
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methane synthesis from CO2 and H2O using electrochemical cells with polymer electrolyte membranes and Ru catalysts at around 120 °C: a comparative study to a phosphate-based electrolyte cell†
Raisei
Sagara
,
Rika
Hayashi
,
Aika
Hirata
,
Shintaroh
Nagaishi
and
Jun
Kubota
 *
*
Department of Chemical Engineering, Fukuoka University, 8-19-1, Nanakuma, Jonan-ku, Fukuoka 814-0180, Japan. E-mail: jkubota@fukuoka-u.ac.jp
First published on 16th October 2023
Abstract
Electrochemical cells with fluorine-based polymer electrolyte membranes (PEMs) and Ru catalysts have been investigated for the production of methane (CH4) from CO2 and H2O by using electricity at 120 °C. CH4 was synthesized with a rate of 12 nmol s−1 cm−2 at a current density of 10 mA cm−2 with a CO2 flow of 0.055 mlSTP min−1 in the cathode vessel and with an Ar + H2O flow of 10 mlSTP min−1 + 10 μLliquid min−1 in the anode vessel (STP; standard temperature and pressure at 0 °C and 101.3 kPa, respectively). This rate was corresponding to the current efficiency of ca. 85%, and unreacted H2 and subproducts of CO were obtained with current efficiencies of ca. 14 and 1%, respectively. The properties of temperature and current density dependence were discussed in this article. The present system exhibits significantly higher selectivity in synthesizing CH4 compared to electrochemical CO2 reduction systems at the electrode/electrolyte interfaces. Electrochemical CO2 reduction did not take place in the present system, and the importance of the combination of water electrolysis and catalytic methanation at solid/gas interfaces was proposed. A comparative study between phosphate (CsH2PO4/SiP2O7) at 250 °C and polymer electrolytes at 120 °C was performed and the possibility for practical applications of both systems was discussed.
Introduction
The conversion of renewable electricity generated from natural power sources such as solar photovoltaics, solar thermal, wind turbines, geothermal, and ocean energies into chemical fuels is a critical technology for achieving a carbon-neutral society.1–5 The current capacity of rechargeable batteries is insufficient for large-scale power storage, making the conversion of such power into chemical fuels a more favourable option.1–5 CH4 is one of the promising energy carriers that can be synthesized from sustainable electricity and atmospheric species such as CO2 and water, due to the abundance of natural gas infrastructure. This type of CH4 is a carbon-neutral fuel. Conventional technology can be used to produce sustainable CH4 through the combination of water electrolysis to produce hydrogen and the catalytic hydrogenation of CO2, known as the Sabatier reaction. This process has been extensively examined in Germany, and many projects on methanation from renewable energy are currently underway worldwide.4,5However, the combination of water electrolysis and catalytic reactions has a significant disadvantage when coupled with unsteady power supplies such as solar and wind. Water electrolysis essentially needs to follow fluctuations in the unsteady power supplies, and as seen from fuel cells, electrochemical cells have the potential to follow the unsteady operation. However, the catalytic reactors need heat management to maintain the reaction temperature, so they strongly prefer the steady operation. If water electrolysis and the catalytic reaction are operated independently, it is hard to synchronize the operation of both processes. Thus, a hydrogen storage system is likely required between water electrolysis and catalytic reactions, but there are many issues associated with hydrogen storage.6,7 In addition to eliminating the need for hydrogen storage, the one-step process has another advantage. If water electrolysis and catalytic hydrogenation are performed in the same system at the same temperature with thermal equilibrium, there are thermodynamic advantages, such as lowering the thermoneutral potential which is the practical cell voltage. CO2 methanation is a highly exothermic reaction with a ΔrH° of −41 kJ mol−1-H2.8,9 On the other hand, water and steam electrolysis requires heat corresponding to a TΔrS° of 49 and 13 kJ mol−1-H2, respectively.8,9 If water electrolysis and CO2 methanation are performed in the same system at a common temperature, these exothermic and endothermic heats can compensate for each other, reducing heat loss. In other words, if both CO2 methanation and water electrolysis are conducted at the same temperature, the exothermic heat produced by CO2 methanation can be fully utilized for the water electrolysis process. The theoretical standard thermoneutral potential of steam electrolysis is 1.19 V, while that for CH4 synthesis from CO2 and steam is 1.02 V.8,9 It is, therefore, favourable for the methanation catalysts to be assembled in a water electrolyzer.
There have been numerous attempts to perform electrochemical reduction of CO2 in aqueous or polymer electrolytes at low temperatures below 100 °C.10–16 However, there have been few studies that have successfully synthesized CH4 or such hydrocarbons at realistic current efficiencies and selectivity suitable for practical applications.10–16 CO and formic acid are relatively selectively synthesizable compounds,11 but there are numerous difficulties in utilizing them directly within existing fuel networks. Even when using polymer electrolyte membrane (PEM) cells, the resulting product is either diversified or becomes CO.17–19 Our group primarily investigated synthesizing CH4 using electrochemical devices that combine an electrochemical cell and a catalytic reactor at the same temperature, mainly using phosphate electrolytes in the range of 200–270 °C.8,9 Our studies are focused on the combination of water electrolysis and the Sabatier reaction, rather than the electrochemical reduction of CO2 at the electrolyte/cathode interface which often results in the production of various products without selectivity. The Sabatier reaction can convert CO2 to CH4 with >95% conversion and low amounts of subproducts (<1%) are obtained. For electrochemical CO2 reduction at an electrolyte/cathode interface, a successful example with high selectivity is only that CO can be obtained from electrochemical CO2 reduction.10–19 Typical examples show the production of CH4, which may also include significant amounts of CO, ethylene, formic acid, hydrogen, and other by products.8 Our group has investigated the electrochemical cells with phosphate-based electrolyte at around 270 °C and a maximum current efficiency of ∼93% for CH4 synthesis was reported.8,9 However, catalytic methanation of CO2 by using H2 at a gas/catalyst surface readily occurs at lower temperatures, and PEM electrolyzers seems to be applicable to methanation. Using PEM electrolyzers, selective synthesis for a specific hydrocarbon or alcohol has not been achieved.10–19
The strategy of our study is that it focuses on tuning catalyst performance for matching the electrolysis conditions. If CO2 reduction is carried out electrochemically at the electrolyte/electrode interface, it can lead to the production of various products. Combining CO2 methanation on the gas/solid surfaces with hydrogen evolution through water electrolysis appears to enable the synthesis of methane with high selectivity. In the present study, the conditions are determined to match the electrolysis conditions, and optimizing the catalyst under these particular conditions is required.
In this study, we have found that PEM electrolyzers can be used for CH4 synthesis by increasing the operating temperature from 80 to 120 °C and combining the cathode with Ru methanation catalysts. This short article describes the properties of electrochemical devices equipped with polymer electrolytes and Ru catalysts, and discusses the feasibility of using this electrochemical system for converting surplus sustainable electricity into CH4.
Experimental
The polymer electrolyte membrane electrochemical cell was similar to that reported in our previous studies with phosphate electrolytes.8,9 The illustrations of both cells are shown in Fig. 1. Aquivion® was picked up among a variety of polymer electrolytes such as Nafion® and other perfluorosulfonic acids, because of the high glass-transition temperature of 140 °C. An ionomer membrane of Aquivion® E98-05 50 μm thick (Sigma-Aldrich) was cut into a diameter of 25 mm. A drop of suspension of Aquivion® D83-24B (Sigma-Aldrich) was coated on both surfaces of the ionomer sheet, and hot-pressed at 150 °C between the cathode and anode plates. The cathode was that 50 wt%-Pt/C (TEC10E50E, Tanaka Kikinzoku Kogyo K.K.) of 2 mg-Pt cm−2 was pasted with suspension of Aquivion® on a carbon paper (TGP-H-120, Toray). On the backside of the cathode, 10 wt%-Ru/ZrO2 was placed. The electrochemical area was defined by the geometric area of the cathode of 3.14 cm−2. 10 wt%-Ru/ZrO2 was synthesized by the impregnation method of ZrO2 (Catalysis Society of Japan, reference catalysts, JRC-ZRO-9) into tetrahydrofuran solution of Ru3(CO)12 (Tanaka Kikinzoku Kogyo K.K.), and detailed information on the catalysts is given in the ESI.† The Ru/ZrO2 catalysts were pressed and crushed to form granules of 500–800 μm and 800 mg of Ru/ZrO2 was placed on the opposite face of the Pd–Ag cathode from the electrolyte. The substrate of the anode was a sintered-Ti metal fiber plate 20 mm in diameter and 1 mm thick (Bekaert Japan), and 3.2 mg cm−2 of IrO2 powder (Tanaka Kikinzoku Kogyo K.K.) was pasted as an ink with a suspension of Aquivion® on the substrate. Cathode and anode vessels were made of stainless used steel (SUS304) and titanium, respectively.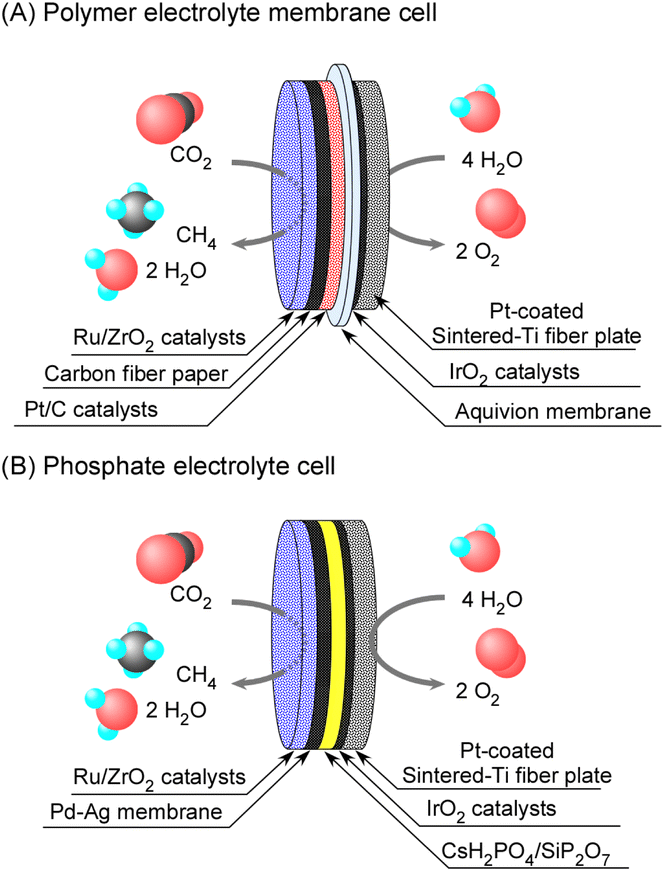 | ||
| Fig. 1 Schematic illustration of polymer electrolyte membrane (A) and phosphate electrolyte (B) electrochemical cells for methane synthesis from CO2 and H2O. | ||
The phosphate electrolyte cell was similar to that in our previous reports, and the electrolyte was a composite of CsH2PO4 and SiP2O7 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio.8,9 A hydrogen-permeable Pd–Ag (3:1 atomic ratio) membrane 25 mm in diameter and 100 μm thick was used to penetrate hydrogen to avoid immersion of melted phosphate into the Ru catalyst layer. An IrO2-coated sintered-Ti metal fiber plate was used even for the phosphate electrolyte cells.
:2 molar ratio.8,9 A hydrogen-permeable Pd–Ag (3:1 atomic ratio) membrane 25 mm in diameter and 100 μm thick was used to penetrate hydrogen to avoid immersion of melted phosphate into the Ru catalyst layer. An IrO2-coated sintered-Ti metal fiber plate was used even for the phosphate electrolyte cells.
The gas from the cathode was analyzed by using a gas chromatograph equipped with a thermal conductivity detector. The concentration of respective species was quantitatively estimated with respect to the amount of introduced CO2, assuming carbon-balance.
Results and discussion
The temperature-dependence of the formation rate and current efficiency for CH4 synthesis is shown in Fig. 2. The current efficiency for CH4, CO, and H2 formations (CECH4, CECO, and CEH2, respectively) was estimated as:

where rCH4, rCO, and rH2 are the rates of formation of CH4, CO, and H2, respectively, in mol cm−2 s−1. F and j are the faradaic constant (96
 500 s A mol−1) and current density in A cm−2, respectively. It should be noted that the current efficiency here is expressed in terms of the total number of electrons, including the portion of H2O generated as a byproduct.
500 s A mol−1) and current density in A cm−2, respectively. It should be noted that the current efficiency here is expressed in terms of the total number of electrons, including the portion of H2O generated as a byproduct.
 | ||
| Fig. 2 Formation rates and the current efficiencies of CH4, H2, and CO as a function of temperature at 30 mA cm−2 for the polymer electrolyte membrane cell with 10 wt%-Ru/ZrO2. The flow rate of CO2 for the cathode vessel was 0.165 mlSTP min−1 and that of the mixture of Ar and H2O for the anode vessel was 10 mlSTP min−1 and 10 μlLiquid min−1, respectively. | ||
As shown in Fig. 2, no CH4 production was detected below 70 °C. However, even in the absence of CH4 and CO production, the current efficiency for H2 formation was estimated to be only ca. 63% at these temperatures. The discrepancy from 100% in the total current efficiency was notable at lower temperatures (<100 °C). At these temperatures, there may have been issues related to cross-leakage of gases between the cathode and anode due to insufficient gas sealing. Additionally, the discrepancy from 100% can be attributed to the relatively lower absolute accuracy of the formation rate measurements. The rate of formation was estimated based on the CO2 feeding rate to the cell assuming the carbon balance, which had a flow rate of only 0.165 mlSTP min−1, stoichiometrically matched with a current density of 30 mA cm−2. The control and measurement of such a small flow rate posed significant challenges, resulting in an accuracy of approximately ±10% or more in many cases.
As the temperature increased from 80 to 120 °C, the current efficiency for CH4 formation increased to 70%. However, in the same temperature range, the current efficiency for H2 production decreased from 65% to 25%. At temperatures between 100 and 120 °C, CO was detected at approximately 0.4% of the current efficiency. This finding suggests that the polymer electrolyte membrane cell is capable of synthesizing CH4 at 120 °C with a current efficiency of around 70%. Unfortunately, operating the cell at 120 °C resulted in the degradation of its characteristics within a few days. Consequently, further testing at higher temperatures was not conducted.
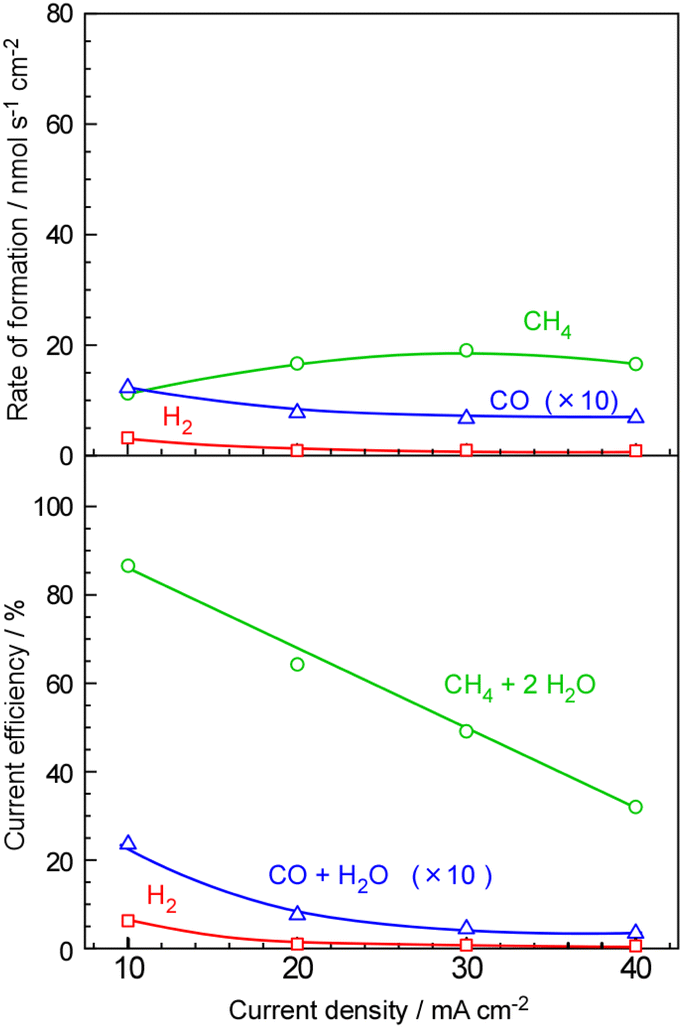
The dependence of current density on the formation rate and current efficiency for CH4 synthesis is illustrated in Fig. 3. As the current density increased from 10 mA cm−2 to 40 mA cm−2, both CH4 and H2 formation rates increased. However, the rate of H2 formation exhibited a significantly steeper slope compared to that of CH4, indicating inadequate catalytic activity for CH4 formation at higher current densities. Thus, the current efficiency for unreacted H2 increased from 14% to 35% with increasing current density from 10 mA cm−2 to 40 mA cm−2. The formation of CO was as high as 1.2% at 10 mA cm−2. The reason for the increase in CO production with decreasing current density is unclear. The formation rate of CO remained constant at approximately 0.5–0.6 nmol s−1 cm−2 regardless of the current density, leading to a higher current efficiency for CO at lower current densities. The insufficient mixing between the fed CO2 and evolved H2 in the catalyst layer may be a potential cause.
 | ||
| Fig. 3 Formation rates and the current efficiencies of CH4, H2, and CO as a function of current densities at 120 °C for the polymer electrolyte membrane cell with 10 wt%-Ru/ZrO2. The flow rate of CO2 for the cathode vessel was 0.055 mlSTP min−1 per 1 mA cm−2 and that of the mixture of Ar and H2O for the anode vessel was 10 mlSTP min−1 and 10 μlLiquid min−1, respectively. | ||
The cell voltage was measured during CH4 synthesis at various current densities, as shown in Fig. 4. The experiments were conducted over a period of several days. The cell voltage was 1.9 V at 10 mA cm−2 and 2.4–2.5 V at 40 mA cm−2. The theoretical standard thermoneutral potential for CH4 synthesis from CO2 and steam is 1.02 V, which corresponds to 54% of the voltage observed at 10 mA cm−2 (1.9 V). Considering the current efficiency of approximately 85% under these conditions, the estimated energy efficiency for the conversion of electricity to CH4 was 46%. Lowering the cell voltage is crucial for achieving higher energy efficiency.
 | ||
| Fig. 4 Cell voltage and current density as a function of the elapsed time in the measurements for Fig. 3. The operating temperature was 120 °C and the cell was operated in a constant current mode. | ||
Summarizing the results of the polymer electrolyte membrane cell, CH4 was synthesized with ca. 10% of current efficiency for unreacted H2 at a current density of 10 mA cm−2, suggesting that ca. 90% of current was used for CH4 synthesis. This indicated that the polymer electrolyte membrane cell had feasible potential for CH4 production from CO2 and H2O with electricity. To achieve practical application, it is necessary to attain a target current density of several hundred mA cm−2. This can be accomplished by either enhancing the activity of Ru catalysts by more than tenfold or increasing the operating temperature by a few tens of degrees Celsius. In any case, the present system exhibits significantly higher selectivity in synthesizing CH4 compared to electrochemical CO2 reduction systems at the electrode/electrolyte interfaces. This conclusion suggests that the system holds great potential as a practical conversion technology for transforming surplus electricity into CH4.
For comparison, we conducted CH4 synthesis using a cell with a phosphate-based electrolyte, which our group has been investigating. The results are presented in Fig. 5. At a low current density of 10 mA cm−2, the current efficiency for H2 and CO formation was 6.1% and 0.23%, respectively, indicating that 94% of the current was utilized for CH4 production. In practical terms, CH4 production was calculated as 87% of current efficiency, with the deviation of the total current efficiency from 100% falling within the margin of error.
 | ||
| Fig. 5 Formation rates and the current efficiencies of CH4, H2, and CO as a function of current densities at 250 °C for the phosphate electrolyte cell with 10 wt%-Ru/ZrO2. The flow rate of CO2 for the cathode vessel was 0.055 mlSTP min−1 per 1 mA cm−2 and that of the mixture of Ar and H2O for the anode vessel was 10 mlSTP min−1 and 10 μlLiquid min−1, respectively. | ||
As the current density increased, the rate of CH4 formation did not consistently increase, and the rates for H2 and CO decreased. Furthermore, the overall current efficiencies for CH4, H2, and CO decreased with higher current densities. The deficit in total current efficiency, falling short of 100%, was most likely due to cross-leakage of evolved gases between the cathode and anode. In the case of the phosphate electrolyte, a composite of CsH2PO4 and SiP2O7, the mechanical strength of the electrolyte was insufficient, and cross-leakage of gas between the cathode and anode was frequently observed at higher current densities.
Unlike phosphate electrolytes, polymer membranes such as Nafion® and Aquivion® used in fuel cells and water electrolysis typically do not encounter issues like cross-leakage. However, a notable drawback observed in phosphate electrolytes is a decrease in overall current efficiency, particularly under high current conditions. We had previously believed that it would be difficult to directly synthesize methane using polymer electrolytes due to their low operating temperature, and thus, we thought that phosphate-based electrolytes should be used. However, the results of this study demonstrate that direct methane synthesis is indeed possible with polymer electrolytes. This suggests the potential for achieving practical levels of methane synthesis at sufficient conversion rates with further catalyst improvements, opening up new avenues for research and development.
There are essentially two reaction pathways both in the polymer electrolyte membrane cells and the phosphate electrolyte cells. H+ ions are reduced to H on Pt or Pd–Ag. These H atoms desorb as H2 molecules into the gas phase and react with CO2 on the Ru surface. In another pathway, H atoms spill over through the support surface to the Ru surface to directly react with CO2. In the proposed cells, the catalyst layer is 1.5 mm thick, making long-distance diffusion via spill-over unlikely, and therefore, the majority of reactions occur in the gas phase pathway.
Efficient contact between CO2 and hydrogen significantly impacts conversion efficiency. To achieve efficient reactions, methods such as recycling the cathode outlet gas, which includes unreacted hydrogen, to increase residence time can be employed. Another approach is to promote the reaction pathway through spill-over by increasing the contact surface area between the electrolyte and catalyst by moderate mixing instead of separating them. In our concept, it is crucial to preserve Ru surfaces that remain uncovered by the electrolyte and can catalytically activate CO2. Therefore, it is essential to generate Pt sites that facilitate proton reduction in contact with the electrolyte and Ru sites that activate CO2 independent of contact with the electrolyte. The polymer electrolyte cells can be developed to such cell structures because the use of hydrogen-permeable membranes is unnecessary.
This result is highly unique in the field of synthesizing methane from water and CO2, with very few comparable research examples available for comparison.10–16 There is a research example where the temperature difference between the water electrolysis cell and the methanation catalyst reactor is as much as 100 °C, but it is difficult to consider them as a single electrochemical device.20 While there have been numerous studies on electrochemical CO2 reduction, there are hardly any instances where a single hydrocarbon is selectively synthesized through electrochemical CO2 reduction, as demonstrated in this study.10–16 Most reports on hydrocarbon production through electrochemical CO2 reduction involve the concomitant generation of olefins, alcohols, carboxylic acids, and other byproducts. This underscores the significance of utilizing catalytic hydrogenation reactions at the gas–solid interface rather than conducting CO2 reduction at the electrode–electrolyte interface.
Through this research, the possibility of creating a device that combines water electrolysis and methane synthesis using a polymer electrolyte in the temperature range of approximately 100 to 120 °C has been demonstrated. This device could produce CH4 from CO2 and H2O in a single process. At a current density of around 10 mA cm−2, the catalyst activity was sufficiently close to optimal, leading to approximately 90% of the current being converted to CH4, with the remainder being transformed into H2 and CO. Approaching practical current density ranges, nearing several hundred mA cm−2, the efficiency of converting current to CH4 decreases, and H2 production becomes more prominent. The development of higher-activity catalysts is identified as a key factor in achieving the desired outcomes at higher practical current densities. Considering the utilization of power from renewable sources such as solar and wind energy, the polymer electrolyte cells with lower operating temperature can be more easily adapted to fluctuating operation than phosphate electrolyte cells. We hope that these methods will continue to be developed as promising approaches for directly producing chemical fuels such as CH4 from surplus electricity originating from renewable energy sources.
Conclusions
Polymer electrolyte membrane cells were examined for CH4 synthesis from CO2 and H2O with electricity. Utilizing water electrolysis at electrode/electrolyte interfaces and methanation at the solid/gas interface, CH4 was perfectly synthesized with a subproduct of <1% of CO and 15% of unreacted H2 at a current efficiency of 10 mA cm−2 and 120 °C. With increasing current density, the portion of unreacted H2 increased significantly. It was found that electrolysis at 100–120 °C was required for synthesis of CH4. In comparison with the phosphate-based electrochemical system which was reported in previous literature studies, it was found that the catalytic activity for CH4 synthesis in the polymer electrolyte cells at 120 °C was insufficient, while using the phosphate-based electrolyte system at 250 °C provided more favorable results. To make CH4 synthesis from CO2 and H2O by using polymer electrolyte membrane cells more practical, key factors include improving the catalyst performance and increasing the operating temperature. Unlike the electrochemical reduction of CO2 at the electrode/electrolyte interface, where various products can form, the process described in this study results in almost exclusively CH4 and H2O as products, with only minimal byproducts of unreacted H2 and a small amount of CO. CO2 reduction at the electrode/electrolyte interface and the process demonstrated here are at completely different levels of practicality and applicability.Author contributions
Sagara conducted all practical experiments for electrochemical cell operations, while Hayashi and Hirata contributed to the synthesis of catalysts and electrolyte materials. Nagaishi and Kubota contributed to all categories in CRediT, including supervision.Conflicts of interest
There are no conflicts to declare.References
- S. Mucci, A. Mitsos and D. Bongartz, Comput. Chem. Eng., 2023, 175, 108260 CrossRef CAS.
- M. J. Palys and P. Daoutidis, Comput. Chem. Eng., 2022, 165, 107948 CrossRef CAS.
- J. Gong, N. J. English, D. Pant, G. R. Patzke, S. Protti and T. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 7179–7181 CrossRef CAS.
- M. Held, D. Schollenberger, S. Sauerschell, S. Bajohr and T. Kolb, Chem. Ing. Tech., 2020, 92, 595–602 CrossRef CAS.
- E. Kötter, L. Schneider, F. Sehnke, K. Ohnmeiss and R. Schröer, J. Energy Storage, 2016, 5, 113–119 CrossRef.
- T. Amirthan and M. S. A. Perera, J. Nat. Gas Sci. Eng., 2022, 108, 104843 CrossRef CAS.
- M. R. Usman, Renewable Sustainable Energy Rev., 2022, 167, 112743 CrossRef CAS.
- J. Kubota, T. Okumura and R. Hayashi, Sustainable Energy Fuels, 2022, 6, 1362–1372 RSC.
- J. Kubota and T. Okumura, Sustainable Energy Fuels, 2021, 5, 935–940 RSC.
- M. Gattrell, N. Gupta and A. Co, J. Electroanal. Chem., 2006, 594, 1–19 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- R. J. Lim, M. Xie, M. A. Sk, J.-M. Lee, A. Fisher, X. Wang and K. H. Lim, Catal. Today, 2014, 233, 169–180 CrossRef CAS.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439–456 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 044508 CrossRef.
- S. Garg, M. Li, A. Z. Weber, L. Ge, L. Li, V. Rudolph, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- M. Sato, H. Ogihara and I. Yamanaka, ISIJ Int., 2019, 59, 623–627 CrossRef CAS.
- H. Ogihara, T. Maezuru, Y. Ogishima and I. Yamanaka, Electrocatal, 2018, 9, 220–225 CrossRef CAS.
- N. Gutiérrez-Guerra, L. Moreno-López, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas-Consuegra, Appl. Catal., B, 2016, 188, 272–282 CrossRef.
- H. Nakajima, A. Shima, M. Inoue, T. Abe, H. Matsumoto, O. S. Mendoza-Hernandez and Y. Sone, Electrochem, 2022, 90, 067008 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00985h |
| This journal is © The Royal Society of Chemistry 2023 |