
Highly selective semiconductor photocatalysis for CO2 reduction
Shan
Yao
ab,
Jiaqing
He
ab,
Feng
Gao
ab,
Haowei
Wang
ab,
Jiahui
Lin
ab,
Yang
Bai
ab,
Jingyun
Fang
c,
Feng
Zhu
 e,
Feng
Huang
abd and
Mengye
Wang
*ab
e,
Feng
Huang
abd and
Mengye
Wang
*ab
aSchool of Materials, Sun Yat-Sen University, Shenzhen 518107, China. E-mail: wangmengye@mail.sysu.edu.cn
bState Key Laboratory of Optoelectronic Materials and Technologies, Sun Yat-Sen University, Guangzhou 510275, China
cGuangdong Provincial Key Laboratory of Environmental Pollution Control and Remediation Technology, School of Environmental Science and Engineering, Sun Yat-Sen University, Guangzhou 510275, China
dGanjiang Innovation Academy, Chinese Academy of Sciences, Ganzhou 341000, PR China
eShenzhen Shiage Electronic Technology Co., Ltd, Shenzhen 518107, China
First published on 10th January 2023
Abstract
Over the past few decades, photocatalytic CO2 reduction has remained a prominent and growing research field due to the efficient conversion of CO2 to value-added chemicals. Among the various photocatalytic performances, product selectivity has garnered considerable attention, which is the focus of this review. Herein, we first introduce the general background of photocatalytic CO2 reduction, then according to the sequence of the entire reaction process, the adsorption and activation of reactants, the formation and stabilization of intermediates, and the desorption of products are summarized. After introducing each of the above steps that could mediate the final products, several modification techniques to improve the product selectivity are highlighted, including noble metal decoration, metal and non-metal doping, vacancy engineering, facet engineering, composite construction, and hydroxyl modification. Finally, current challenges and opportunities of interest in this rich field are discussed.
 Shan Yao | Shan Yao received her B.E. in polymer materials and engineering from Guangdong University of Technology. She is currently pursuing her PhD under the supervision of Prof. Mengye Wang in the School of Materials at Sun Yat-Sen University. Her current research focuses on advanced materials for photocatalysis and environmental applications. |
 Jiaqing He | Jiaqing He received her B.S. degree from Sun Yat-sen University. She is currently pursuing her PhD under the supervision of Prof. Mengye Wang in Sun Yat-sen University. Her current research focuses on advanced materials for piezocatalysis and environmental applications. |
 Feng Gao | Feng Gao received his B.S. degree from Sun Yat-Sen University. He is currently pursuing his PhD under the supervision of Prof. Mengye Wang in Sun Yat-Sen University. His current research focuses on advanced materials for electrocatalysis and energy-related applications. |
 Haowei Wang | Haowei Wang is pursuing his master's degree in the school of materials at Sun Yat-Sen University. He obtained his B.E. in polymer materials and engineering from Wuhan University of Technology in 2021. His current research focuses on advanced materials for electrocatalysis. |
 Jiahui Lin | Jiahui Lin is pursuing her PhD in the School of Materials at Sun Yat-Sen University. She obtained her B.S. degree from Sun Yat-Sen University in 2022. Her current research focuses on advanced materials for electrocatalysis and environmental applications. |
 Mengye Wang | Mengye Wang is an Associate Professor in the School of Materials at Sun Yat-Sen University. She received her PhD in Physical Chemistry from Xiamen University. Her research interests include advanced materials for environmental and energy-related applications. |
10th anniversary statementJournal of Materials Chemistry A is one of the most important journals that concentrates on material applications in energy and sustainability and covers a wide range of specific content. In past studies, we focused on advanced materials for photocatalysis, piezocatalysis and electrocatalysis, including hydrogen production, nitrogen fixation, CO2 reduction and degradation of organic pollutants. Our research interests include designing efficient catalysts via morphology control, vacancy engineering, size adjustment and modification. The source of synthetic reagents can be either chemical reagents or solid waste. Making solid waste into catalysts can alleviate the environmental problems caused by waste residue disposal sites. Moreover, we also concentrate on the catalytic mechanism using advanced spectroscopic techniques. On the occasion of the tenth anniversary of Journal of Materials Chemistry A, we sincerely celebrate the 10th anniversary of Journal of Materials Chemistry A and look forward to making further progress with the development of the journal in the future. |
1. Introduction
The industrial revolution has triggered the rapid development of science and technology,1–10 however, the massive consumption of fossil fuels has caused a sharp increase in CO2 concentration.11–17 As of December 2020, CO2 concentration in the atmosphere has reached 414.02 ppm, far higher than 270 ppm in the early 1800s, which is considerably higher than the safe value of atmospheric CO2 concentration (i.e., 350 ppm).18 As a consequence, a series of natural disasters, such as desertification, global warming and ocean acidification, have occurred frequently.19–21 Therefore, it has become an urgent issue to both alleviate the pollution caused by CO2 and replace fossil fuels with clean and renewable energy sources.22–28The photocatalytic conversion of CO2 into hydrocarbon fuels of small molecular weight, such as CH4, CH3OH and C2H5OH, might kill two birds with one stone in terms of protecting the environment and saving energy.29–32 In the past several decades, tremendous efforts have been made toward CO2 reduction initiated by solar light,33–38 and thus far, the activity of photocatalytic CO2 reduction has greatly improved. Although the research and reports on the regulation of photocatalytic activity have been quite thorough, CO2 reduction still has an obvious drawback, which is low selectivity and can be mainly attributed to the following reasons. First, as the energy barrier of CO2 reduction to target products is comparable to that of hydrogen evolution (eqn (1)–(7) at pH 7 in aqueous solution), the side reaction (i.e., hydrogen generation) is inevitable.39,40
| CO2 + 2H+ + 2e → CO + H2O E0 = −0.52 V vs. NHE | (1) |
| CO2 + 2H+ + 2e → HCOOH E0 = −0.61 V vs. NHE | (2) |
| CO2 + 4H+ + 4e → HCHO + H2O E0 = −0.51 V vs. NHE | (3) |
| CO2 + 8H+ + 8e → CH4 + 2H2O E0 = −0.24 V vs. NHE | (4) |
| CO2 + 6H+ + 6e → CH3OH + H2O E0 = −0.38 V vs. NHE | (5) |
| CO2 + 12H+ + 12e → C2H4 + 4H2O E0 = −0.34 V vs. NHE | (6) |
| 2H+ + 2e → H2 E0 = −0.42 V vs. NHE | (7) |
Thus, CO2 reduction exhibits limited selectivity in aqueous solutions due to the critical challenge of overcoming the competition with the hydrogen evolution reaction (HER). In addition, the reduction potentials of converting CO2 to different products are similar (eqn (1)–(6)).41 This phenomenon results in an undesirable mixture of products, which is hard to separate and utilize. Therefore, the improvement in selectivity is of key importance for photocatalytic CO2 reduction.42–45
This review aims to summarize recent impressive developments in the highly selective photocatalytic CO2 reduction. After a brief discussion that motivates the research on high product selectivity, the reaction mechanism of photocatalytic CO2 reduction is introduced to better understand the obstacle of reaction selectivity and the specific steps involved in the entire photocatalytic procedure. Then, the promotion of selective reaction at different reaction stages is summarized, including the adsorption and activation of reactants (i.e., CO2 adsorption and H2 evolution inhibition, electron supply and others), the formation and stabilization of intermediates (i.e., the formation energy of crucial intermediates and stability of intermediates), and the desorption of products. Particularly, modification methods of photocatalysts to boost selective reactions are discussed, including noble metal decoration, metal and non-metal doping, vacancy engineering, facet engineering, composite construction, hydroxyl and ion modification and other decoration techniques (Fig. 1). Lastly, a perspective on the challenges and future research directions of photocatalytic CO2 reduction possessing enhanced photocatalytic selectivity is proposed.
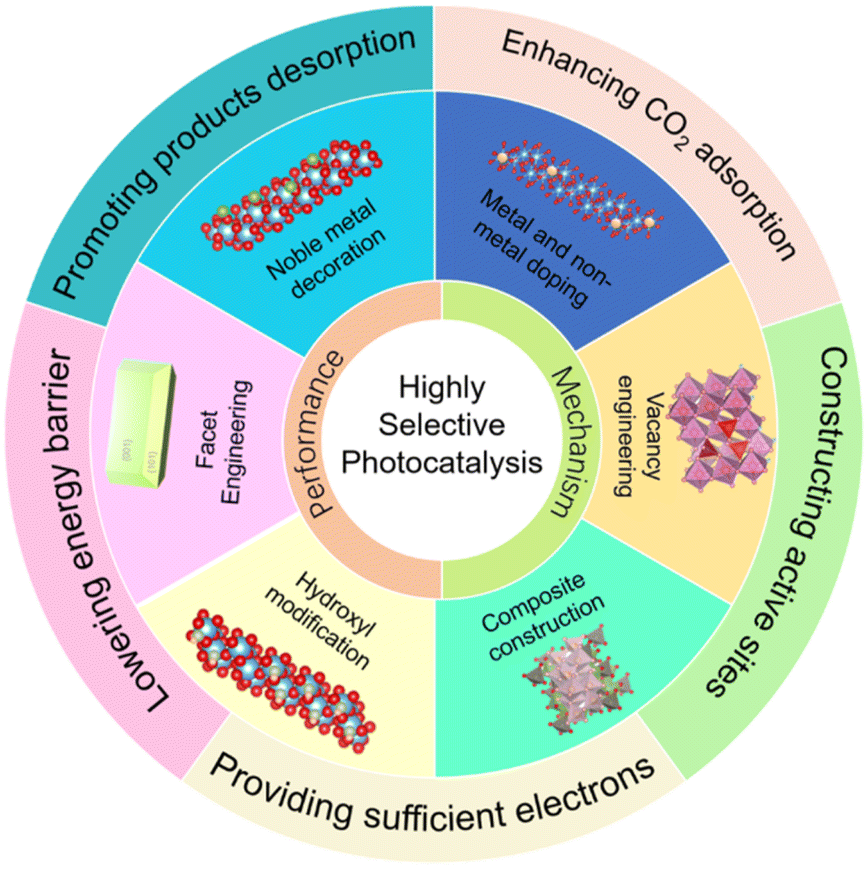 | ||
| Fig. 1 Schematic illustration of photocatalyst modification and mechanisms in this review. | ||
2. Mechanism of photocatalytic CO2 reduction
Specific reaction processes of photocatalytic CO2 reduction are displayed in Fig. 2. Under light irradiation, the semiconductor photocatalyst absorbs photons. If the photon energy is equal to or larger than the band gap (Eg) of the photocatalyst, electrons and holes of the semiconductor are generated. The photogenerated electrons and holes then migrate to the surface of the photocatalyst.46 Photogenerated electrons are capable of reduction reactions that reduce CO2 on the surface of the semiconductor be reduced, while the holes oxidize H2O to O2. Photo-excited electrons and holes are prone to recombine both during the migration to photocatalyst surface and after reaching the surface, which significantly reduces the photocatalytic efficiency. Some parameters affect the reaction selectivity, including the reduction potentials of CO2 to products, the adsorption and desorption of different substances (i.e., CO2, intermediates and products), and the utilization efficiency of photogenerated electrons.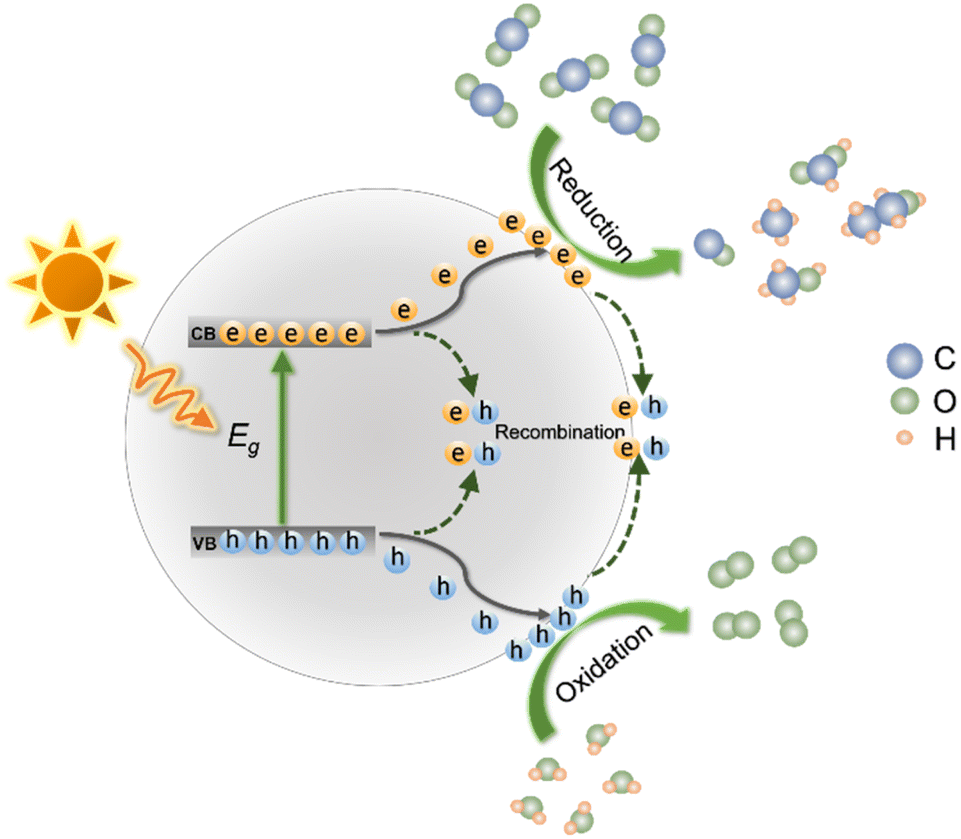 | ||
| Fig. 2 Schematic illustration of photocatalytic CO2 reduction. | ||
The standard reduction potentials of CO2 are summarized in eqn (1)–(6) and eqn (8).47 As shown in eqn (8), CO2˙− formation through the single-electron reduction of CO2 is unfavorable due to its very negative redox potential (i.e., −1.90 V vs. NHE).
| CO2 + e− → CO2˙− E0 = −1.90 V vs. NHE | (8) |
Therefore, multi-proton-assisted CO2 reduction reactions are prone to occur, possessing a relatively low thermodynamic barrier and bypassing the formation of CO2˙− (eqn (1)–(6)).48 From a thermodynamic point of view, the potentials of CO2 reduction (eqn (1)–(6)) are comparable to the potentials of hydrogen evolution (eqn (7)), which leads to a large amount of H2 as the by-product.49 According to the less negative redox potentials, it seems that the photoconversion of CO2 to CH3OH and CH4 is more favorable than the H2 evolution ([eqn (4)], [eqn (5)] and [eqn (7)]). Nevertheless, the requirement of more electrons to accomplish highly selective formation of CH3OH and/or CH4 rather than H2 is a great challenge.50 Therefore, in order to realize the highly selective photoconversion of CO2, the photocatalysts should be carefully designed to suppress H2 evolution. In addition to the interference of H2, the small differences between the thermodynamic potentials of different CO2 reduction products also make it tough to obtain single desirable products.40
Typically, the photocatalytic CO2 reduction process involves three major steps: (i) the chemical capture and adsorption of CO2 on the surface of the photocatalyst; (ii) activation and breaking of C–O bonds and the formation of C–H bonds via electron transfer and proton migration; (iii) the configuration rearrangement of products and desorption from the photocatalysts.51 Each reaction step can be regulated by modifying photocatalysts or reaction conditions. In step (i), if the CO2 adsorption is enhanced and H2O adsorption is inhibited, the efficiency of CO2 reduction will be largely increased and the H2 evolution will be reduced.52 Similarly, in step (ii), to obtain the products such as CH4, CH3OH and C2H4, a mass of electrons is needed. Thus, a sufficient electron supply and proper suppression of electron–hole recombination are beneficial for their generation.53 During the configuration rearrangement and intermediate conversion process, the formation and stabilization of crucial intermediates determine the direction of the subsequent reactions, promoting the selective generation of the target products. Finally, the easy desorption of obtained products can further enhance the selectivity.
3. Typical methods to enhance the selectivity and mechanism
Diverse strategies, including noble metal decoration, metal and non-metal doping, vacancy engineering, the exposure of highly active crystal facets, construction of complex materials, surface modification, morphology control and other methods, have been explored to manipulate the selectivity of the photocatalytic CO2 reduction. These techniques significantly increase the selectivity in terms of kinetics, adsorption and desorption capacity, electron supply, and intermediate stability. Diverse modification methods will affect different steps of CO2 reduction. Oxygen vacancies, noble metal and non-metal particles can not only affect the adsorption of CO2 but can also act as electron sinks, providing sufficient electrons for products. Composite construction, which can improve the CO2 capture capability, also facilitates efficient photo-excited electron transfer and accumulation. Morphology control and facet engineering mainly affect the adsorption and desorption process during CO2 reduction. We will introduce typical methods in the following sections. However, most of the work only briefly explains the reasons for the improvement of selectivity, rarely involving the essential reasons. The modified highly selective photocatalysts are listed in Table 1.| Photocatalyst | Light source | Experimental conditions | Main products and selectivity | Ref. |
|---|---|---|---|---|
| Ag/CaTiO3 | 100 W Hg lamp | 0.3 g of catalysts, NaHCO3 aqueous solution (1.0 M) | CO (180 μmol g−1 h−1), 94% | 54 |
| TiO2–Pd@Au | 300 W Xe lamp | 15 mg of catalysts, CO2 and 5 mL H2O | CH4 (48.2 μmol g−1 h−1), 93.5% | 55 |
| (Pt/TiO2)@rGO | 300 W Xe lamp | Certain amounts of catalysts, CO2 and 2 mL H2O | CH4 (41.3 μmol g−1 h−1), 99.1% | 56 |
| Pt@Ag–TiO2 | 350 W Xe lamp | Certain amounts of catalysts, CO2 and Na2SO4 aqueous solution (0.5 M) | CH4 (160.3 μmol g−1 h−1), 87.9% | 57 |
| Pt/TiO2 | 300 W Xe lamp | 50 mg of catalysts, CO2 and 50 mL H2O | CH4 (150.04 μmol g−1 h−1), nearly 100% | 58 |
| N-doped C dot/CoAl-LDH/C3N4 | 300 W Xe lamp | 50 mg of catalysts, CO2 and 300 μL H2O | CH4 (25.69 μmol g−1 h−1), 99% | 59 |
| N–TiO2 | 200 W Hg lamp | Gas mixture of CO2 and H2 with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
CO (56.30 μmol g−1 h−1), 96.3% | 60 |
| Cu–TiO2 | 300 W Xe lamp | 60 mg of catalysts, 1.60 g NaHCO3 and 5 mL H2SO4 solution (5.0 M) | CH4 (150.9 μmol g−1 h−1), 85% | 61 |
| WO3−x | UV-Vis-NIR light | 5 mg of catalysts, CO2 and 0.2 mL H2O | C2H4 (61.6 μmol g−1 h−1), 89.3% | 62 |
| BiMoO6 | 300 W Xe lamp | 50 mg of catalysts, 1.50 g NaHCO3 and 5 mL H2SO4 (4 M) | CH4 (2.01 μmol g−1 h−1), 96.7% | 63 |
| Defective CeO2 | 300 W Xe lamp | 50 mg of catalysts, CO2 and 0.2 mL H2O | CO (7 μmol g−1 h−1), nearly 100% | 64 |
| g-C3N4/FeWO4 | Visible light (λ > 420 nm) | 40 mg of catalysts, CO2 and H2O | CO (6 μmol g−1 h−1), 99% | 65 |
| NiAl-LDH/Ti3C2 | 300 W Xe lamp | 100 mg of catalysts, CO2 and 0.4 mL H2O | CO (11.82 μmol g−1 h−1), 92% | 66 |
| Ag-loaded Ga2O3/ZnGa2O4 | 400 W Hg lamp | 1 g of catalysts, CO2 and 1 L H2O | CO, nearly 100% | 67 |
| Pt/HAP/TiO2 | 300 W Xe lamp | 20 mg of catalysts, CO2 and 40 mL H2O | CH4 (4.64 μmol g−1 h−1), 99.1% | 68 |
| C3N4/Pd9Cu1Hx | Visible light | 15 mg of catalysts, CO2 and H2O | CH4 (1.20 μmol g−1 h−1), nearly 100% | 69 |
| Cl−/Bi2WO6 | 300 W Xe lamp | 20 mg of catalysts, CO2 and H2O | CH4 (3.21 μmol g−1 h−1), 94.98% | 70 |
| Pt@h-BN | 300 W Xe lamp | 10 mg of catalysts, CO2 and 0.5 mL H2O | CH4 (184.7 μmol g(Pt)−1 h−1), 99.1% | 71 |
| Ultrathin Bi4O5Br2 | 300 W Xe lamp | 20 mg of catalysts, CO2 and H2O | CO (31.565 μmol g−1 h−1), 99.5% | 72 |
| Au–Pd alloying loaded TiO2 | UV light | 10 mg of catalysts, CO2 and H2O | Hydrocarbon fuels (CH4, C2H4, and C2H6) (25.06 μmol g−1 h−1), 85% | 73 |
| V-defective BiVO4 | 300 W Xe lamp | 100 mg of catalysts, CO2 and H2O | CH3OH (398.3 μmol g−1 h−1) | 74 |
| Defective C3N4 | 350 W Xe lamp | 100 mg of catalysts, CO2 and H2O | CH4, CH3OH, and CH3CH2OH (12.07 μmol g−1 h−1), 91.5% | 75 |
| W-doped g-C3N4 | 300 W Xe lamp | 5 mg of catalysts, CO2 and H2O | CH4 and C2H4 (11.91μmol g−1 h−1), 83% | 76 |
| CuxIn5S8–CuySe | 300 W Xe lamp | 10 mg of catalysts, CO2 and H2O | CH3OH (5.25 μmol g−1 h−1), about 100% | 77 |
| Microwave-synthesised carbon-dots | 300 W Xe lamp | 10 mg of catalysts, CO2 and H2O | CH3OH (13.9 μmol g−1 h−1), 99.6% | 78 |
| C3N4-supported CoS | 300 W Xe lamp | 10 mg of catalysts, CO2 and H2O | CH3OH (97.3 μmol g−1 h−1), 87.2% | 79 |
3.1. Adsorption and activation of reactants
For example, nearly 100% selective CO generation was achieved on the surface of spindle-like oxygen-vacancy rich (VO-rich) Pt–Ga2O3 (Fig. 3a).81 The oxygen vacancies served as the main sites for CO2 adsorption.81 Meanwhile, the hydrogen formed on Pt nanoparticles in the process of photocatalytic water splitting could quickly reduce the adsorbed CO2.81 Besides Pt, other noble metals also showed similar effects. After anchoring Au–Pd alloy on the {101}n facets of TiO2, the Au–Pd alloy provided abundant sites for CO2 adsorption and activation.73 Remarkably, the optimal sample achieved a high selectivity of 85% for hydrocarbons (71%: CH4, 14%: C2H4 and C2H6, Fig. 3b).73
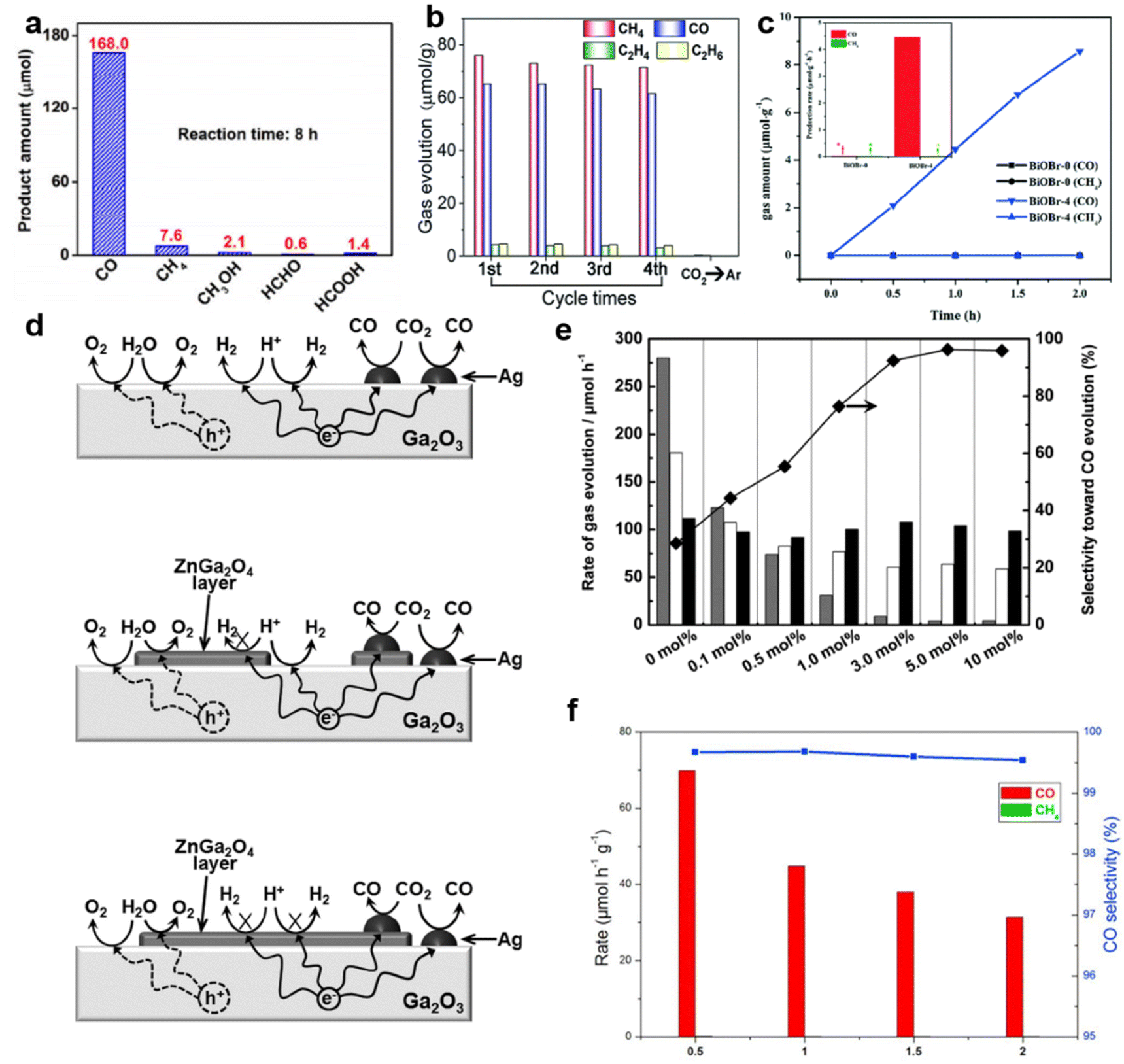 | ||
| Fig. 3 (a) The amount of products over VO-enriched Pt–Ga2O3 after irradiation for 8 h. Reproduced with permission from ref. 81 copyright 2018 Springer. (b) Photocatalytic yield and stability of Au–Pd-alloy-decorated TiO2. Reproduced with permission from ref. 73. copyright 2019 Royal Society of Chemistry. (c) Production rates of CH4 and CO over BiOBr photocatalysts under Xe light irradiation. Reproduced with permission from ref. 83 copyright 2017 Royal Society of Chemistry. (d) A proposed mechanism for the photocatalytic conversion of CO2 in H2O over Ag-loaded Ga2O3, Ag-loaded Zn-modified Ga2O3 with a low Zn content, and Ag-loaded Zn-modified Ga2O3 with a high Zn content. (e) Evolution rates of CO (black), O2 (white), and H2 (gray) in the photocatalytic conversion of CO2 over Ag-loaded ZnGa2O4-modified Ga2O3 containing different amounts of ZnGa2O4. Reproduced with permission from ref. 67. Copyright 2016 Royal Society of Chemistry. (f) Evolution rates and selectivity over Bi4O5Br2 and Bi4O5Br2-UN under UV-vis illumination. Reproduced with permission from ref. 72. Copyright 2017 Elsevier. | ||
Some special groups are also capable of the adsorption and activation of CO2. For instance, the –OH groups on the Cu2O surface facilitated the selective catalytic CO2 reduction by suppressing the hydrogen evolution.84 Besides, in the noble-metal-free SiO2–TiO2 system, the enhanced CO2 photoreduction selectivity was assigned to the rational hydrophobic modification of the TiO2–SiO2 surface by replacing Si–OH with hydrophobic Si–F bonds.85 This kind of modification changed the hydrophilicity and hydrophobicity of the photocatalyst surface and thus mediated the reaction process.85 This improved selectivity was attributed to the efficient CO2 adsorption, triggering efficient CO2 photoreduction.85
Different crystal faces possess different CO2 adsorption capacities.86 ZnO nanomaterials with a large ratio of {0001} facets could enhance the CO production selectivity and the exposed facets were terminated with a high density of oxygen atoms.87 Therefore, oxygen vacancies were prone to form on the surface of ZnO.87 These vacancies could preferentially capture CO2 molecules and work as reduction sites for CO2.87 As a consequence, the CO molecules could be produced as the main products.87 In addition, BiOBr nanosheets exposing {001} facets were successfully synthesized in the presence of nitric acid.83 Compared to the BiOBr nanosheets prepared in the absence of nitric acid (BiOBr-0), the {001} facets-dominated BiOBr nanosheets exhibited more efficient CO2 absorption and activation, selectively converting CO2 to CO (Fig. 3c).83
The adsorption and activation capacity can also be improved by changing the structure of the material. Typically, g-C3N4-based composite catalysts display strong CO2 capture ability, which promotes the generation of CO rather than H2.88
The selectivity enhancement of CO production also appeared in other semiconductor composites, such as LDH/Ti3C2 and Ga2O3/ZnGa2O4.46,67 Taking the Ga2O3/ZnGa2O4 catalyst as an example, the ZnGa2O4 layer could suppress the reduction of H+.67 The proposed mechanism is displayed in Fig. 3d. Since a large number of active sites on the surface of Ga2O3 could capture and reduce H+, the main reaction on the surface of Ag–Ga2O3 was hydrogen production (Fig. 3d).67 After the growth of the ZnGa2O4 layer on the surface of Ga2O3, it blocked the active sites that are conducive to hydrogen evolution. Therefore, with the amount of ZnGa2O4 increasing from 0.1 to 10.0 mol%, the generation of H2 was significantly suppressed. Finally, CO generation with nearly 100% selectivity was achieved over Ga2O3/ZnGa2O4 heterostructures (Fig. 3e).67 Crafting ultrathin two-dimensional semiconductor nanomaterials is another popular technique for achieving high photocatalytic selectivity.89–92 Bai found that compared with the bulk counterpart, the ultrathin Bi4O5Br2 (Bi4O5Br2-UN) exhibited increased CO generation of over 99.5% through an enhanced CO2 adsorption capacity (Fig. 3f).72
In summary, by constructing composites, introducing metal sites and constructing oxygen vacancies, the adsorption and activation capabilities of photocatalysts in the first step of the CO2 reduction reaction can be significantly improved. Therefore, the target reaction occurs instead of the competitive hydrogen evolution. However, in previous reports about the first step of the CO2 reduction reaction, there is a lack of the selective generation of different carbon reduction products. In addition, how CO2 adsorption mediates carbon products needs to be further explored.
Pan et al. reported that after decoration by Pt nanoparticles, LaPO4 reached a 5.6 times enhancement in CH4 yield compared to pure LaPO4 (Fig. 4a).53 The selectivity of CH4 production increased from the original 58.6% to 100% when the amount of modified Pt nanoparticles increased from 1 wt% to 3 wt% (Fig. 4a).53 In this research, Pt nanoparticles functioned as the photogenerated electron sink, which thus accelerated the eight-electron reduction of CO2 to CH4.53 This mechanism was proved in another report, where Pt/Cu2O nanoparticles trapped sufficient photogenerated electrons and ensured the multielectron photocatalytic reactions to form CH4 (Fig. 4b–d).97 It is noteworthy that the electron sink effect of Pt also exists in other catalysts. The selectivity of CH4 formation was improved when Pt@Ag core@shell structures were decorated on TiO2 nanoparticles (Fig. 4e).57 The Pt core served as a sink for photogenerated electrons, and the Ag shell suppressed the competitive photocatalytic water-splitting process. By tuning the ratio of Pt to Ag, 1.95 wt.‰ Pt@Ag1.0–TiO2 achieved the outstanding photocatalytic performance with a CH4 formation rate of 160.3 mmol g−1 h−1 and a CO2 conversion selectivity of 87.90%.57
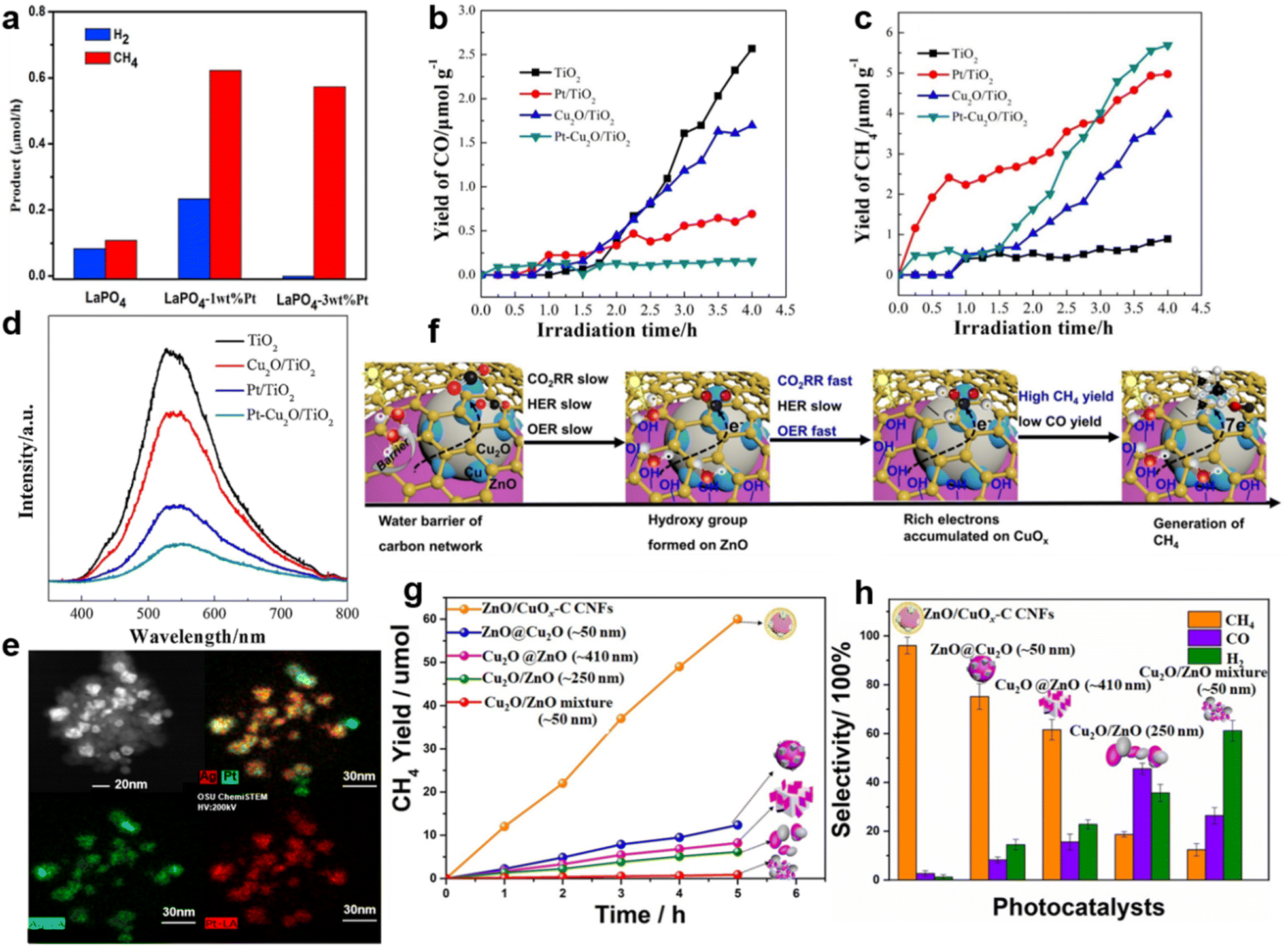 | ||
| Fig. 4 (a) Generation rates of CH4 and H2 over the LaPO4 and LaPO4–Pt samples. Reproduced with permission from ref. 98 copyright 2015 Elsevier. (b) CO yields, (c) CH4 yields and (d) PL spectra of TiO2, Cu2O/TiO2, Pt/TiO2 and Pt–Cu2O/TiO2. Reproduced with permission from ref. 97 copyright 2017 Elsevier. (e) STEM mapping images of Pt@Ag1.0–TiO2. Reproduced with permission from ref. 57 copyright 2018 Elsevier. (f) The proposed photocatalytic mechanism of highly selective CH4 production over ZnO/CuOx–C CNFs. (g) The CH4 yield and (h) the product selectivity of ZnO/CuOx–C CNFs, ZnO@CuO, Cu2O@ZnO, ZnO/Cu2O and Cu2O/ZnO. Reproduced with permission from ref. 99 copyright 2021 Elsevier. | ||
Besides noble metals, doped common metals could also perform as electron traps and active sites for the highly selective CO2 photoreduction.104,105 For example, Cu doping effectively provided TiO2 with electron traps, leading to a higher methanol yield.93,106 The Cu sites of ZnO/CuOx–C (the abbreviation C stands for Cu doping) carbon nanofibers (CNFs), which worked as electron traps, could also generate enough electrons for CH4 formation (Fig. 4f).99 Among ZnO–Cu2O hybrid nanoparticles (ZnO@Cu2O), Cu2O cube–ZnO heterostructures (Cu2O@ZnO), Cu2O/ZnO nanocomposites (Cu2O/ZnO) and Cu2O/ZnO mixtures, ZnO/CuOx–C CNFs demonstrated the highest CH4 generation rate of 241.6 μmol h−1 g−1 with the selectivity of ∼96% (Fig. 4g and h).99 Besides the introduction of metallic Cu, highly selective CH4 synthesis was achieved by introducing metal Mg.100 CO2 photoreduction rates of pure TiO2 and TiO2 decorated by different amounts of Mg are shown in Fig. 5a. Mg–TiO2 promoted a large enhancement of electron trap sites, thereby ensuring sufficient electron supply for the selective reduction of CO2 to CH4.100 Moreover, the doping or co-doping of other metals, such as Mo and Ce, could improve the photocatalytic conversion selectivity of CO2 to high value-added products by increasing electron traps.107,108 However, not all metals have the function of electron sink. Due to high CO2 adsorption, In–TiO2 nanoparticles achieved the highest CO selectivity of 94.39% with In doping of 10% (Fig. 5b) but with the limited electron supply, it was unable to achieve a high CH4 yield.101 Therefore, we need to select the appropriate metal to modify the photocatalyst according to the designed product target.101
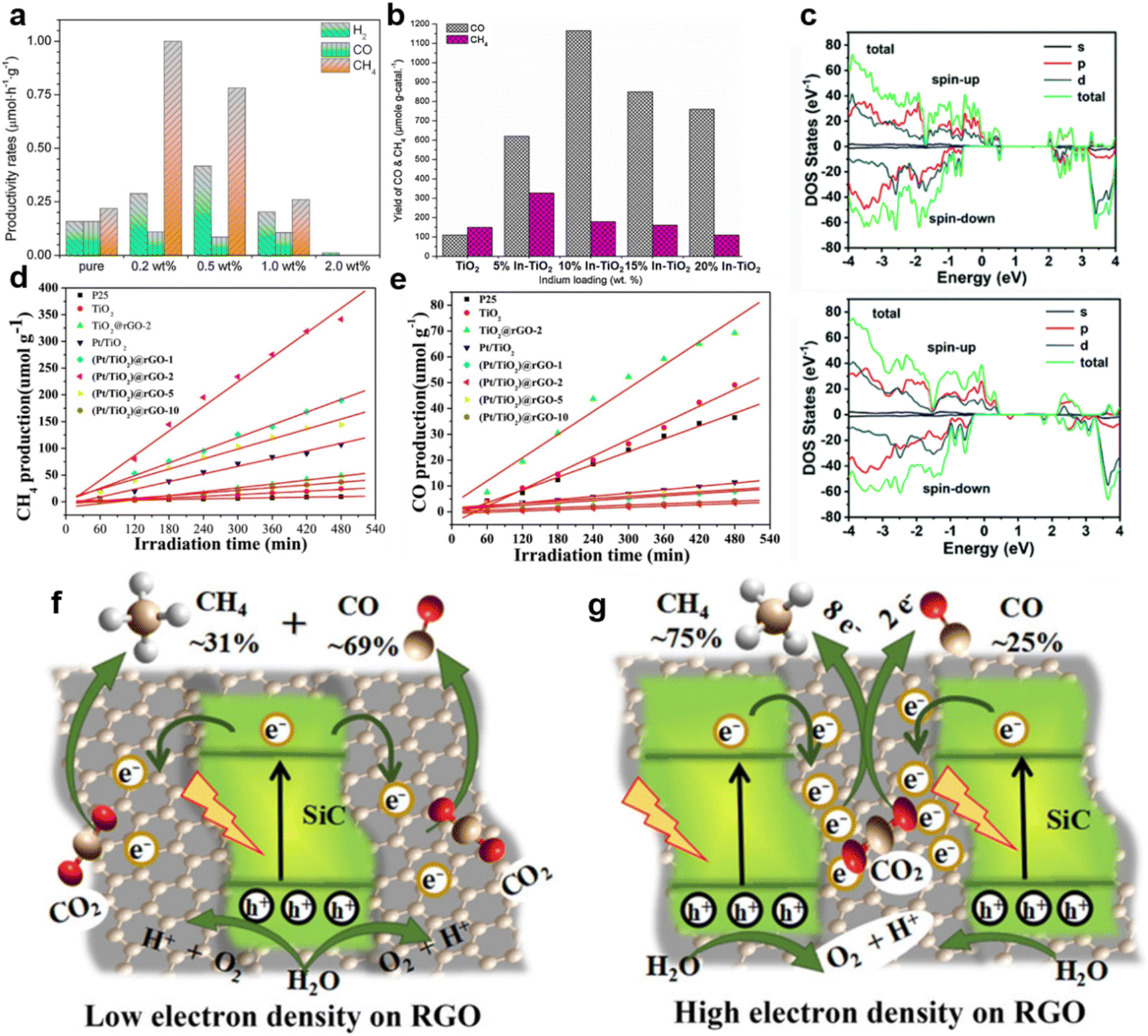 | ||
| Fig. 5 (a) The production rates of pure TiO2 and Mg–TiO2 containing different amounts of Mg doping, reproduced with permission from ref. 100 copyright 2014 Elsevier. (b) The yield of products of In–TiO2 with different amounts of In doping, reproduced with permission from ref. 101 copyright 2013 Elsevier. (c) Calculated total DOS (TDOS) and partial DOS (PDOS) plots of HC-NiCo-LDH without (upper) and with (lower) oxygen vacancies and Ni vacancies. Reproduced with permission from ref. 102 copyright 2018 Royal Society of Chemistry. (d) CH4 production and (e) CO production over TiO2, Pt/TiO2, TiO2@rGO-2 and (Pt/TiO2)@rGO-n catalysts. Reproduced with permission from ref. 56 copyright 2018 Elsevier. A schematic illustration of the electron density-dependent CH4 selective production over SiC/rGO heterojunctions with (f) low electron density and (g) high electron density on rGO. Reproduced with permission from ref. 103 copyright 2018 Wiley. | ||
The selective preparation of high-value-added products can be also achieved by creating oxygen vacancies.111 Therefore, defect-rich Bi6Mo2O15 sub-microwires with abundant oxygen vacancies, which could capture sufficient photo-generated electrons, are favorable for the photocatalytic reduction of CO2 to CH4.93 Oxygen vacancies and metal vacancies can act synergistically to jointly improve the reaction selectivity.102 Both oxygen and metal Ni vacancies were detected in NiCo-layered double hydroxide hollow nanocages (HC-NiCo-LDH), and the introduction of vacancies could create a new defect level in the middle of the band gap, causing a decreased band gap and enhanced charge transfer of HC-NiCo-LDH (Fig. 5c);102 this guaranteed enough photo-induced electrons to reduce CO2 molecules.102 Finally, the CH4 selectivity was increased from 8.92% to 62.66%.102
g-C3N4-based semiconductor composites could also selectively obtain CH4 or other fuels.112,113 Truc reported that compared with pure Cu2V2O7 (i.e., 0) and g-C3N4 (i.e., 0), the 50Cu2V2O7/50g-C3N4 photocatalysts possessed more active electrons in the CB of the g-C3N4.114 Enough electron supply of 50Cu2V2O7/50g-C3N4 accomplished the selective generation of CH4 (i.e., 1696 mmol g−1 cat. h−1).114 Additionally, in the g-C3N4/ZnO system, the electrons of ZnO recombined with the photo-generated holes of g-C3N4, thereby retaining the electrons with strong reduction capability in g-C3N4.115 Sufficient electrons in the CB of g-C3N4 boosted the conversion of CO2 to CH3OH.115 ZnO nanorod arrays@-carbon fiber (ZnO NA@CF) composites showed highly selective CH3OH production during the photocatalytic CO2 reduction.116 Owing to the interaction between ZnO nanocrystals and carbon fiber, photo-generated electrons could transfer from ZnO to the surface of the carbon fiber, thus preventing the recombination of electron–hole pairs.116 Photo-induced holes in the VB of ZnO reacted with water to produce O2 and H+, while CO2 molecules were selectively reduced to CH3OH by sufficient electrons on the carbon fiber.116 rGO is another appealing photocatalyst that can accumulate electrons of high energy to accelerate CH4 generation via an eight-electron catalytic process.117 Bare rGO is not active for the photocatalytic conversion of CO2 to CH4 (ref. 56) but rGO-wrapped Pt/TiO2 ((Pt/TiO2)@rGO) photocatalysts achieved excellent selectivity (i.e., 99.1%) of CH4 evolution (Fig. 5d and e).56 Pt nanoparticles in (Pt/TiO2)@rGO-n photocatalysts functioned as an accumulator for electron transfer in the TiO2–Pt–rGO ternary system.56 Due to the strong electron-withdrawing capacity of the rGO shell, the photogenerated electrons could further transfer from Pt to rGO.56 Thus, the photo-generated electrons were transferred from TiO2 to Pt and finally to the rGO shell (i.e., TiO2 → Pt → rGO).56 The photogenerated electrons accumulated on the rGO shell and subsequently reacted with the adsorbed CO2 to produce CH4.56 The photo-excited electron transfer and accumulation process was also observed in SiC/rGO composites.103 The electron density of the pure rGO surface was low.103 In the SiC/rGO heterojunctions, the SiC served as the source of photogenerated electrons, while the rGO helped to quickly transfer energetic electrons for subsequent CO2 reduction, resulting in a high CH4 selectivity.103 It is worth noting that by choosing a suitable ratio of SiC to RGO, a high electron density could be formed on the surface, thereby promoting the selective formation of CH4 (Fig. 5f and g).103 Additionally, crafting semiconductor composites might enhance photocatalytic selectivity via inducing oxygen vacancies.109 Devi reported that the considerable carbon reduction selectivity of the In2O3–2wt% rGO nanocomposite (i.e. CH4 generation rate of 953.72 μmol h−1 g−1 with the selectivity of ∼74%) could mainly be attributed to the induced oxygen vacancy defects by the addition of rGO (Fig. 6a–c).109
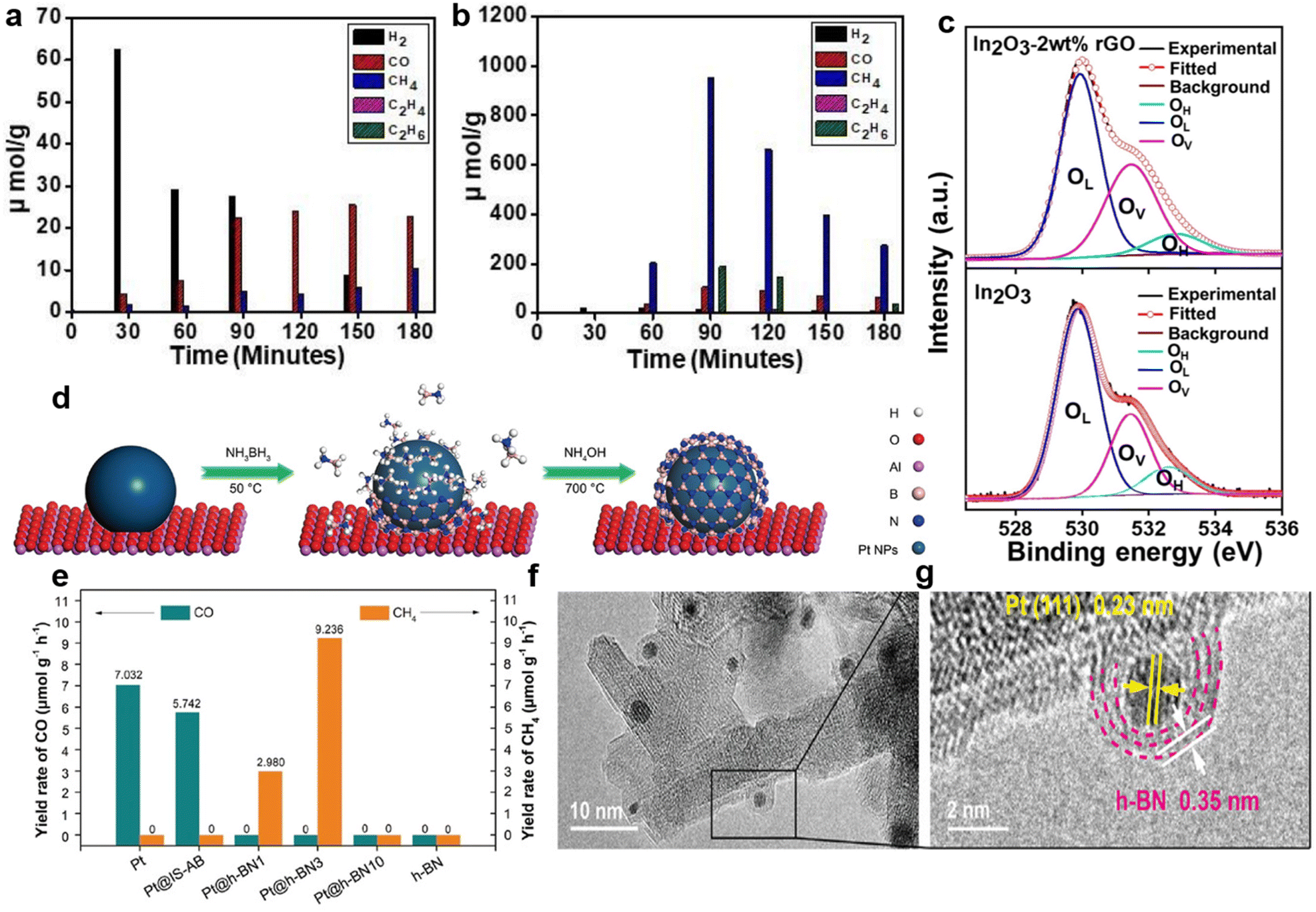 | ||
| Fig. 6 Generation efficiency of various products over (a) In2O3 and (b) In2O3-2 wt% rGO. (c) High-resolution XPS spectra of O 1s of In2O3-2 wt% rGO nanocomposites (upper) and In2O3 nanostructures (lower). Reproduced with permission from ref. 109 copyright 2021 Elsevier. (d) Schematic diagram of the synthesis process of Pt@IS-AB and Pt@h-BN series catalysts originating from Pt. (e) The photocatalytic CO2 conversion rate of Pt, Pt@IS-AB, Pt@h-BN series catalysts and pure h-BN. (f and g) HRTEM images of Pt@h-BN3. Reproduced with permission from ref. 110 copyright 2021 Wiley. | ||
The adjustment of the nanostructure and morphology of the materials can also change the accumulation of electrons, thereby facilitating the enhancement of photocatalytic performance in terms of selectivity. Pt-coated hexagonal boron nitride nanoreactors (Pt@h-BN) were synthesized by a two-step technique (Fig. 6d).110 Among pure Pt, pure BN and the intermediate state BN before obtaining Pt-coated BN nanoreactors (Pt@IS-AB, middle part of Fig. 6d), Pt@h-BN with the Pt:B molar ratio of 1:3 (Pt@h-BN3) demonstrated the best photocatalytic activity and achieved a nearly 100% selectivity of CO2-to-CH4 (Fig. 6e).110 It was revealed that the special nanostructure composed of Pt as the core and h-BN as the shell (Fig. 6f and g) accelerated the electron mobility to produce the key intermediate CO2− species on the surface of Pt@h-BN.110 Compared to pure BiVO4, the lamellar BiVO4 showed the selective generation of CH3OH due to efficient electron capture to form CO2˙− radical anions.118 These provided researchers with a new method to construct highly selective photocatalysts by the delicate design of the architectures of photocatalysts.
Adequate electron supply is the most common mechanism to explain the selective reduction of CO2 to CH4 and other high-value-added products.
For example, non-metals and metals can be co-doped into the lattice of the photocatalyst in order to accomplish high-efficiency selectivity.119 The C and N co-doped TiO2 nanotubes with Na+ ions intercalated between the titanate layers were prepared by an alkaline hydrothermal method and achieved an increase in CH4 yield.120 Na+ ions introduced on the surface of C–N co-doped TiO2 nanotubes during the synthesis process were proposed to act as the active sites for effective CO2 absorption and further increase the conversion of CO2 to CH4.120 Similarly, compared with pure TiO2, V and N co-doped TiO2 nanocube arrays exhibited nearly four times improvement of photocatalytic CO2 to CH4 conversion.121 In addition to the ions just mentioned, Bi3+ ions adsorbed on the surface of TiO2 nanosheets favoured highly selective CH4 photocatalytic production via stimulating the reduction of intermediate CO.122 In this study, isolated Bi3+ ions were confined on the surface of 2D TiO2 (TiO2–Bi).122 Compared with the pure TiO2 counterpart, the TiO2–Bi exhibited a higher photocatalytic selectivity and efficiency of CH4 (Fig. 7a and b).122 The mechanism was the consecutive transfer of protons and electrons to intermediate CO, finally producing CH4, where the introduced Bi3+ was responsible for this fast conversion.122 Furthermore, halogen element (i.e., Cl, Br and I) doping can improve the catalytic selectivity.123 XPS analysis of I-doped TiO2 revealed that I5+ substituted for Ti4+ in the lattice (Fig. 7c).124 As a result, Ti3+ was generated to balance the charge (Fig. 7c).124 The high CO selectivity of I–TiO2 was achieved after the I5+ doping.124 We speculate that the low-valent titanium metal ions here are very likely to be used as active sites to promote the selective occurrence of the reaction, but direct experimental verification is needed.
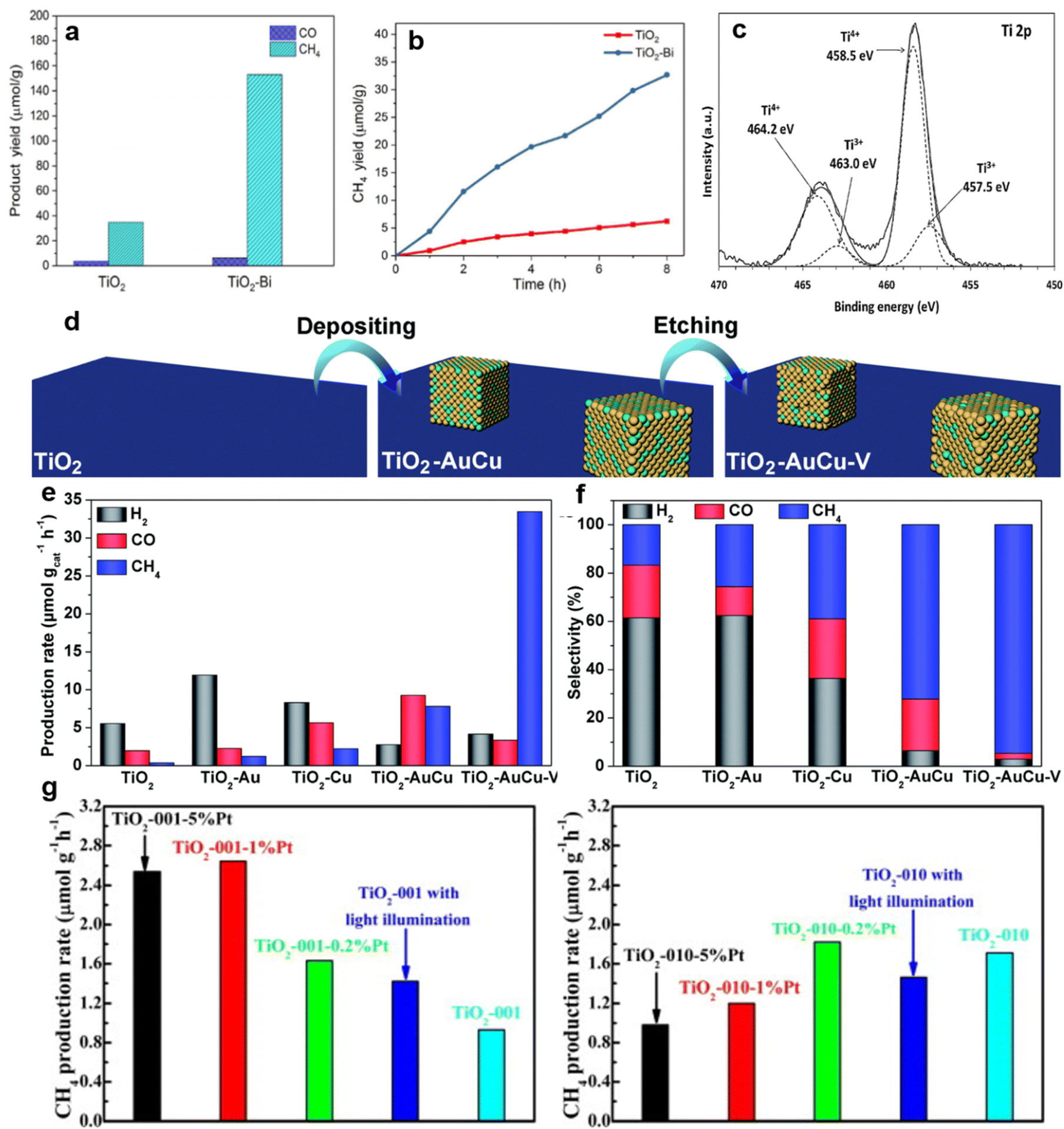 | ||
| Fig. 7 (a) Product yields of TiO2 and TiO2–Bi after photocatalytic CO2 reduction for 14 h. (b) Photocatalytic CH4 generation of TiO2 and TiO2–Bi. Reproduced with permission from ref. 122. Copyright 2018 Springer. (c) A high-resolution XPS spectrum of Ti 2p of 10% I–TiO2. Reproduced with permission from ref. 124. Copyright 2011 Elsevier. (d) The crafting process of TiO2–AuCu–V. (e) Yields and (f) selectivity of CH4, CO, H2 over TiO2-based photocatalysts. Reproduced with permission from ref. 125 copyright 2019 Royal Society of Chemistry. (g) Photocatalytic reduction activity of CO2 to CH4 over TiO2-001 and TiO2-010 and their corresponding 1 wt% or 5 wt% Pt-loaded products under UV-light irradiation for 4 h. Reproduced with permission from ref. 130 copyright 2014 Elsevier. | ||
Liu et al. loaded the Au–Cu alloy on TiO2 nanosheets (TiO2–AuCu) by ultrasonic and hydrothermal treatment and then obtained photocatalysts with Cu vacancies (TiO2–AuCu–V) by etching (Fig. 7d).125 In comparison with TiO2, TiO2–Au, TiO2–Cu and TiO2–AuCu, the CH4 generation selectivity of TiO2–AuCu–V was dramatically elevated to 94.7% (Fig. 7e and f).125 The removal of Cu atoms at the surface of TiO2–AuCu–V enhanced the electron trapping ability, consequently providing sufficient electrons for the generation of CH4 as mentioned above.125 Moreover, the experimental results also showed that there is a strong correlation between the yield of CH4 and the low-coordination Cu atoms near the vacancies, indicating that these Cu atoms could be used as active sites for the conversion of CO2 to CH4, further enhancing the selectivity of CH4 generation.125
Interestingly, although some metals have been proven to be able to improve selectivity in research, the same kind of metal loaded on different crystal faces of a special catalyst may exhibit different site activities. Taking the commonly used anatase TiO2 catalyst as an example, the theoretical surface energy order of the low index facets is {101} (i.e., 0.44 J m−2) < {010} (i.e., 0.53 J m−2) < {001} (i.e., 0.90 J m−2).126,127 This means that the most stable {101} facets exhibit the lowest catalytic reactivity for the CO2 reduction. Thus, in order to manipulate products and achieve high photocatalytic efficiency, ratios of {001} to {010} TiO2 exposed facets were tuned.128 Due to the strong interaction between CO2 and the {010} surface of TiO2, anatase TiO2 rods with dominant {010} facets accomplished the efficient photoreduction of CO2 to CH4.128 Ma reported that after Pt loading, anatase TiO2 exposing {001} facets exhibited improved CH4 evolution rates.129 After modifying Pt on {001}TiO2 facets, uniform Pt nanoparticles acted as active sites and led to a higher CH4 yield.130 However, when decorated on the {010} TiO2 facets, Pt nanoparticles agglomerated, exerting a negative effect on the photocatalytic reaction (Fig. 7g).130
To sum up, the introduction of metals, vacancies, and different ions to the photocatalysts might serve as active sites for the selective conversion of CO2 to the target products. However, the relationship between the active sites and the high reaction selectivity is not clear. The roles of some special ions, such as low-valence metal ions (Ti3+) generated together with the Vo (Fig. 7c) during the selective CO2 reduction reaction have not been fully assessed. Many of the discussions about the active sites just put forward conjectures that need to be further investigated by experiment.
3.2. Intermediates
Cu-nanocluster-decorated brookite TiO2 quasi-nanocubes (Cu-BTN) exhibited good activity and selectivity (Fig. 8a).61 When using 1.5% Cu-BTN as photocatalysts, the main products were CH4, accounting for about 85% of the total amount of final products.61 The reason for this high photocatalytic selectivity of Cu-BTN was explained as follows. When CO32− ions were intermediates, CO would be the main product, while more CH4 would be produced if HCO3− ions were intermediates.61 With the introduction of Cu nanoclusters and the gradual increase within the appropriate range, the formation of HCO3− on the surface of Cu-BTN would become easier (Fig. 8b); finally, CH4 would be formed selectively.61
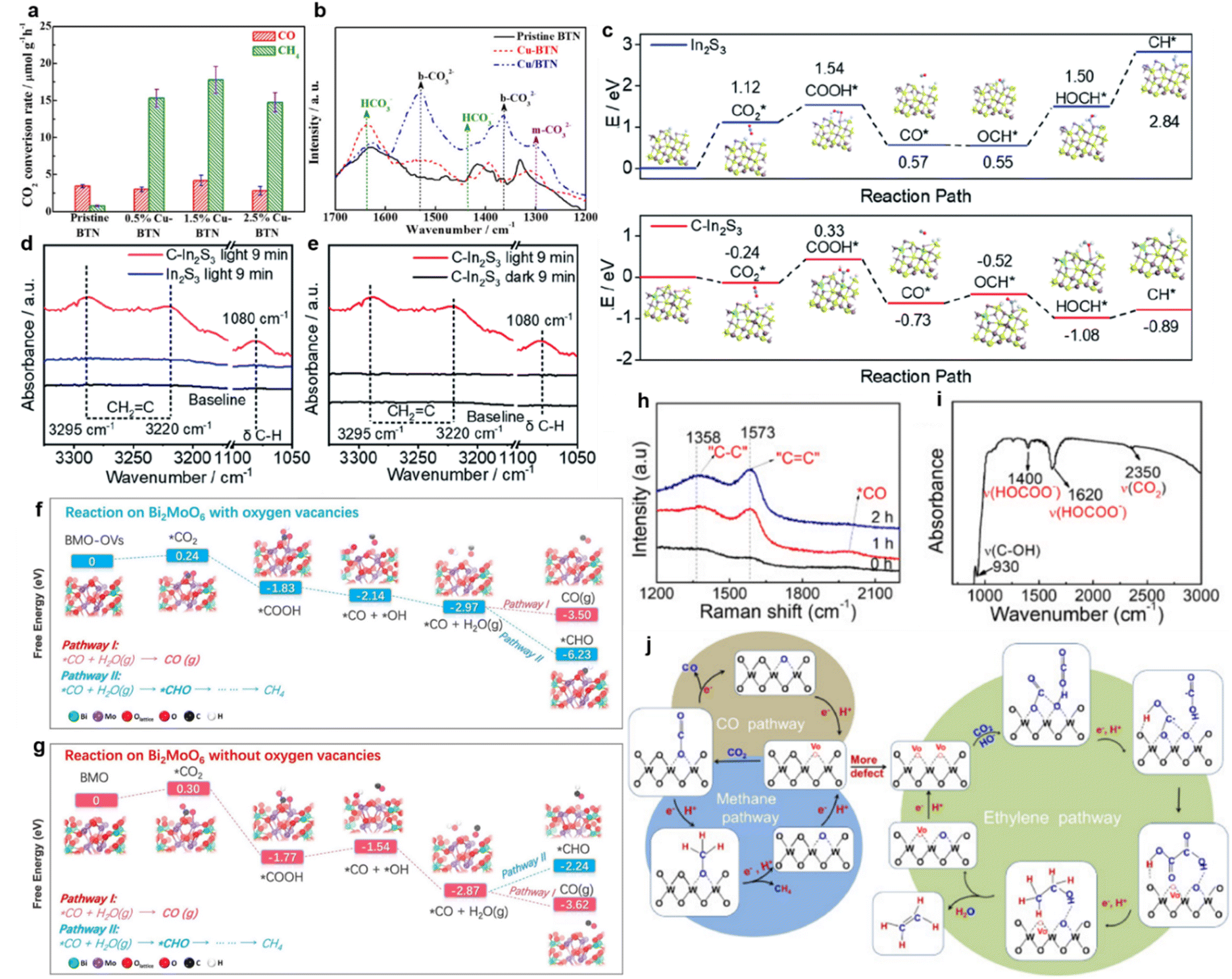 | ||
| Fig. 8 (a) CO2 photoreduction rate over pristine BTN and Cu-BTN with different amounts of Cu decoration. (b) In situ DRIFTS IR spectra of pristine BTN, Cu-BTN and Cu/BTN (obtained by depositing Cu on the as-prepared brookite TiO2 nanocubes). Reproduced with permission from ref. 61 copyright 2017 Wiley. (c) Calculated reaction energy diagrams of CO2 to CH* over the H-terminated surfaces of pristine In2S3 and C–In2S3. In situ DRIFTS spectra of (d) In2S3 and (e) C–In2S3. Reproduced with permission from ref. 133 copyright 2020 Royal Society of Chemistry. Reaction pathways of CO2 reduction over BiMoO6 (f) with oxygen vacancies and (g) without oxygen vacancies. Reproduced with permission from ref. 63 copyright 2019 Elsevier. (h) Raman spectra of WO3−x during the photocatalytic CO2 reduction. (i) IR spectrum of WO3−x after photocatalysis. (j) Schematic diagram of possible pathways of C2H4, CH4 and CO generation via photocatalytic CO2 reduction over WO3−x. Reproduced with permission from ref. 62 copyright 2020 Elsevier. | ||
In addition to the experimental characterization of the formation of key intermediates, the free energy of intermediate formation was studied through theoretical calculations. Wang achieved a C2H4 production selectivity close to 50% over C-doped In2S3 nanosheets.133 According to the calculated reaction energy, the reaction Gibbs free energies from OCH* to HOCH* and from HOCH* to CH* on C–In2S3 were −0.56 and 0.19 eV, respectively, which were much lower than the corresponding energies on pure In2S3 (i.e., 0.95 and 1.34 eV), respectively (Fig. 8c).133 It was indicated that OCH* could be further hydrogenated to form CH* on C–In2S3 (Fig. 8d and e).133 Meanwhile, unsaturated *CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, one of the important intermediates to produce C2H4, could be observed on the surface of C–In2S3 in the in situ DRIFTS spectra, which was not detected on pure In2S3 (Fig. 8d and e).133
C, one of the important intermediates to produce C2H4, could be observed on the surface of C–In2S3 in the in situ DRIFTS spectra, which was not detected on pure In2S3 (Fig. 8d and e).133
Similarly, Yang et al. prepared Bi2MoO6 nanosheets containing oxygen vacancies via a facile one-step solvothermal process.63 According to the calculated stepwise Gibbs free energy (Fig. 8f and g), the further hydrogenation of the intermediate CO* to the *CHO over Bi2MoO6 with oxygen vacancies was thermodynamically supported, compared to Bi2MoO6 without oxygen vacancies.63 Since *CHO was the prerequisite for the synthesis of CH4, it could be accompanied by the subsequent hydrogenation steps, ultimately realizing the highly selective CH4 production of 96.7% under visible light irradiation.63 The tuning of the energy barrier by oxygen vacancies was further confirmed on the surface of Ni–TiO2.136 Besides the theoretical calculations, the important intermediate formation could be identified by experiments.62 For instance, the photocatalytic selectivity of WO3-x micro-rods reducing CO2 to C2H4 increased to 89.3% after introducing oxygen vacancies.62 It could be observed in Raman and Infrared (IR) spectra that essential intermediates, including CC, C–C, adsorbed CO and HOCOO, appeared (Fig. 8h and i). It was indicated that adjacent oxygen vacancies provided active sites for C–C coupling to generate C2H4. The CO2 reduction pathway on the WO3-x was proposed to be CO2 → ·COOH → (COOH)2 → CH3COOH → CH3CH2OH → CH2CH2via a hydrogenation and dehydration process (Fig. 8j).62 Apart from the reactions mentioned above, CO2 could also be selectively reduced to methanol, formic acid and other hydrocarbons.74,75,137 For example, the energy barrier of the rate-limiting step (CO2 → HCOO) on the surface of MXene (0.53 eV) for the reduction of CO2 to HCOOH is much lower than that of anatase (0.87 eV), which was prone to producing HCCOH.97
Besides, metal vacancies of semiconductors can adjust the formation energy of intermediates, thereby affecting the photocatalytic reaction.134 Since ZnS samples possessing Zn vacancies could be obtained by acid etching over commercial ZnS powders, different pH values of sulfuric acid were used to craft ZnS containing various amounts of Zn vacancies and obtained different HCOOH formation selectivities (Fig. 9a and b).134 Acid-etched ZnS attained the highest selectivity of 86.6% for HCOOH production at pH = 0.2.134 As displayed in Fig. 9c, with the existence of Zn vacancies, ZnS required a lower energy barrier for the reduction of CO2 to HCOOH.134 Moreover, hydroxyl groups on the surface might bind closely with CO2 molecules to form key intermediates during the photocatalytic CO2 reduction.135 The proposed CH4 formation mechanism on the hydroxylated mesoporous TiO2 surface is shown in Fig. 10d.135 According to the experimental results, on the hydroxyl-abundant surface of TiO2, the –OH groups could convert absorbed CO2 molecules to carbonate or bicarbonate species (Fig. 9d).135 The bidentate carbonate (b-CO32−) species sequentially received 2 electrons and 2 protons and were transformed into CH4 through the sequence of Ti–OOCH2 → Ti–O–CH3 → CH4 (Fig. 9d).135 Meanwhile, the carboxylic species experienced the process of Ti–COOH → Ti–CHO → Ti–CH2OH → Ti–CH3 → CH4 to produce CH4 (Fig. 9e).135
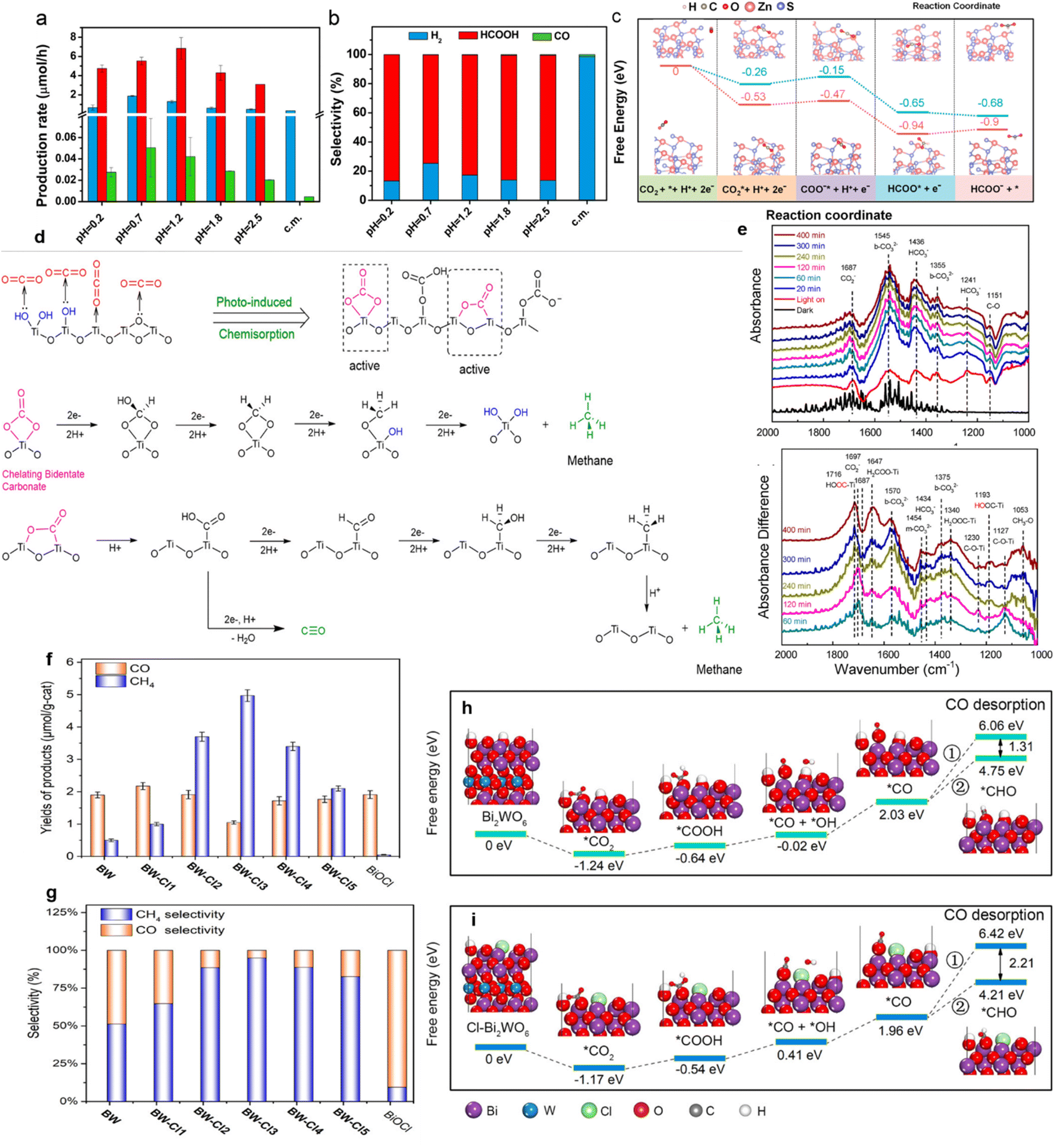 | ||
| Fig. 9 (a) Yields and (b) selectivity of HCOOH, CO, H2 over ZnS samples etched by sulfuric acid of different pH values. (c) Free energy diagram for the pathways of CO2 conversion to formate on a perfect ZnS (blue) and VZn-ZnS (pink) surface. Reproduced with permission from ref. 134 copyright 2019 American Chemical Society. (d) Proposed CH4 and CO formation mechanism on the hydroxylated surface of TiO2. (e) The DRIFT spectra of adsorbed and transformed species on the hydroxylated surface of TiO2 after different light irradiation times. Reproduced with permission from ref. 135 copyright 2020 American Chemical Society. (f) Yields and (g) selectivity of CO and CH4 over BW, BiOCl, and BW–Clx (x = 1–5) after photocatalytic CO2 reduction for 3 h. Free energy diagrams of CO2 photoreduction over (h) Bi2WO6 and (i) Cl−-modified Bi2WO6. Reproduced with permission from ref. 70 copyright 2020 American Chemical Society. | ||
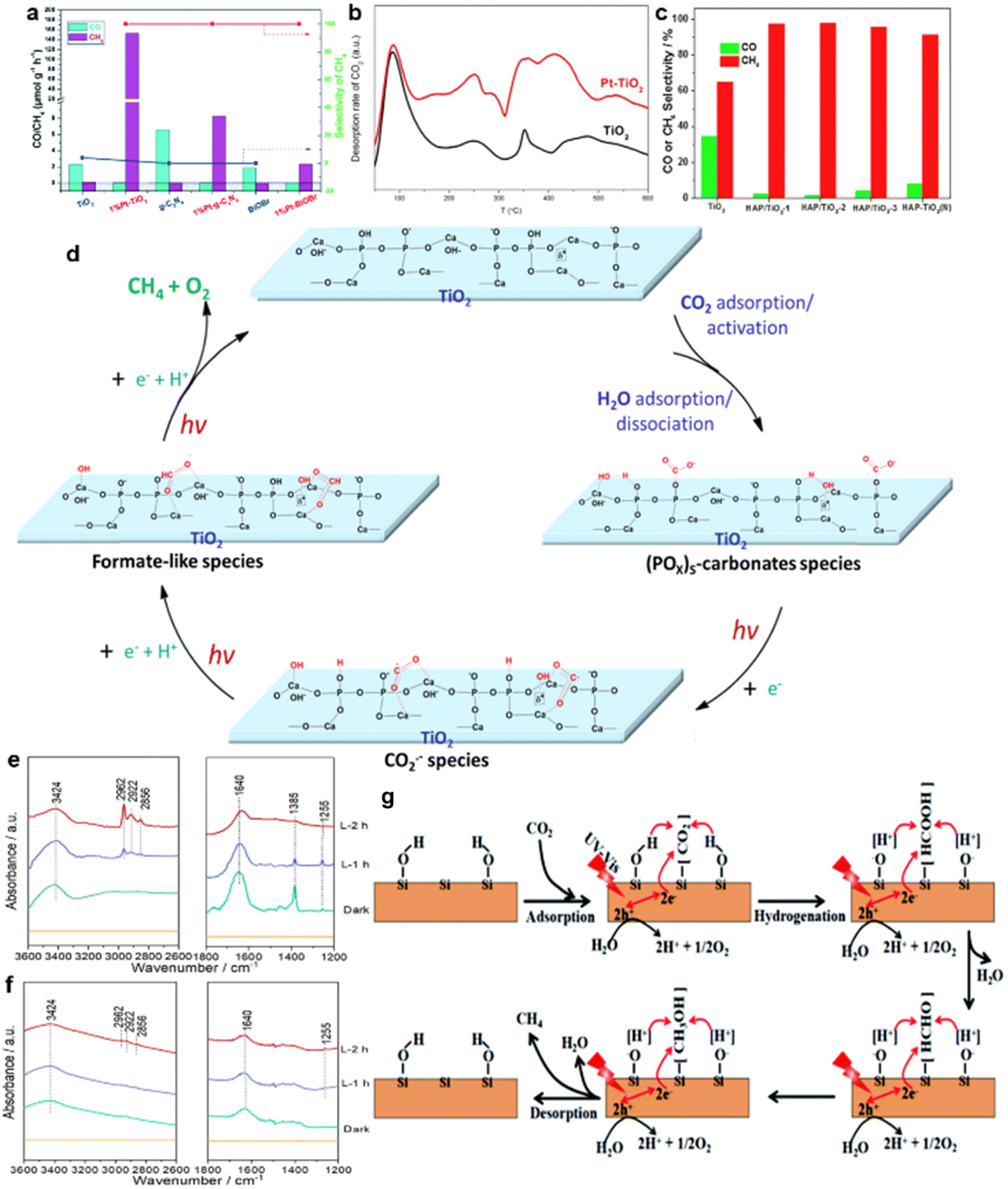 | ||
| Fig. 10 (a) Photocatalytic activities and selectivities of CO2 reduction over TiO2, g-C3N4 and BiOBr with or without Pt loading. (b) CO-TPD of TiO2 and Pt–TiO2. Reproduced with permission from ref. 58 copyright 2018 Royal Society of Chemistry. (c) The evolution rates and the selectivity of CO and CH4 over TiO2 and various HAP/TiO2 photocatalysts. (d) A possible pathway for the conversion of CO2 to CH4 over Pt/HAP/TiO2 in the presence of H2O. In situ IR spectra of (e) Pt/HAP/TiO2 and (f) Pt/TiO2.68 Reproduced with permission from ref. 68 copyright 2018 Elsevier. (g) Schematic illustration of the photocatalytic CO2 conversion to CH4 over hydroxylated SiC nanosheets. Reproduced with permission from ref. 141 copyright 2019 Royal Society of Chemistry. | ||
Ion modification can also improve the selectivity of CO2 reduction by adjusting the formation energy of key intermediates.138 Li prepared Bi2WO6 (BW) nanosheets loaded with different amounts of Cl− ions, which were marked as BW–Clx (x = 1–5).70 A maximum selectivity of 94.98% for CH4 generation was achieved on the surface of BW–Cl3 (Fig. 9f and g).70 In the presence of Cl− ions, the half-reaction (i.e., oxygen evolution) of Bi2WO6 nanosheets was greatly increased.70 Simultaneously, the produced protons facilitated the formation of CH4.70 Moreover, DFT calculations (Fig. 9h and i) confirmed that the presence of Cl− ions on Bi2WO6 nanosheets was capable of lowering the energy barrier for the generation of crucial intermediate (i.e., *CHO), thus enhancing CH4 production.70
In the above reports, the existence of key intermediates in some highly selective systems was verified by the experimental data, and the formation energy of the target intermediates was studied through theoretical calculations. The difficulty in producing intermediates and the stability of the as-obtained intermediates will be discussed in the next section.
For instance, the introduction of noble metals could reduce the intermediate desorption capacity of the photocatalysts and ensure that the intermediates are adsorbed on the surface of the catalysts for subsequent reactions.58 As shown in Fig. 10a, compared with pure semiconductor photocatalysts, nearly 100% selectivity of CH4 generation was achieved by loading 1% Pt on the TiO2, C3N4 and BiOBr photocatalysts, respectively.56,58,140 The temperature-programmed desorption (TPD) results confirmed that CO demonstrated an especially strong adsorption capacity on the Pt clusters (Fig. 10b).58 Meanwhile, since only physical adsorption occurred between CH4 and the Pt clusters, CH4 exhibited low adsorption energy on the Pt clusters, leading to the easy desorption of generated CH4 from the Pt surface.58 Thus, most CO products produced during the photocatalytic process were anchored on the catalyst surfaces, and CH4 molecules were desorbed from the photocatalyst surface, eventually achieving enhanced selectivity of CH4 generation.58
Through compound modification of the catalysts, the stability of some key intermediates can also be improved. Hydroxyapatite (HAP)-decorated TiO2 could achieve 99.1% selectivity of CH4 generation (Fig. 10c).68 The formation of much more stable intermediates over HAP/TiO2 nanorods was responsible for this selectivity enhancement.68 A possible pathway for the conversion of CO2 to CH4 over Pt/HAP/TiO2 in the presence of H2O is proposed in Fig. 10d.68 Specifically, the formate-like species (HCOO−) was identified as the crucial intermediate for CH4 production.68 Compared with that of TiO2, IR peaks at 3000–2800 cm−1 of Pt/HAP/TiO2 were obviously enhanced, indicating that the HAP effectively improved the stability of HCOO− (Fig. 10e and f).68
Besides, as the cocatalyst of P25, surface alkalization of Ti3C2 could dramatically enhance the evolution rate of CH4 (16.61 μmol g−1 h−1),142 in which surface hydroxyls on selective CH4 generation over SiC nanosheets have gained much attention.141,143 Hydroxyl groups on the surface of SiC nanosheets boosted the photoreduction of CO2 into CH4, achieving the CH4 generation selectivity of about 80%.141 The mechanism is depicted in Fig. 10g.141 Specifically, –OH groups on the surface of SiC could provide sufficient protons to CO2, which is critical to the CO2 activation.141 Moreover, the intermediates could be stabilized by forming hydrogen bonds between the intermediates and –OH groups. These effects improved the selective CH4 generation.141
Although the stability of the intermediates remarkably affects the reaction path of photocatalytic CO2 reduction, the relative investigations are limited in scope. More attention should be paid to the generation process and stabilization of key intermediates to make the study on CO2 reduction more comprehensive and convincing.
3.3. Desorption of products
As one critical step in the entire CO2 reduction process, the desorption of the product from the surface of photocatalysts affects the selectivity. The rapid release of the product prevents the subsequent reaction, thereby maintaining the selective and continuous yield of this product. Various types of vacancies and crystal facets cause varied adsorption capabilities of products, which results in the adjustment of the final products.Metal vacancies of semiconductors play a significant role in manipulating the photocatalytic selectivity. Different reduction products can be obtained by introducing various metal vacancies.144 Selective CO generation could be achieved on the surface of BiOBr ultrathin nanosheets containing abundant Bi vacancies (VBi).144 Compared with BiOBr nanosheets without VBi, CO could be more easily desorbed from the surface of VBi–BiOBr, leading to increasing the amount of CO production.144
In addition to the introduction of vacancies, some facets of photocatalysts may exhibit the unique ability of CO2 adsorption and product desorption.145–147 These beneficial effects can enhance the photocatalytic selectivity. As shown in Fig. 11a, distinguished from chemisorbed CO with a larger endothermic value of 0.45 eV on the {100} facet, the exposed {110} facet of the InVO4 atomic layer was confirmed to weakly bind the generated CO, leading to the quick desorption of CO molecules from the catalyst surface.148 Therefore, by the facet engineering technique, ultrathin InVO4 nanosheets possessing the {110} facet could gain much higher CO formation selectivity (i.e., 98%) as compared with regular InVO4 nanocubes showing the (100) facet and the bulk InVO4 obtained by a conventional solid-state reaction (SSR, Fig. 11b and c).148 Due to the favorable CO2 absorption and easy desorption of CO, hexagonal Co3O4 nanoplatelets exposing {112} facets could realize the selectivity of 77.1% for CO generation (Fig. 11d).149
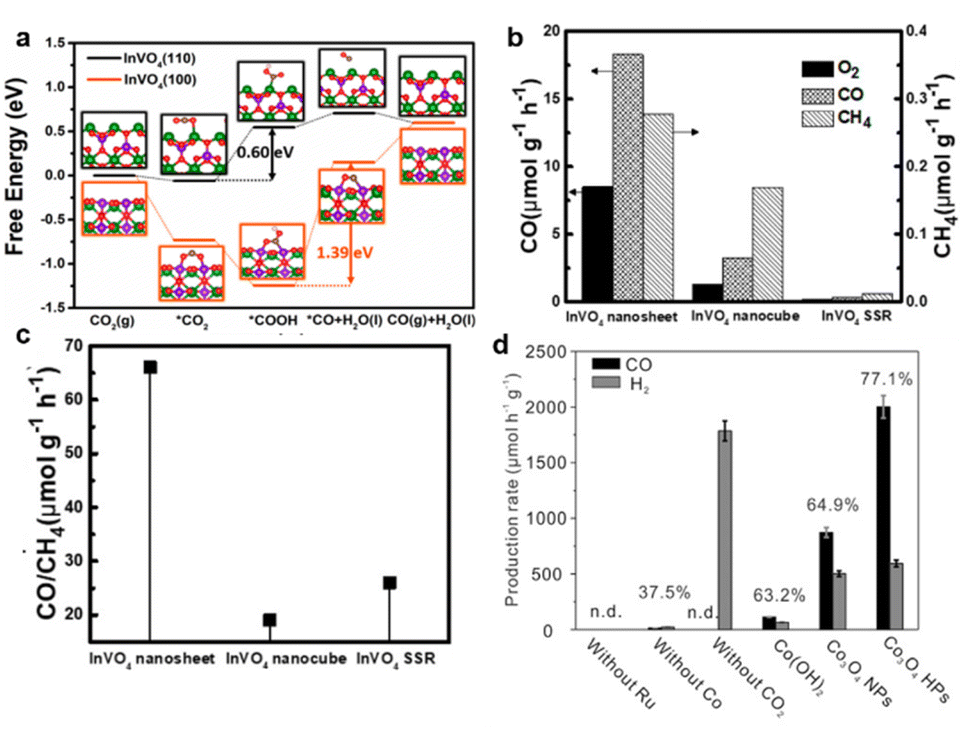 | ||
| Fig. 11 (a) The photocatalytic activity and (b) the production rates of CO to CH4 over the ultrathin InVO4 nanosheets, nanocubes, and obtained by SSR. (c) Calculated Gibbs free energy profiles of photocatalytic CO2 reduction to CO over InVO4 of the (110) and (100) planes, respectively. Reproduced with permission from ref. 148 copyright 2019 American Chemical Society. (d) Photocatalytic production rate using β-Co(OH)2, Co3O4 nanoparticles (Co3O4 NPs), or Co3O4 hexagonal platelets (Co3O4 HPs) as catalysts under visible light (λ > 420 nm) irradiation. Reproduced with permission from ref. 150 copyright 2016 Wiley. | ||
In general, both metal vacancies and crystal facets have selective desorption capacity. However, it is unknown whether other modification methods could mediate the desorption ability of products. There is limited discussion in this area, and further exploration is expected.
4. Summary and outlook
There has been widespread interest in the fact that photocatalytic product selectivity is one of the crucial factors limiting the application of photocatalytic CO2 reduction. The importance of photocatalytic CO2 reduction to energy utilization and environmental protection is summarized at the beginning of this review. We introduced different reaction steps that could increase the selectivity of the reaction, that is, the adsorption and activation of reactants (including CO2 adsorption and H2 evolution inhibition, electron supply and others), the formation and stabilization of intermediates (including formation energy of crucial intermediates and stability of intermediates), and the desorption of products. The corresponding modification methods for achieving selective improvement at each stage are summarized, including noble metal decoration, metal and non-metal doping, vacancy engineering, facet engineering, composite construction, hydroxyl modification and other decoration techniques. Although this research field has been developed for several decades, the photocatalytic conversion of CO2 to high-value products selectively is still in its infancy. A lot of effort should be made to achieve a great breakthrough.Firstly, although many materials have been successfully fabricated for highly selective photocatalytic CO2 reduction, the photocatalytic mechanism of high selectivity remains unclear. Most mechanisms have been proposed based on the simulation results instead of experimental data. In addition, plenty of intriguing phenomena, such as the formation of low-valent metal ions near the oxygen vacancies, and the production of surface hydroxyl groups, have been described but no explanations are given in detail. Therefore, more in situ characterization techniques, such as FITR, Raman, XRD and NMR, should be conducted to uncover the underlying photocatalytic process. To design highly selective catalysts on purpose, a deep understanding and exploration of the reaction must be gained.
Secondly, more investigations should be focused on the oxidation reaction during photocatalytic CO2 reduction. Since main products are obtained via reducing CO2, most research is concentrated on the reduction reaction caused by the photo-generated electrons. However, as a half-reaction of the entire CO2 reduction, how photo-induced holes participate in the photocatalytic reaction might dramatically affect the catalytic performance of semiconductors. Once the oxidation reaction is restricted, it will be tough for the entire process to proceed. A quick oxidation half-reaction can suppress the inverse reactions of CO2 reduction. In addition, the oxidation products might participate in the reaction of CO2 reduction. Therefore, to have a comprehensive understanding of the entire CO2 reduction process, the oxidation half-reaction involving photo-generated holes requires more in-depth investigations.
Thirdly, precise modification techniques of photocatalysts should be further improved. The introduction of impurities into the lattice of semiconductors can increase the effective capture of electrons and thus improve the selectivity of CH4 generation. However, there is always an optimal doping concentration. Similarly, the construction of vacancies can bring about an increase in selectivity but excessive vacancies will destroy the bulk structure and reduce the catalytic performance. More delicate methods should be developed to craft decorated photocatalysts.
Fourthly, based on this review, it was found that each step during the entire CO2 reduction can affect selectivity, so different modification methods could be combined to synergistically boost the reaction selectivity. For example, after oxygen vacancies were introduced into Pt-loaded Ga2O3, the selectivity of the reaction increased to nearly 100%.81 The oxygen vacancies served as the main sites for CO2 adsorption, while Pt nanoparticles used the hydrogen formed in the photocatalytic decomposition process to reduce the adsorbed CO2.81 This synergy increased photocatalytic production selectivity.81 Besides, the photocatalytic conditions of the catalytic system can influence the production selectivity, including temperature, pressure, gas flow rate, reaction solution, etc. Thus, more explorations can be concentrated on adjusting the composition of photocatalysts and the reaction parameters.
In summary, photocatalytic CO2 conversion to fuel products can not only alleviate carbon dioxide emissions but also provide clean chemical energy using green solar light. To realize high catalytic efficiency and achieve value-added products of high selectivity, the above four points require numerous efforts to explore. More advanced in situ characterization techniques, including solid-state NMR, isotope research, FITR, Raman, and XRD should be applied to study the chemical evolution of catalysts during the catalytic process. To understand the entire reaction process more comprehensively, more studies should be conducted that pay attention to both the reduction half-reaction and the oxidation half-reaction since how photo-induced electrons and holes participate in the photocatalytic reaction will affect the catalytic performance. Due to the different effects of various modification strategies on the reaction process, the modification process should be controlled more accurately, which requires more advanced synthesis equipment and more delicate fabrication strategies. Different modification techniques could be combined to synergistically achieve higher efficiency and selectivity.
Author contributions
M. W. conceived the idea. M. W., S. Y. designed the conceptualization. S. Y., J. H., F. G., H. W., J. L., Y. B. performed the investigation and data curation. J. F., F. H., F. Z. conducted the supervision. M. W., S. Y. wrote the paper. All authors participated in the review and editing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Mengye Wang gratefully acknowledges the financial support from the National Natural Science Foundation of China (No. 21905317), the Young Elite Scientists Sponsorship Program by CAST (2019QNRC001), and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (76180-31620007).Notes and references
- I. L. T. M. S. Dresselhaus, Nature, 2001, 414, 332–337 CrossRef.
- F. Liu, M. Wang, X. Liu, B. Wang, C. Li, C. Liu, Z. Lin and F. Huang, Nano Lett., 2021, 21, 1643–1650 CrossRef CAS PubMed.
- Q. Liu, X. Hong, X. You, X. Zhang, X. Zhao, X. Chen, M. Ye and X. Liu, Energy Storage Mater., 2020, 24, 541–549 CrossRef.
- S. Yao, J. Liu, F. Liu, B. Wang, Y. Ding, L. Li, C. Liu, F. Huang, J. Fang, Z. Lin and M. Wang, Environ. Sci. Nano, 2022, 9, 1996–2005 RSC.
- S. Han, Q. Wan, K. Zhou, A. Yan, Z. Lin, B. Shu and C. Liu, ACS Appl. Nano Mater., 2021, 4, 8273–8281 CrossRef CAS.
- Y. Li, L. Li, F. Liu, B. Wang, F. Gao, C. Liu, J. Fang, F. Huang, Z. Lin and M. Wang, Nano Res., 2022, 15, 7986–7993 CrossRef CAS PubMed.
- J. He, F. Gao, H. Wang, F. Liu, J. Lin, B. Wang, C. Liu, F. Huang, Z. Lin and M. Wang, Environ. Sci. Nano, 2022, 9, 1952–1960 RSC.
- Z. Wang, J. Zheng, M. Li, Q. Wu, B. Huang, C. Chen, J. Wu and C. Liu, Appl. Phys. Lett., 2018, 113, 122101 CrossRef.
- Y. Wang, Z. Wang, K. Huang, X. Liang, C. Liu, C. Chen and C. Liu, Appl. Phys. Lett., 2020, 116, 141604 CrossRef CAS.
- X. Liang, L. Liu, G. Cai, P. Yang, Y. Pei and C. Liu, J. Phys. Chem. Lett., 2020, 11, 2765–2771 CrossRef CAS PubMed.
- D. Moreira and J. C. M. Pires, Bioresour. Technol., 2016, 215, 371–379 CrossRef CAS PubMed.
- F. Barzagli, C. Giorgi, F. Mani and M. Peruzzini, J. CO2 Util., 2017, 22, 346–354 CrossRef CAS.
- W. P. Wang Zeyan, Y. Liu, Z. Zheng, H. Cheng and B. Huang, J. Synth. Cryst., 2021, 50, 685–707 Search PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Wang, B. Wang, F. Huang and Z. Lin, Angew. Chem., Int. Ed. Engl., 2019, 58, 7526–7536 CrossRef CAS PubMed.
- X. Zhang, F. Xie, X. Li, H. Chen, Y. She, C. Wang, Z. Mo, W. Yang, P. Hou, C. Wu, H. Xu and H. Li, Appl. Surf. Sci., 2021, 542, 148619 CrossRef CAS.
- Y. Wang, S. Z. F. Phua, G. Dong, X. Liu, B. He, Q. Zhai, Y. Li, C. Zheng, H. Quan, Z. Li and Y. Zhao, Chem, 2019, 5, 2775–2813 CAS.
- C. W. W. Ng, R. Tasnim and J. L. Coo, Eng. Geol., 2018, 242, 108–120 CrossRef.
- D. R. Feldman, W. D. Collins, P. J. Gero, M. S. Torn, E. J. Mlawer and T. R. Shippert, Nature, 2015, 519, 339–343 CrossRef CAS PubMed.
- J. Cai, J. Shen, X. Zhang, Y. H. Ng, J. Huang, W. Guo, C. Lin and Y. Lai, Small Methods, 2019, 3, 1800184 CrossRef.
- J. Xiong, J. Luo, J. Di, X. Li, Y. Chao, M. Zhang, W. Zhu and H. Li, Fuel, 2020, 261, 116448 CrossRef CAS.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- M. Wang, L. Cai, Y. Wang, F. Zhou, K. Xu, X. Tao and Y. Chai, J. Am. Chem. Soc., 2017, 139, 4144–4151 CrossRef CAS PubMed.
- M. Wang, Y. Zuo, J. Wang, Y. Wang, X. Shen, B. Qiu, L. Cai, F. Zhou, S. P. Lau and Y. Chai, Adv. Energy Mater., 2019, 9, 1901801 CrossRef CAS.
- Q. Wang, J. Cai, G. V. Biesold-McGee, J. Huang, Y. H. Ng, H. Sun, J. Wang, Y. Lai and Z. Lin, Nano Energy, 2020, 78, 105313 CrossRef CAS.
- F. He, X. You, H. Gong, Y. Yang, T. Bai, W. Wang, W. Guo, X. Liu and M. Ye, ACS Appl. Mater. Interfaces, 2020, 12, 6442–6450 CrossRef CAS.
- C. He, B. Han, S. Han, Q. Xu, Z. Liang, J. Y. Xu, M. Ye, X. Liu and J. Xu, J. Mater. Chem. A, 2019, 7, 26884–26892 RSC.
- G. Feng, H. Jiaqing, W. Haowei, L. Jiahui, C. Ruixin, Y. Kai, H. Feng, L. Zhang and W. Mengye, Nano Res. Energy, 2022, 1, e9120029 CrossRef.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- Y. Chen, G. Jia, Y. Hu, G. Fan, Y. H. Tsang, Z. Li and Z. Zou, Sustain. Energy Fuels, 2017, 1, 1875–1898 RSC.
- S. Zhu, Q. Wang, X. Qin, M. Gu, R. Tao, B. P. Lee, L. Zhang, Y. Yao, T. Li and M. Shao, Adv. Energy Mater., 2018, 8, 1802238 CrossRef.
- K. Hua, X. Liu, B. Wei, S. Zhang, H. Wang and Y. Sun, Acta Phys.-Chim. Sin., 2020, 2009098 Search PubMed.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS.
- G. Xiaoya, L. Jiaofu and Z. Zicheng, Nano Research Energy, 2022, 1, e9120036 CrossRef.
- X. O. Bin Han, Z. Zhong, S. Liang, Y. Xu, H. Deng and Z. Lin, Appl. Catal., B, 2021, 283119594 Search PubMed.
- J.-N. Zhang, N. K. Niazi, J. Qiao, L. Li, I. M. u. Hasan, R. He, L. Peng, N. Xu and F. Farwa, Nano Research Energy, 2022, 1, e9120015 Search PubMed.
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Sci. Bull., 2020, 65, 987–994 CrossRef CAS PubMed.
- J. Z. Han Li, J. Yu and S. Cao, Trans. Tianjin Univ., 2021, 27, 338–347 CrossRef.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- C. W. Kim, M. J. Kang, S. Ji and Y. S. Kang, ACS Catal., 2018, 8, 968–974 CrossRef CAS.
- Z. Wu, S. Guo, L.-H. Kong, A.-F. Geng, Y.-J. Wang, P. Wang, S. Yao, K.-K. Chen and Z.-M. Zhang, Chinese J. Catal., 2021, 42, 1790–1797 CrossRef CAS.
- D.-C. Liu, D.-C. Zhong and T.-B. Lu, EnergyChem, 2020, 2, 100034 CrossRef.
- A. Touqeer, L. Shuang, S. Muhammad, L. Ke, A. Mohsin, L. Liang and C. Wei, Nano Research Energy, 2022, 1, e9120021 CrossRef.
- B. H. Weiyi Chen, C. Tian, X. Liu, S. Liang, H. Deng and Z. Lin, Appl. Catal., B, 2019, 244, 996–1003 CrossRef.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- W. Zhang, A. R. Mohamed and W. J. Ong, Angew. Chem., Int. Ed. Engl., 2020, 59, 22894–22915 CrossRef CAS PubMed.
- R. Schlogl, Angew. Chem., Int. Ed. Engl., 2015, 54, 3465–3520 CrossRef PubMed.
- A. Li, Q. Cao, G. Zhou, B. Schmidt, W. Zhu, X. Yuan, H. Huo, J. Gong and M. Antonietti, Angew. Chem., Int. Ed. Engl., 2019, 58, 14549–14555 CrossRef CAS PubMed.
- B. Pan, S. Luo, W. Su and X. Wang, Appl. Catal., B, 2015, 168–169, 458–464 CrossRef CAS.
- A. Anzai, N. Fukuo, A. Yamamoto and H. Yoshida, Catal. Commun., 2017, 100, 134–138 CrossRef CAS.
- X. Cai, J. Wang, R. Wang, A. Wang, S. Zhong, J. Chen and S. Bai, J. Mater. Chem. A, 2019, 7, 5266–5276 RSC.
- Y. Zhao, Y. Wei, X. Wu, H. Zheng, Z. Zhao, J. Liu and J. Li, Appl. Catal., B, 2018, 226, 360–372 CrossRef CAS.
- Y. Wang, Q. Lai, Y. He and M. Fan, Catal. Commun., 2018, 108, 98–102 CrossRef CAS.
- Z. Ma, P. Li, L. Ye, L. Wang, H. Xie and Y. Zhou, Catal. Sci. Technol., 2018, 8, 5129–5132 RSC.
- W.-K. Jo, S. Kumar and S. Tonda, Composites, Part B, 2019, 176, 107212 CrossRef CAS.
- B. Tahir, M. Tahir and N. A. Saidina Amin, Mal. J. Fund. Appl. Sci., 2015, 11, 114–117 Search PubMed.
- J. Jin, J. Luo, L. Zan and T. Peng, Chemphyschem, 2017, 18, 3230–3239 CrossRef CAS PubMed.
- C. Lu, J. Li, J. Yan, B. Li, B. Huang and Z. Lou, Appl. Mater. Today, 2020, 20, 100744 CrossRef.
- X. Yang, S. Wang, N. Yang, W. Zhou, P. Wang, K. Jiang, S. Li, H. Song, X. Ding, H. Chen and J. Ye, Appl. Catal., B, 2019, 259, 118088 CrossRef CAS.
- D. Jiang, W. Wang, E. Gao, S. Sun and L. Zhang, Chem. Commun., 2014, 50, 2005–2007 RSC.
- R. Bhosale, S. Jain, C. P. Vinod, S. Kumar and S. Ogale, ACS Appl. Mater. Interfaces, 2019, 11, 6174–6183 CrossRef CAS PubMed.
- Q. Shi, X. Zhang, Y. Yang, J. Huang, X. Fu, T. Wang, X. Liu, A. Sun, J. Ge, J. Shen, Y. Zhou and Z. Liu, J. Energy Chem., 2021, 59, 9–18 CrossRef CAS.
- Z. Wang, K. Teramura, Z. Huang, S. Hosokawa, Y. Sakata and T. Tanaka, Catal. Sci. Technol., 2016, 6, 1025–1032 RSC.
- R. Chong, Y. Fan, Y. Du, L. Liu, Z. Chang and D. Li, Int. J. Hydrogen Energy, 2018, 43, 22329–22339 CrossRef CAS.
- L. Zhao, F. Ye, D. Wang, X. Cai, C. Meng, H. Xie, J. Zhang and S. Bai, ChemSusChem, 2018, 11, 3524–3533 CrossRef CAS PubMed.
- Y.-Y. Li, J.-S. Fan, R.-Q. Tan, H.-C. Yao, Y. Peng, Q.-C. Liu and Z.-J. Li, ACS Appl. Mater. Interfaces, 2020, 12, 54507–54516 CrossRef CAS PubMed.
- W. Bi, Y. Hu, H. Jiang, L. Zhang and C. Li, Adv. Funct. Mater., 2021, 31, 2010780 CrossRef CAS.
- Y. Bai, P. Yang, L. Wang, B. Yang, H. Xie, Y. Zhou and L. Ye, Chem. Eng. J., 2019, 360, 473–482 CrossRef CAS.
- Q. Chen, X. Chen, M. Fang, J. Chen, Y. Li, Z. Xie, Q. Kuang and L. Zheng, J. Mater. Chem. A, 2019, 7, 1334–1340 RSC.
- S. Gao, B. Gu, X. Jiao, Y. Sun, X. Zu, F. Yang, W. Zhu, C. Wang, Z. Feng, B. Ye and Y. Xie, J. Am. Chem. Soc., 2017, 139, 3438–3445 CrossRef CAS PubMed.
- P. Xia, M. Antonietti, B. Zhu, T. Heil, J. Yu and S. Cao, Adv. Funct. Mater., 2019, 29, 1900093 CrossRef.
- Y. Liang, X. Wu, X. Liu, C. Li and S. Liu, Appl. Catal., B, 2022, 304, 120978 CrossRef CAS.
- H. Zhao, J. Duan, Z. Zhang and W. Wang, ChemCatChem, 2021, 14, e2021017 Search PubMed.
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa, S. A. Shevlin, J. R. Durrant, Z. Guo and J. Tang, Nat. Commun., 2020, 11, 2531 CrossRef CAS PubMed.
- M. Ma, Z. Huang, R. Wang, R. Zhang, T. Yang, Z. Rao, W. Fa, F. Zhang, Y. Cao, S. Yu and Y. Zhou, Green Chem., 2022, 24, 8791–8799 RSC.
- W. Zhang, M. Jiang, S. Yang, Y. Hu, B. Mu, Z. Tie and Z. Jin, Nano Research Energy, 2022, 1, e9120033 CrossRef.
- Y.-X. Pan, Z.-Q. Sun, H.-P. Cong, Y.-L. Men, S. Xin, J. Song and S.-H. Yu, Nano Res., 2016, 9, 1689–1700 CrossRef CAS.
- B. Han, X. Ou, Z. Zhong, S. Liang, H. Deng and Z. Lin, Small, 2020, 16, 2002985 CrossRef CAS PubMed.
- D. Wu, L. Ye, H. Y. Yip and P. K. Wong, Catal. Sci. Technol., 2017, 7, 265–271 RSC.
- P. Yang, Z. J. Zhao, X. Chang, R. Mu, S. Zha, G. Zhang and J. Gong, Angew. Chem., Int. Ed. Engl., 2018, 57, 7724–7728 CrossRef CAS PubMed.
- C. Dong, M. Xing and J. Zhang, J. Phys. Chem. Lett., 2016, 7, 2962–2966 CrossRef CAS PubMed.
- K. Song, S. Liang, X. Zhong, M. Wang, X. Mo, X. Lei and Z. Lin, Appl. Catal., B, 2022, 309, 121232 CrossRef CAS.
- X. Liu, L. Ye, S. Liu, Y. Li and X. Ji, Sci. Rep., 2016, 6, 38474 CrossRef CAS PubMed.
- M. Li, L. Zhang, X. Fan, M. Wu, M. Wang, R. Cheng, L. Zhang, H. Yao and J. Shi, Appl. Catal., B, 2017, 201, 629–635 CrossRef CAS.
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC.
- P. Tao, S. Yao, F. Liu, B. Wang, F. Huang and M. Wang, J. Mater. Chem. A, 2019, 7, 23512–23536 RSC.
- J. Xiong, J. Di and H. Li, J. Mater. Chem. A, 2021, 9, 2662–2677 RSC.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K.-Q. Zhang, S. S. Al-Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 RSC.
- P. Li, Y. Zhou, W. Tu, R. Wang, C. Zhang, Q. Liu, H. Li, Z. Li, H. Dai, J. Wang, S. Yan and Z. Zou, CrystEngComm, 2013, 15, 9855–9858 RSC.
- Z. Zhao, J. Zhang, M. Lei and Y. Lum, Nano Research Energy, 2023, 2, e9120044 Search PubMed.
- G. Chen, M. Guo, X. Li, W. Wang, F. Liu, C. Ning, G. Yuan, J. Chen, S. Deng and C. Liu, IEEE Trans. Electron Devices, 2022, 69, 2430–2435 CAS.
- C. Chen, B. R. Yang, G. Li, H. Zhou, B. Huang, Q. Wu, R. Zhan, Y. Y. Noh, T. Minari, S. Zhang, S. Deng, H. Sirringhaus and C. Liu, Adv. Sci., 2019, 6, 1801189 CrossRef PubMed.
- Z. Xiong, Z. Lei, C.-C. Kuang, X. Chen, B. Gong, Y. Zhao, J. Zhang, C. Zheng and J. C. S. Wu, Appl. Catal., B, 2017, 202, 695–703 CrossRef CAS.
- B. Pan, S. Luo, W. Su and X. Wang, Appl. Catal., B, 2015, 168–169, 458–464 CrossRef CAS.
- Y. Dou, A. Zhou, Y. Yao, S. Y. Lim, J.-R. Li and W. Zhang, Appl. Catal., B, 2021, 286, 119876 CrossRef CAS.
- M. Manzanares, C. Fàbrega, J. Oriol Ossó, L. F. Vega, T. Andreu and J. R. Morante, Appl. Catal., B, 2014, 150–151, 57–62 CrossRef CAS.
- M. Tahir and N. S. Amin, Appl. Catal., A, 2013, 467, 483–496 CrossRef CAS.
- J. An, T. Shen, W. Chang, Y. Zhao, B. Qi and Y.-F. Song, Inorg. Chem. Front., 2021, 8, 996–1004 RSC.
- C. Han, Y. Lei, B. Wang and Y. Wang, ChemSusChem, 2018, 11, 4237–4245 CrossRef CAS PubMed.
- I. H. Tseng and J. C. S. Wu, Catal. Today, 2004, 97, 113–119 CrossRef CAS.
- K. Teramura, Z. Wang, S. Hosokawa, Y. Sakata and T. Tanaka, Chemistry, 2014, 20, 9906–9909 CrossRef CAS PubMed.
- I. H. Tseng, J. C. S. Wu and H.-Y. Chou, J. Catal., 2004, 221, 432–440 CrossRef CAS.
- S. Feng, J. Zhao, Y. Bai, X. Liang, T. Wang and C. Wang, J. CO2 Util., 2020, 38, 1–9 CrossRef CAS.
- D. Luo, Y. Bi, W. Kan, N. Zhang and S. Hong, J. Mol. Struct., 2011, 994, 325–331 CrossRef CAS.
- P. Devi and J. P. Singh, J. CO2 Util., 2021, 43, 101376 CrossRef CAS.
- W. Bi, Y. Hu, H. Jiang, L. Zhang and C. Li, Adv. Funct. Mater., 2021, 31, 2010780 CrossRef CAS.
- Y. X. Pan, Y. You, S. Xin, Y. Li, G. Fu, Z. Cui, Y. L. Men, F. F. Cao, S. H. Yu and J. B. Goodenough, J. Am. Chem. Soc., 2017, 139, 4123–4129 CrossRef CAS PubMed.
- Y. He, Y. Wang, L. Zhang, B. Teng and M. Fan, Appl. Catal., B, 2015, 168–169, 1–8 CAS.
- H. Shi, G. Chen, C. Zhang and Z. Zou, ACS Catal., 2014, 4, 3637–3643 CrossRef CAS.
- N. T. Thanh Truc, N. T. Hanh, M. V. Nguyen, N. T. P. Le Chi, N. Van Noi, D. T. Tran, M. N. Ha, D. Q. Trung and T.-D. Pham, Appl. Surf. Sci., 2018, 457, 968–974 CrossRef CAS.
- W. Yu, D. Xu and T. Peng, J. Mater. Chem. A, 2015, 3, 19936–19947 RSC.
- L. Liu, Ceram. Int., 2016, 42, 12516–12520 CrossRef CAS.
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed.
- J. Mao, T. Peng, X. Zhang, K. Li and L. Zan, Catal. Commun., 2012, 28, 38–41 CrossRef CAS.
- X. Y.-z. Zhao Lin, R. Chen and Y. Diao, J. Synth. Cryst., 2018, 47, 2663–2668 Search PubMed.
- S. K. Parayil, A. Razzaq, S.-M. Park, H. R. Kim, C. A. Grimes and S.-I. In, Appl. Catal., A, 2015, 498, 205–213 CrossRef CAS.
- M. Z. Dandan Lu, Z. Zhang, Q. Li, X. Wang and J. Yang, Nanoscale Res. Lett., 2014, 272, 9 Search PubMed.
- X. Li, W. Bi, Z. Wang, W. Zhu, W. Chu, C. Wu and Y. Xie, Nano Res., 2018, 11, 3362–3370 CrossRef CAS.
- Q. Zhang, T. Gao, J. M. Andino and Y. Li, Appl. Catal., B, 2012, 123–124, 257–264 CrossRef CAS.
- Q. Zhang, Y. Li, E. A. Ackerman, M. Gajdardziska-Josifovska and H. Li, Appl. Catal., A, 2011, 400, 195–202 CrossRef CAS.
- Q. Liu, Q. Chen, T. Li, Q. Ren, S. Zhong, Y. Zhao and S. Bai, J. Mater. Chem. A, 2019, 7, 27007–27015 RSC.
- J. Pan, G. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed. Engl., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- J. Pan, X. Wu, L. Wang, G. Liu, G. Q. Lu and H. M. Cheng, Chem. Commun., 2011, 47, 8361–8363 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- J. Mao, L. Ye, K. Li, X. Zhang, J. Liu, T. Peng and L. Zan, Appl. Catal., B, 2014, 144, 855–862 CrossRef CAS.
- B. Han, X. Ou, Z. Deng, Y. Song, C. Tian, H. Deng, Y. J. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- L. Wang, B. Zhao, C. Wang, M. Sun, Y. Yu and B. Zhang, J. Mater. Chem. A, 2020, 8, 10175–10179 RSC.
- H. Pang, X. Meng, P. Li, K. Chang, W. Zhou, X. Wang, X. Zhang, W. Jevasuwan, N. Fukata, D. Wang and J. Ye, ACS Energy Lett., 2019, 4, 1387–1393 CrossRef CAS.
- A. K. Kharade and S.-m. Chang, J. Phys. Chem. C, 2020, 124, 10981–10992 CrossRef CAS.
- T. Billo, F. Y. Fu, P. Raghunath, I. Shown, W. F. Chen, H. T. Lien, T. H. Shen, J. F. Lee, T. S. Chan, K. Y. Huang, C. I. Wu, M. C. Lin, J. S. Hwang, C. H. Lee, L. C. Chen and K. H. Chen, Small, 2018, 14, 1702928 CrossRef PubMed.
- X. Zhang, Z. Zhang, J. Li, X. Zhao, D. Wu and Z. Zhou, J. Mater. Chem. A, 2017, 5, 12899–12903 RSC.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- X. Qian, W. Yang, S. Gao, J. Xiao, S. Basu, A. Yoshimura, Y. Shi, V. Meunier and Q. Li, ACS Appl. Mater. Interfaces, 2020, 12, 55982–55993 CrossRef CAS PubMed.
- Q. Lang, W. Hu, P. Zhou, T. Huang, S. Zhong, L. Yang, J. Chen and S. Bai, Nanotechnology, 2017, 28, 484003 CrossRef PubMed.
- C. Han, Y. Lei, B. Wang, C. Wu, X. Zhang, S. Shen, L. Sun, Q. Tian, Q. Feng and Y. Wang, Chem. Commun., 2019, 55, 1572–1575 RSC.
- M. Ye, X. Wang, E. Liu, J. Ye and D. Wang, ChemSusChem, 2018, 11, 1606–1611 CrossRef CAS PubMed.
- Y. Peng, L. Wang, Q. Luo, Y. Cao, Y. Dai, Z. Li, H. Li, X. Zheng, W. Yan, J. Yang and J. Zeng, Chem, 2018, 4, 613–625 CAS.
- J. Di, C. Chen, C. Zhu, P. Song, J. Xiong, M. Ji, J. Zhou, Q. Fu, M. Xu, W. Hao, J. Xia, S. Li, H. Li and Z. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 30786–30792 CrossRef CAS PubMed.
- Y. Liu, B. Huang, Y. Dai, X. Zhang, X. Qin, M. Jiang and M.-H. Whangbo, Catal. Commun., 2009, 11, 210–213 CrossRef CAS.
- X. Zhu, A. Yamamoto, S. Imai, A. Tanaka, H. Kominami and H. Yoshida, Appl. Catal., B, 2020, 274, 119085 CrossRef CAS.
- P. Li, Y. Zhou, Z. Zhao, Q. Xu, X. Wang, M. Xiao and Z. Zou, J. Am. Chem. Soc., 2015, 137, 9547–9550 CrossRef CAS PubMed.
- Q. Han, X. Bai, Z. Man, H. He, L. Li, J. Hu, A. Alsaedi, T. Hayat, Z. Yu, W. Zhang, J. Wang, Y. Zhou and Z. Zou, J. Am. Chem. Soc., 2019, 141, 4209–4213 CrossRef CAS PubMed.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |