatalysis achieving a high yield of 1.0 kg PCPC gcat−1. The depolymerization of PCPC involved simple random chain cleavage and unchaining pathways. This intriguing monomer–polymer–monomer cycle demonstrates the potential of organocatalysis in plastic recycling.Moreover, researchers have been exploring the use of field effects as an energy source to catalyze the decomposition of waste plastics. The thermal effect of microwave fields, for instance, enhances the efficiency of hydrogen production from waste PE. The Fe1Co1Al2 catalyst's structure benefits from the field effect during the reaction process, facilitating charge transfer from hydrogen to iron within the waste plastic.104 When the catalyst is capable of absorbing microwaves, the deconstruction rate and selectivity of the plastic molecules are significantly enhanced. Cao et al. employed MAX (Ti3AlC2) as a microwave acceptor catalyst, resulting in the generation of high-purity hydrogen and highly selective products during the treatment of various types of waste plastics.105 Inexpensive iron-based catalysts are also commonly used as microwave catalysts due to their strong microwave absorption capability, enabling the conversion of waste plastic powder into hydrogen and carbon nanotubes in as little as 30–90 seconds.106 It is conceivable that other field effects, such as piezoelectric and ultrasonic fields, could serve as additional energy sources for catalytic degradation of waste plastics.
4. Photochemical strategies for the resourcefulness of waste plastics
Light plays a crucial role in supporting life within Earth's living systems, as many organisms depend on the products of photosynthesis for sustenance. When substances are exposed to ultraviolet or visible light and absorb light energy, photochemical reactions such as photosynthesis and photolysis can take place.107,108 For example, UV radiation weakens the internal bonding energy of waste plastics, leading to the breaking of bonds by free radicals generated from the combined effects of light and oxygen.109 Consequently, the plastic gradually degrades into biodegradable microplastics with lower molecular weights, ultimately resulting in the production of carbon dioxide (CO2) and water (H2O). Photochemical degradation significantly contributes to the aging of waste plastics in the environment. However, the natural degradation process through light alone is typically lengthy and uncontrollable.110–113By harnessing the principles of photocatalysis, the degradation of waste plastics can be addressed through a green and energy-efficient strategy. Photocatalytic degradation has proven to be highly effective in treating environmental pollutants and can be extended to the treatment and recycling of waste plastics.114–116 Photocatalysts are mostly semiconductor materials. When the absorbed energy is greater than or equal to the band gap photon, the photocatalyst is excited to produce photogenerated electron–hole pairs that leap to the conduction band and remain in the valence band. The photogenerated electron–hole pairs migrate to the catalyst surface under the action of an electric field and react with the adsorbed material on the surface in reduction and oxidation reactions respectively.117 The fundamental concept of photocatalysis involves utilizing light to generate electron–hole pairs in semiconductor materials, which subsequently produce active radicals for various desired redox reactions. Moreover, the photogenerated radicals can be incorporated into macromolecular polymers, allowing the production of plastic products that facilitate photodegradation.118 Accordingly, photocatalysis offers a comprehensive approach to tackle plastic pollution, encompassing both end-of-pipe recycling and source treatment challenges.119,120
4.1 Photocatalytic degradation of microplastics
In their natural state, waste plastics undergo environmental aging and are degraded by sunlight into microplastics (MPs) and nanoplastics (NPs), which then enter the soil or water.121 Solar irradiation causes certain waste plastics to break down into dissolved organic carbon (DOC), which can potentially be assimilated by bacteria.122 However, not all waste plastics can be completely degraded by natural light alone. Small-size waste plastic molecules (1–100 μm) can only be fully mineralized by specific microorganisms. Otherwise, they remain as micro-pollutants that are difficult to detect by humans. Liu et al. conducted an analysis of the aging process of PS plastics under light exposure using ionophore mass spectrometry (spICP-MS).123 The findings revealed that PS produces significant amounts of MPs and NPs during aging, posing a long-term chronic toxicity risk to organisms and a “hidden” ecotoxicological threat. Additionally, the natural photodegradation of plastics by light also generates substantial quantities of volatile organic chemical pollutants (VOCs), contributing to air pollution (Fig. 5a and b).124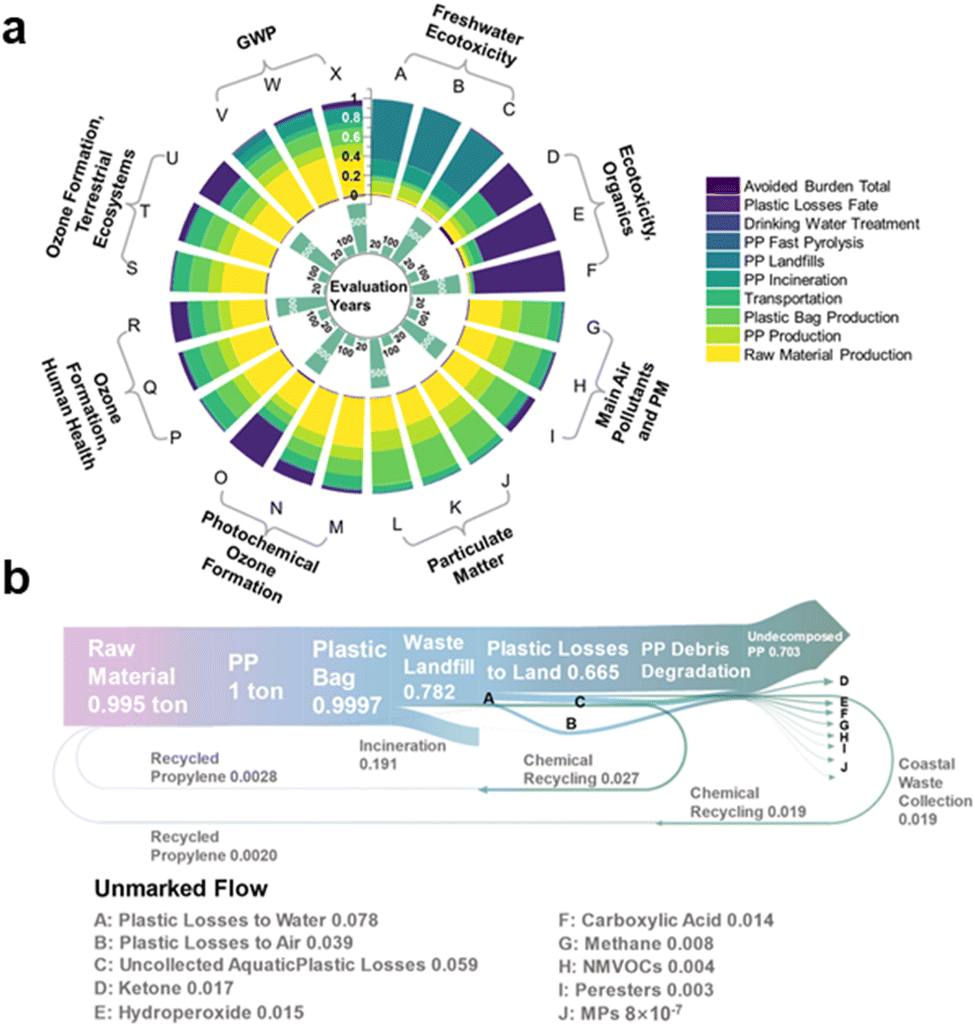 | ||
| Fig. 5 (a) Environmental profile based on various environmental indicators in 20, 100, and 500 years. (b) Mass flow distribution across the entire life cycle in 500 years shows the mass flow rates in tons from raw material and resource extraction to the photo-degradation products as graves. Reproduced with permission from ref. 124 Copyright 2023, ACS. | ||
The natural photodegradation of waste plastics primarily relies on the activity of reactive oxygen species (ROS), particularly oxidative free radicals.125 In photocatalytic reactions, a diverse array of photogenerated radicals is commonly employed for the mineralization of organic pollutants.126,127 To facilitate the rapid and complete photodegradation of waste plastics, photocatalysis can be employed to treat microscopic plastic particles.128 Uheida and colleagues utilized glass fibers embedded with zinc oxide (ZnO) nanorods (NRs) to capture and degrade PP microplastic particles.129 Under visible light, ZnO NRs catalyze the efficient formation of carbonyl and hydroxyl groups on waste plastics, effectively reducing the particle size of PP microplastic waste. While natural aging of waste plastics primarily occurs under UV light, the choice of light for the photocatalytic degradation process is typically based on the photoresponse of the photocatalyst. Ma et al. designed FeOx/g-C3N4 and Fe2O3/g-C3N4 photocatalysts with visible light activity for PET degradation.130 These catalysts exhibit a significant breaking effect on the ester bonds present in PET over a broad wavelength range of 200–800 nm.
4.2 Value-added conversion of waste plastics under photocatalysis
The light-driven reprocessing of waste plastics for recycling can be referred to as photo-reforming. The catalysts that facilitate this process are mostly precious metal or cadmium (Cd)-based photocatalysts.131 Uekert et al. conducted a study where they prepared CdS/CdOx quantum dots for efficient photo-reformation of three commonly used plastic molecules: poly(lactic acid) (PLA), PET, and PU.132 They achieved this in alkaline aqueous solutions, resulting in high-purity H2 as well as organic products such as formate, acetate, and pyruvate. To address the photo-corrosive properties of CdS, they designed a carbon nitride/nickel phosphide (CNx/Ni2P) photocatalyst, which controlled the toxicity and cost of the catalyst.133 Under alkaline conditions, CNx/Ni2P was capable of converting PET and PLA into H2 and various organic small molecule compounds. The catalytic recovery reaction system was scalable up to 60 times and could be extended to polyester fibers and plastic waste contaminated with oil (Fig. 6a and c). | ||
| Fig. 6 (a) Long-term photo reforming of polyester microfibers, a PET bottle, and an oil-coated PET bottle. (b) Upscaled photo reforming of polyester microfibers; the sample was purged every 24 h. (c) Photograph of the batch reactor in use. Reproduced with permission from ref. 133 Copyright 2019, ACS. (d) EPR spectra from in situ irradiation at 405 nm (1 mW LED) of PS and a pTsOH·H2O solution with 4-oxo-TMP and DMPO spin traps (1:4). Center: experimental spectra measured after 10 min (red) and 50 min (blue) irradiation. Left and Right: Simulated spectra of three separate components of nitroxyl, a carbon-centered DMPO adduct, and an oxygen-centered DMPO adduct. Reproduced with permission from ref. 135 Copyright 2022, ACS. (e) Proposed reaction pathway of polystyrene photo–oxidation reaction. Reproduced with permission from ref. 139 Copyright 2020, Springer Nature. (f) Direct microstructure fabrication on bulk TCP. Reproduced with permission from ref. 142 Copyright 2022, Wiley. | ||
Cd-based photocatalysts can also be modified by defective and heterojunction means. This modification led to the development of d-NiPS3/CdS waste plastic photo reformation catalysts with high H2 yields (∼40 mmol gcat−1 h−1) and excellent stability over 100 hours.134 The photocatalytic decomposition of waste plastics is feasible in both alkaline and acidic environments as long as the photocatalyst can produce stable reactive oxygen radicals. Huang et al. developed an acid-catalyzed system for the degradation of plastic waste. They achieved visible light selective acid-catalyzed oxidation of PS without a photosensitizer by adjusting the ratio of acid and solvent.135 The [PS-acid] adduct formed during the acid-catalyzed reaction between PS and acid was found to act as the true photosensitizer. In the presence of light, this adduct generated singly linear oxygen (1O2) to oxidize PS, forming formic acid, benzoic acid, or acetophenone (Fig. 6d). Additionally, other radicals with oxidizing properties could be employed for the decomposition of waste plastics. Oh et al. utilized chlorine radicals generated by FeCl3 under white light irradiation to capture hydrogen atoms from waste plastics, resulting in the degradation of PS into benzoyl compounds.136 They demonstrated the feasibility of this method for gram-scale waste plastics treatment using flow chemistry.
Compared to thermal catalysis, photocatalysis provides the ability to recycle plastics and convert them into high-value chemicals. The key difference lies in the milder reaction conditions and the special high selectivity of photocatalysis for waste plastic products.137,138 Cao et al. achieved the oxidation of PS to aromatic compounds by g-C3N4 in visible light catalysis, showing a high selectivity for benzoic acid in the liquid phase product (Fig. 6e).139 This selectivity was attributed to the precise activation of C–H and C–C bonds on PS by photogenerated radicals, leading to the further oxidation of hydroxyl and carbonyl groups on its main chain, forming aromatic oxygenated compounds. In terms of efficiency, composite photocatalysts prepared using the porous MOFs material UiO66-NH2 encapsulated with ZnO contribute to improved electron–hole separation, enhancing photocatalytic efficiency.140,141 This, in turn, accelerates the catalytic conversion of PLA and PVC to acetic acid. The widespread application of light has also encouraged the photocatalytic cracking of plastics beyond the recycling of waste plastics, even extending to lithography processes. Samal et al. capitalized on the controlled reaction conditions of photocatalysis to directly build subcellular microstructures on PS using deep ultraviolet light in a short period. This simple lithography technique does not require expensive equipment or specific reagents, making it easily accessible even to non-experts (Fig. 6f).142 It significantly expands the possibilities of photocatalytic degradation in plastics.
4.3 Photo-thermal/electro/biocatalytic synergy
The efficiency of carrier separation, light utilization, and quantum efficiency in photocatalytic reactions has been a significant barrier to the industrialization of photocatalysis.120,143 To enhance the conversion of light energy into chemical energy, thermocatalysis, electrocatalysis, and biocatalysis are often integrated into the photocatalytic process, leveraging synergistic effects to improve the cleavage efficiency of waste plastic molecules.144,145 Moreover, the addition of photocatalysis can enhance the selectivity of degradation products in waste plastic, complementing the aforementioned catalytic technologies. In a study by Liu et al., integrated Co single site (Co SSCs) catalysts were introduced into a photothermal catalytic system to promote the conversion of carbonyl groups in waste polyester plastics and optimize the nucleophilic addition elimination process.146 The photothermal synergy significantly increased the conversion of PET and the yield of bis(2-hydroxyethyl) terephthalate by 5.4 and 6.6 times, respectively, compared to thermal catalysis alone. Electrocatalytic enrichment, on the other hand, offers even greater effectiveness in separating photogenerated electron–hole pairs due to the directional charge transfer, thereby enhancing the performance of photocatalytically degraded plastics.147 And to conserve energy resources, piezoelectric materials can be utilized as a source of electricity.148,149 The combination of piezoelectric-photocatalysts has demonstrated the ability to inhibit the rapid recombination of photogenerated charges, exhibiting unique advantages in plastic waste treatment.150The integration of biocatalysis and photocatalysis holds great promise for achieving complete degradation of waste plastics, transforming large molecules into smaller ones and ultimately achieving complete mineralization. Ye et al. developed a bio-photocatalyst system by assembling Methanosarcina barkeri (M. b.) and carbon dot functionalized polymerized carbon nitride (CDPCN) catalysts.151 This system successfully degraded PLA into CH4 in five consecutive cycles, achieving a selectivity of up to 100%. Furthermore, Kim et al. combined electrocatalysis, biocatalysis, and photocatalysis in a photoelectrochemical biosynthesis reaction of PET waste plastic (Fig. 7a).152 In this system, Zr-doped hematite served as a photoanode to extract electrons from the waste PET and transfer them to the bioelectrocatalytic site, while carbon material acted as a cathode to receive the electrons and activate hydrolytic enzymes for the plastic cracking reaction. This comprehensive approach involving all three technologies presents a more promising and efficient pathway for the treatment and recycling of plastics.
 | ||
| Fig. 7 (a) CFP-based cathodes reduce O2 to H2O2 for biocatalytic oxyfunctionalization and NAD+ to NADH for enzymatic amination and asymmetric hydrogenation. Reproduced with permission from ref. 152 Copyright 2022, Springer Nature. (b) Synthetic schemes of the two-step photopolymerization and post-modification. Reproduced with permission from ref. 155 Copyright 2020, ACS. | ||
4.4 Photochemical green preparation of degradable plastics
Plastics continue to play a significant role in manufacturing and daily life, creating a growing need for biodegradable plastics to replace environmentally harmful alternatives.153 Photoexcited photocatalysts generate oxidation-reduction intermediates that aid in the synthesis of plastics through a process called reversible deactivation of radical polymerization (RDRP).154 Simply put, RDRP allows for precise control over the molecular weight, molecular weight distribution, and polymer structure of plastics when there is a uniform and adequate light source.Jiang and colleagues have introduced a groundbreaking technique called photoorganocatalytic reversible deactivation, which applies to vinyl chlorotrifluoroethylene (CTFE) and vinyl ethers (VE) in alternating radical copolymerization (Fig. 7b).155 This method enables the controlled synthesis of main-chain fluorinated alternating copolymers at room temperature and atmospheric pressure. The “activation–deactivation” cycle allows for continuous switching of CTFE molecules, facilitating chain growth and enabling free growth and post-synthesis modification of the copolymer. By incorporating photosensitizers into the polymerization system, recyclable plastics with photodegradable properties can be obtained, thus establishing a more comprehensive recycling chain for the environmentally friendly use of plastics.
5. Summary and perspectives
Polymers are the building blocks of plastics, forming long, complex molecular structures with a carbon skeleton and various functional groups branching out from the carbon elements. These versatile materials have revolutionized consumer goods, but unfortunately, they have also led to the persistent problem of plastic pollution that can endure for decades or even centuries. Waste plastics, which are difficult to degrade, exist in a multitude of forms, ranging from hard to soft, and they resist easy melting and transformation. Presently, waste plastics pervade our environment, infiltrating the soil, infiltrating rivers, and spreading through untouched regions with rainfall. Urgent action is required to address this issue and find suitable methods for treating and recycling waste plastics. Conventional disposal approaches like landfilling, incineration, and mechanical cracking are no longer viable in the face of the current plastic pollution crisis.Catalytic technology offers a promising solution for recycling and reusing plastics, but it is not without its challenges (Table 2). Thermocatalytic processes can effectively lower the thermodynamic requirements for breaking down waste plastics through thermochemical cracking. Ideally, waste plastics should be crackable at lower temperatures and atmospheric pressure, while also enhancing selectivity for desired products. Electrocatalysis, commonly used for energy conversion, can also contribute to waste plastic treatment. However, its industrial implementation necessitates the optimization of reaction cells, and the environmental impact of power supply greening must be considered. Enzyme-driven biocatalytic processes closely mimic natural degradation of waste plastics, with autogenous enzyme activity and catalyst efficiency being crucial factors. Photocatalysis, on the other hand, faces challenges in terms of catalytic efficiency. Enhancing the utilization of visible light and the oxidation capacity of photocatalysts through catalyst modification proves to be an effective strategy. It is vital to effectively prevent secondary pollution during the treatment of waste plastic, making green photocatalysis technology crucial in this regard. Thus, based on photocatalysis, harnessing multiple catalysts working in synergy to leverage their respective strengths may emerge as a powerful approach for rapidly eliminating plastic pollution.
| Catalytic technology | Driving force | Eco-friendliness | Economy |
|---|---|---|---|
| Thermocatalysis | Thermal energy | Low | Middle |
| Electrocatalysis | Electrical energy | Middle | High |
| Biocatalysis | Bioenergy | High | Middle |
| Photocatalysis | Light | High | High |
| Microwave catalysis | Microwave field | Middle | Middle |
| Organic catalysis | Free energy | Middle | High |
Author contributions
Yao Chen: conceptualization; writing-original draft & editing; Lele Bai: resources; writing-original draft; Dening Peng: resources; writing-original draft; Xinru Wang: resources; writing-original draft; Meijun Wu: resources; writing-original draft; Zhenfeng Bian: writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2020YFA0211004), the National Natural Science Foundation of China (22176128, 22236005), Supported by Innovation Program of Shanghai Municipal Education Commission, Sponsored by Program of Shanghai Academic Research Leader (21XD1422800), Shanghai Government (22dz1205400), Chinese Education Ministry Key Laboratory and International Joint Laboratory on Resource Chemistry, and Shanghai Eastern Scholar Program. “111 Innovation and Talent Recruitment Base on Photochemical and Energy Materials” (No. D18020), Shanghai Engineering Research Center of Green Energy Chemical Engineering (18DZ2254200). Shanghai Frontiers Science Center of Biomimetic Catalysis.References
- M. J. B. Amorim and J. J. Scott-Fordsmand, Environ. Pollut., 2021, 271, 116363 CrossRef CAS.
- M. Cormann, Global Plastics Outlook, OECD, French, 2022 Search PubMed.
- C. Amaneesh, S. Anna Balan, P. S. Silpa, J. W. Kim, K. Greeshma, A. Aswathi Mohan, A. Robert Antony, H. P. Grossart, H. S. Kim and R. Ramanan, Environ. Sci. Technol., 2023, 57, 5–24 CrossRef CAS.
- L. M. Alkema, C. J. Van Lissa, M. Kooi and A. A. Koelmans, Environ. Sci. Technol., 2022, 56, 15552–15562 CrossRef CAS.
- N. Cohen and A. Radian, Environ. Sci. Technol., 2022, 56, 17635–17642 CrossRef CAS PubMed.
- K. Bhubalan, A. M. Tamothran, S. H. Kee, S. Y. Foong, S. S. Lam, K. Ganeson, S. Vigneswar, A. Amirul and S. Ramakrishna, Environ. Res., 2022, 213, 113631 CrossRef CAS.
- A. L. Lusher and S. Primpke, Environ. Sci. Technol., 2023, 57, 6033–6039 CrossRef CAS PubMed.
- K. Min, J. D. Cuiffi and R. T. Mathers, Nat. Commun., 2020, 11, 727 CrossRef CAS.
- J. Gigault, H. El Hadri, B. Nguyen, B. Grassl, L. Rowenczyk, N. Tufenkji, S. Feng and M. Wiesner, Nat. Nanotechnol., 2021, 16, 501–507 CrossRef CAS.
- Z. Lei, H. Chen, C. Luo, Y. Rong, Y. Hu, Y. Jin, R. Long, K. Yu and W. Zhang, Nat. Chem., 2022, 14, 1399–1404 CrossRef CAS.
- J. Zhou, T. G. Hsu and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300768 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS.
- J. Li, F. Liu, Y. Liu, Y. Shen and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202207105 CAS.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- X. Zhao, B. Boruah, K. F. Chin, M. Dokic, J. M. Modak and H. S. Soo, Adv. Mater., 2022, 34, e2100843 CrossRef.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 CrossRef CAS.
- O. H. Fred-Ahmadu, G. Bhagwat, I. Oluyoye, N. U. Benson, O. O. Ayejuyo and T. Palanisami, Sci. Total Environ., 2020, 706, 135978 CrossRef CAS.
- G. Pathak, M. Nichter, A. Hardon, E. Moyer, A. Latkar, J. Simbaya, D. Pakasi, E. Taqueban and J. Love, Glob. Environ. Change, 2023, 80, 102648 CrossRef.
- F. Zhang, Y. Zhao, D. Wang, M. Yan, J. Zhang, P. Zhang, T. Ding, L. Chen and C. Chen, J. Cleaner Prod., 2021, 282, 124523 CrossRef CAS.
- V. K. Soni, G. Singh, B. K. Vijayan, A. Chopra, G. S. Kapur and S. S. V. Ramakumar, Energy Fuels, 2021, 35, 12763–12808 CrossRef CAS.
- S. Jung, S. Lee, X. Dou and E. E. Kwon, Chem. Eng. J., 2021, 405, 126658 CrossRef CAS PubMed.
- S. Soltanian, S. A. Kalogirou, M. Ranjbari, H. Amiri, O. Mahian, B. Khoshnevisan, T. Jafary, A.-S. Nizami, V. K. Gupta, S. Aghaei, W. Peng, M. Tabatabaei and M. Aghbashlo, Renewable Sustainable Energy Rev., 2022, 156, 111975 CrossRef CAS.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- M.-Q. Zhang, M. Wang, B. Sun, C. Hu, D. Xiao and D. Ma, Chem, 2022, 8, 2912–2923 CAS.
- K. Hu, Y. Yang, Y. Wang, X. Duan and S. Wang, Chem Catal., 2022, 2, 724–761 CrossRef CAS.
- C. Fu, F. Li, J. Zhang, D. Li, K. Qian, Y. Liu, J. Tang, F. Fan, Q. Zhang, X. Q. Gong and W. Huang, Angew. Chem., Int. Ed., 2021, 60, 6160–6169 CrossRef CAS.
- P. Zhou, M. Luo and S. Guo, Nat. Rev. Chem., 2022, 6, 823–838 CrossRef CAS.
- G. Hayes, M. Laurel, D. MacKinnon, T. Zhao, H. A. Houck and C. R. Becer, Chem. Rev., 2022, 123, 2609–2734 CrossRef PubMed.
- F. G. Torres, D. C. Dioses-Salinas, C. I. Pizarro-Ortega and G. E. De-la-Torre, Sci. Total Environ., 2021, 757, 143875 CrossRef CAS PubMed.
- L. P. Manker, G. R. Dick, A. Demongeot, M. A. Hedou, C. Rayroud, T. Rambert, M. J. Jones, I. Sulaeva, M. Vieli, Y. Leterrier, A. Potthast, F. Marechal, V. Michaud, H. A. Klok and J. S. Luterbacher, Nat. Chem., 2022, 14, 976–984 CrossRef CAS PubMed.
- A. Kinnebrew, S. Vincent and M. L. Curry, Acc. Chem. Res., 2023, 56, 1340–1349 CrossRef CAS PubMed.
- S. Kim, M. Kim, S. K. Lim and Y. Park, J. Environ. Chem. Eng., 2017, 5, 1899–1905 CrossRef CAS.
- S. S. A. Athukoralalage, C. A. Bell, A. C. Gemmell, A. E. Rowan and N. Amiralian, J. Mater. Chem. A, 2023, 11, 1575–1592 RSC.
- Synthesis of polymers, Polym. Sci., Ser. A, 2009, 51, 2–13 Search PubMed.
- F. V. Ferreira, L. S. Cividanes, R. F. Gouveia and L. M. F. Lona, Polym. Eng. Sci., 2019, 59, E7–E15 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- B. Molnar and F. Ronkay, Polym. Bull., 2018, 76, 2387–2398 CrossRef.
- Y. Shoji, C. Zhang, T. Higashihara and M. Ueda, Polym. Chem., 2012, 3, 1978 RSC.
- M. Abid, S. Mhiri, A. Bougarech, R. Triki and S. Abid, Des. Monomers Polym., 2020, 23, 16–24 CrossRef CAS.
- M. Chen, J. Bao, M. Zheng, X. Zhang and W. Chen, J. Appl. Polym. Sci., 2021, 139, 51731 CrossRef.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, J. Appl. Polym. Sci., 2014, 131, 39954 CrossRef.
- A. J. P. Queiroz, C. R. da Silva Morais, L. M. R. Lima, J. da Silva Buriti, J. de Lima Sales and F. P. Filho, J. Therm. Anal. Calorim., 2015, 123, 949–953 CrossRef.
- I. Fahim, O. Mohsen and D. ElKayaly, Polymers, 2021, 13, 915 CrossRef CAS PubMed.
- S. L. Wong, N. Ngadi, T. A. T. Abdullah and I. M. Inuwa, Renewable Sustainable Energy Rev., 2015, 50, 1167–1180 CrossRef CAS.
- B. R. T. Simoneit, P. M. Medeiros and B. M. Didyk, Environ. Sci. Technol., 2005, 39, 6961–6970 CrossRef CAS.
- C. I. Idumah, J. Therm. Anal. Calorim., 2021, 147, 3495–3508 CrossRef.
- M. Kusenberg, M. Roosen, A. Zayoud, M. R. Djokic, H. Dao Thi, S. De Meester, K. Ragaert, U. Kresovic and K. M. Van Geem, Waste Manage., 2022, 141, 104–114 CrossRef CAS PubMed.
- G. Kwon, D.-W. Cho, J. Park, A. Bhatnagar and H. Song, Chem. Eng. J., 2023, 464, 142771 CrossRef CAS.
- A. Toktarova, L. Göransson, H. Thunman and F. Johnsson, J. Cleaner Prod., 2022, 374, 133891 CrossRef CAS.
- Q. Hu, Y. S. Ok and C. H. Wang, Environ. Sci. Technol., 2022, 56, 8953–8963 CrossRef CAS PubMed.
- W. A. Wan Mahari, C. T. Chong, C. K. Cheng, C. L. Lee, K. Hendrata, P. N. Yuh Yek, N. L. Ma and S. S. Lam, Energy, 2018, 162, 309–317 CrossRef CAS.
- L. Chibwe, A. O. De Silva, C. Spencer, C. F. Teixera, M. Williamson, X. Wang and D. C. G. Muir, Environ. Sci. Technol., 2023, 57, 3380–3390 CrossRef CAS.
- F. J. Christopher, P. S. Kumar, D.-V. N. Vo, F. C. Christopher and L. Jayaraman, Environ. Chem. Lett., 2021, 20, 223–242 CrossRef.
- D. Zhao, X. Wang, J. B. Miller and G. W. Huber, ChemSusChem, 2020, 13, 1764–1774 CrossRef CAS.
- A. Angyal, N. Miskolczi, L. Bartha, A. Tungler, L. Nagy, L. Vida and G. Nagy, Fuel Process. Technol., 2010, 91, 1717–1724 CrossRef CAS.
- P. Smith, A. G. Obando, A. Griffin, M. Robertson, E. Bounds and Z. Qiang, Adv. Mater., 2023, 35, e2208029 CrossRef PubMed.
- K. M. Wyss, J. T. Li, P. A. Advincula, K. V. Bets, W. Chen, L. Eddy, K. J. Silva, J. L. Beckham, J. Chen, W. Meng, B. Deng, S. Nagarajaiah, B. I. Yakobson and J. M. Tour, Adv. Mater., 2023, 35, e2209621 CrossRef PubMed.
- M. Seitz and S. Schröter, Chem. Ing. Tech., 2022, 94, 720–726 CrossRef CAS.
- Q. Dong, A. D. Lele, X. Zhao, S. Li, S. Cheng, Y. Wang, M. Cui, M. Guo, A. H. Brozena, Y. Lin, T. Li, L. Xu, A. Qi, I. G. Kevrekidis, J. Mei, X. Pan, D. Liu, Y. Ju and L. Hu, Nature, 2023, 616, 488–494 CrossRef CAS.
- E. G. Morado, M. L. Paterson, D. G. Ivanoff, H. C. Wang, A. Johnson, D. Daniels, A. Rizvi, N. R. Sottos and S. C. Zimmerman, Nat. Chem., 2023, 15, 569–577 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- D. Lüke and M. Oetken, Chemkon, 2022 DOI:10.1002/ckon.202100074.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao and H. Duan, ACS Catal., 2022, 12, 9307–9324 CrossRef CAS.
- Y. Yu, B. Gao, Y. Liu and X. B. Lu, Angew. Chem., Int. Ed., 2022, 61, e202204492 CAS.
- X. Yu, G. Chen, M. Widenmeyer, I. Kinski, X. Liu, U. Kunz, D. Schüpfer, L. Molina-Luna, X. Tu, G. Homm and A. Weidenkaff, Appl. Catal., B, 2023, 334, 122838 CrossRef CAS.
- S. Dong, H. Li, I. K. Bloede, A. J. Al Abdulghani, E. A. Lebrón-Rodríguez, G. W. Huber and I. Hermans, Appl. Catal., B, 2023, 324, 122219 CrossRef CAS.
- M. Zare, P. A. Kots, S. Caratzoulas and D. G. Vlachos, Chem. Sci., 2023, 14, 1966–1977 RSC.
- K. McCullough, I. L. Peczak, R. M. Kennedy, Y.-Y. Wang, F. A. Perras, J. Hall, A. J. Kropf, R. Hackler, Y. Shin, J. Niklas, O. Poluektov, J. Wen, W. Huang, A. D. Sadow, K. Poeppelmeier, M. Delferro, M. Ferrandon, J. Lin, X. Wu and A. L. Paterson, J. Mater. Chem. A, 2023, 11, 1216–1231 RSC.
- L. Li, H. Luo, Z. Shao, H. Zhou, J. Lu, J. Chen, C. Huang, S. Zhang, X. Liu, L. Xia, J. Li, H. Wang and Y. Sun, J. Am. Chem. Soc., 2023, 145, 1847–1854 CrossRef CAS PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- C. F. Gallin, W. W. Lee and J. A. Byers, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed.
- Z. Gao, B. Ma, S. Chen, J. Tian and C. Zhao, Nat. Commun., 2022, 13, 3343 CrossRef CAS PubMed.
- G. O'Rourke, T. Hennebel, M. Stalpaert, A. Skorynina, A. Bugaev, K. Janssens, L. Van Emelen, V. Lemmens, R. De Oliveira Silva, C. Colemonts, P. Gabriels, D. Sakellariou and D. De Vos, Chem. Sci., 2023, 14, 4401–4412 RSC.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, e2003297 CrossRef PubMed.
- J. Wang, X. Li, T. Zhang, Y. Chen, T. Wang and Y. Zhao, J. Phys. Chem. Lett., 2022, 13, 622–627 CrossRef CAS.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- T. Zhang, X. Li, J. Wang, Y. Miao, T. Wang, X. Qian and Y. Zhao, J. Hazard. Mater., 2023, 450, 131054 CrossRef CAS.
- N. Li, J. Liu, B. X. Dong and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 20779–20793 CrossRef CAS.
- X. Li, J. Wang, T. Zhang, T. Wang and Y. Zhao, ACS Sustainable Chem. Eng., 2022, 10, 9546–9552 CrossRef CAS.
- Y. Li, Y. Zhao, H. Zhao, Z. Wang, H. Li and P. Gao, J. Mater. Chem. A, 2022, 10, 20446–20452 RSC.
- X. Liu, J. Wang, Z. Fang, S. Gong, D. Xiong, W. Chen, D. Wu and Z. Chen, Appl. Catal., B, 2023, 334, 122870 CrossRef CAS.
- N. Wang, X. Li, M.-K. Hu, W. Wei, S.-H. Zhou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2022, 316, 121667 CrossRef CAS.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- S. Behera, S. Dinda, R. Saha and B. Mondal, ACS Catal., 2022, 13, 469–474 CrossRef.
- Y. Yan, H. Zhou, S. M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. Andre, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS.
- L. Avilan, B. R. Lichtenstein, G. Konig, M. Zahn, M. D. Allen, L. Oliveira, M. Clark, V. Bemmer, R. Graham, H. P. Austin, G. Dominick, C. W. Johnson, G. T. Beckham, J. E. McGeehan and A. R. Pickford, ChemSusChem, 2023, 16, e202202277 CrossRef CAS.
- C. Sonnendecker, J. Oeser, P. K. Richter, P. Hille, Z. Zhao, C. Fischer, H. Lippold, P. Blazquez-Sanchez, F. Engelberger, C. A. Ramirez-Sarmiento, T. Oeser, Y. Lihanova, R. Frank, H. G. Jahnke, S. Billig, B. Abel, N. Strater, J. Matysik and W. Zimmermann, ChemSusChem, 2022, 15, e202101062 CAS.
- S. Kalathil, M. Miller and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202211057 CrossRef CAS PubMed.
- B. Eiamthong, P. Meesawat, T. Wongsatit, J. Jitdee, R. Sangsri, M. Patchsung, K. Aphicho, S. Suraritdechachai, N. Huguenin-Dezot, S. Tang, W. Suginta, B. Paosawatyanyong, M. M. Babu, J. W. Chin, D. Pakotiprapha, W. Bhanthumnavin and C. Uttamapinant, Angew. Chem., Int. Ed., 2022, 61, e202203061 CAS.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef CAS.
- R. Wei, G. von Haugwitz, L. Pfaff, J. Mican, C. P. S. Badenhorst, W. Liu, G. Weber, H. P. Austin, D. Bednar, J. Damborsky and U. T. Bornscheuer, ACS Catal., 2022, 12, 3382–3396 CrossRef CAS PubMed.
- B. Zhu, Q. Ye, Y. Seo and N. Wei, Environ. Sci. Technol. Lett., 2022, 9, 650–657 CrossRef CAS.
- M. Zandieh and J. Liu, Angew. Chem., Int. Ed., 2022, 61, e202212013 CrossRef CAS.
- K. Saito, F. Eisenreich, T. Turel and Z. Tomovic, Angew. Chem., Int. Ed., 2022, 61, e202211806 CAS.
- H. C. Hayes and L. Y. P. Luk, Sci. Rep., 2023, 13, 1267 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, E. Pollet and L. Averous, Waste Manage., 2021, 132, 23–30 CrossRef CAS PubMed.
- M. Valle, M. Ximenis, X. Lopez de Pariza, J. M. W. Chan and H. Sardon, Angew. Chem., Int. Ed., 2022, 61, e202203043 CrossRef CAS.
- U. L. D. I. Kalana, P. P. Datta, R. S. Hewawasam, E. T. Kiesewetter and M. K. Kiesewetter, Polym. Chem., 2021, 12, 1458–1464 RSC.
- L. Mezzasalma, A. P. Dove and O. Coulembier, Eur. Polym. J., 2017, 95, 628–634 CrossRef CAS.
- G. W. Yang, Y. Wang, H. Qi, Y. Y. Zhang, X. F. Zhu, C. Lu, L. Yang and G. P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202210243 CAS.
- W. Li, K. Qian, Z. Yang, X. Ding, W. Tian and D. Chen, Appl. Catal., B, 2023, 327, 122451 CrossRef CAS.
- Q. Cao, H.-C. Dai, J.-H. He, C.-L. Wang, C. Zhou, X.-F. Cheng and J.-M. Lu, Appl. Catal., B, 2022, 318, 121828 CrossRef CAS.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri, J. Dilworth, J. Thomas, T. Xiao and P. Edwards, Nat. Catal., 2020, 3, 902–912 CrossRef CAS.
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- Y. Shi, P. Liu, X. Wu, H. Shi, H. Huang, H. Wang and S. Gao, Water Res., 2021, 195, 116980 CrossRef CAS PubMed.
- F. Li, X. Zhai, M. Yao and X. Bai, Environ. Pollut., 2022, 314, 120307 CrossRef CAS.
- F. Eisenreich, Angew. Chem., Int. Ed., 2023, 62, e202301303 CrossRef CAS PubMed.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- Y. Chen, X. Deng, J. Wen, J. Zhu and Z. Bian, Appl. Catal., B, 2019, 258, 118024 CrossRef CAS.
- X. Deng, Y. Chen, J. Wen, Y. Xu, J. Zhu and Z. Bian, Sci. Bull., 2020, 65, 105–112 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- Y. Chen, M. Xu, J. Wen, Y. Wan, Q. Zhao, X. Cao, Y. Ding, Z. L. Wang, H. Li and Z. Bian, Nat. Sustain., 2021, 4, 618–626 CrossRef.
- Y. Chen, S. Guan, H. Ge, X. Chen, Z. Xu, Y. Yue, H. Yamashita, H. Yu, H. Li and Z. Bian, Angew. Chem., Int. Ed., 2022, 61, e202213640 CAS.
- Y. Zhang, H. Liu, F. Gao, X. Tan, Y. Cai, B. Hu, Q. Huang, M. Fang and X. Wang, EnergyChem, 2022, 4, 100078 CrossRef CAS.
- Z. Ouyang, Y. Yang, C. Zhang, S. Zhu, L. Qin, W. Wang, D. He, Y. Zhou, H. Luo and F. Qin, J. Mater. Chem. A, 2021, 9, 13402–13441 RSC.
- Y. Lee, C. Boyer and M. S. Kwon, Nat. Rev. Mater., 2021, 7, 74–75 CrossRef.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, e1901997 CrossRef.
- J. Chen, J. Wu, P. C. Sherrell, J. Chen, H. Wang, W. X. Zhang and J. Yang, Adv. Sci., 2022, 9, e2103764 CrossRef.
- L. Zhu, S. Zhao, T. B. Bittar, A. Stubbins and D. Li, J. Hazard. Mater., 2020, 383, 121065 CrossRef CAS.
- Z. Liu, Y. Zhu, S. Lv, Y. Shi, S. Dong, D. Yan, X. Zhu, R. Peng, A. A. Keller and Y. Huang, Environ. Sci. Technol. Lett., 2021, 9, 50–56 CrossRef.
- X. Zhao and F. You, Environ. Sci. Technol., 2023, 57, 6506–6519 CrossRef CAS.
- K. Zhu, H. Jia, Y. Sun, Y. Dai, C. Zhang, X. Guo, T. Wang and L. Zhu, Water Res., 2020, 173, 115564 CrossRef CAS PubMed.
- R. Zheng, D. Yang, Y. Chen, Z. Bian and H. Li, J. Environ. Sci., 2022 DOI:10.1016/j.jes.2022.01.042.
- Q. Zhao, Y. Ren, L. Huang, Y. Chen and Z. Bian, Catal. Today, 2023, 410, 309–316 CrossRef CAS.
- M. C. Ariza-Tarazona, J. F. Villarreal-Chiu, J. M. Hernandez-Lopez, J. Rivera De la Rosa, V. Barbieri, C. Siligardi and E. I. Cedillo-Gonzalez, J. Hazard. Mater., 2020, 395, 122632 CrossRef CAS.
- A. Uheida, H. G. Mejia, M. Abdel-Rehim, W. Hamd and J. Dutta, J. Hazard. Mater., 2021, 406, 124299 CrossRef CAS.
- H. Ma, Q. Luo, J. Zhou, N. Reddy, H. Wang and Y. Zhang, Chem. Eng. J., 2023, 454, 140377 CrossRef CAS.
- M. Du, Y. Zhang, S. Kang, X. Guo, Y. Ma, M. Xing, Y. Zhu, Y. Chai and B. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- S. Zhang, H. Li, L. Wang, J. Liu, G. Liang, K. Davey, J. Ran and S. Z. Qiao, J. Am. Chem. Soc., 2023, 145, 6410–6419 CrossRef CAS.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS.
- M. Han, S. Zhu, C. Xia and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- M. Wang, J. Wen, Y. Huang and P. Hu, ChemSusChem, 2021, 14, 5049–5056 CrossRef CAS.
- R. Cao, M. Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- J. Qin, Y. Dou, J. Zhou, V. M. Candelario, H. R. Andersen, C. Hélix-Nielsen and W. Zhang, Adv. Funct. Mater., 2023, 33, 2214839 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro, B. Ferrer and H. Garcia, Chem. Rev., 2022, 123, 445–490 CrossRef PubMed.
- P. Samal, J. R. K. Samal, E. Gubbins, P. Vroemen, C. van Blitterswijk, R. Truckenmuller and S. Giselbrecht, Adv. Mater., 2022, 34, e2200687 CrossRef.
- J. Kou, C. Lu, J. Wang, Y. Chen, Z. Xu and R. S. Varma, Chem. Rev., 2017, 117, 1445–1514 CrossRef CAS.
- C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem Catal., 2021, 2, 52–83 CrossRef.
- X. Li, W. Wang, F. Dong, Z. Zhang, L. Han, X. Luo, J. Huang, Z. Feng, Z. Chen, G. Jia and T. Zhang, ACS Catal., 2021, 11, 4739–4769 CrossRef CAS.
- Y. Liu, X. Wang, Q. Li, T. Yan, X. Lou, C. Zhang, M. Cao, L. Zhang, T. K. Sham, Q. Zhang, L. He and J. Chen, Adv. Funct. Mater., 2022, 33, 2210283 CrossRef.
- J. Wang, X. Li, T. Zhang, X. Qian, T. Wang and Y. Zhao, Appl. Catal., B, 2023, 332, 122744 CrossRef CAS.
- H. Liu, Y. Feng, J. Shao, Y. Chen, Z. L. Wang, H. Li, X. Chen and Z. Bian, Nano Energy, 2020, 70, 104499 CrossRef CAS.
- J. Wen, L. Ling, Y. Chen and Z. Bian, Chin. J. Catal., 2020, 41, 1674–1681 CrossRef CAS.
- F. Liu, X. Zhuang, Z. Du, Y. Dan, Y. Huang and L. Jiang, Appl. Catal., B, 2022, 318, 121897 CrossRef CAS.
- J. Ye, Y. Chen, C. Gao, C. Wang, A. Hu, G. Dong, Z. Chen, S. Zhou and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202213244 CAS.
- J. Kim, J. Jang, T. Hilberath, F. Hollmann and C. B. Park, Nat. Synth., 2022, 1, 776–786 CrossRef.
- X. Fang, N. Tian, W. Hu, Y. Qing, H. Wang, X. Gao, Y. Qin and J. Sun, Adv. Funct. Mater., 2022, 32, 2208623 CrossRef CAS.
- R. Li and Z. An, Angew. Chem., Int. Ed., 2020, 59, 22258–22264 CrossRef CAS.
- K. Jiang, S. Han, M. Ma, L. Zhang, Y. Zhao and M. Chen, J. Am. Chem. Soc., 2020, 142, 7108–7115 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |