
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solventless hydrodeoxygenation of isoeugenol and dihydroeugenol in batch and continuous modes over a zeolite-supported FeNi catalyst†
Zuzana
Vajglová
a,
Olha
Yevdokimova
a,
Ananias
Medina
 a,
Kari
Eränen
a,
Teija
Tirri
a,
Jarl
Hemming
a,
Johan
Lindén
b,
Ilari
Angervo
c,
Pia
Damlin
d,
Dmitry E.
Doronkin
e,
Päivi
Mäki-Arvela
a and
Dmitry Yu.
Murzin
*a
a,
Kari
Eränen
a,
Teija
Tirri
a,
Jarl
Hemming
a,
Johan
Lindén
b,
Ilari
Angervo
c,
Pia
Damlin
d,
Dmitry E.
Doronkin
e,
Päivi
Mäki-Arvela
a and
Dmitry Yu.
Murzin
*a
aÅbo Akademi University, Johan Gadolin Process Chemistry Centre, Henriksgatan 2, 20500, Turku/Åbo, Finland. E-mail: dmurzin@abo.fi
bFaculty of Science and Engineering/Physics, Åbo Akademi University, Henriksgatan 2, 20520, Turku/Åbo, Finland
cWihuri Physical Laboratory, University of Turku, 20014, Turku, Finland
dDepartment of Material Chemistry, University of Turku, 20014, Turku, Finland
eInstitute of Chemical Technology and Polymer Chemistry and Institute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Kaiserstrasse 12, 76131, Karlsruhe, Germany
First published on 8th August 2023
Abstract
A low-cost bimetallic bifunctional 5–5 wt% FeNi/H-Beta-300 catalyst was investigated in solventless hydrodeoxygenation of lignin-derived model compounds isoeugenol or dihydroeugenol in batch and continuous modes. The catalyst was characterized in detail by laser diffraction, scanning electron microscopy–energy-dispersive X-ray microanalysis, inductively coupled plasma–optical emission spectrometry, transmission electron microscopy, Fourier-transform infrared spectroscopy with pyridine, X-ray diffraction, Mössbauer spectroscopy, X-ray absorption spectroscopy, hydrogen temperature programmed reduction, nitrogen physisorption, thermogravimetric analysis, oxygen temperature-programmed oxidation, organic elemental analysis, soluble coke extraction with dichloromethane, and Raman spectroscopy. The composition of the reaction mixture was analysed by GC-FID, GC-MS, SEC and Karl-Fischer titration, while microGC-TCD was used for the analysis of the gas phase. Selectivity of 80% to the desired oxygen-free compounds was obtained at ca. 80% of the initial dihydroeugenol conversion with 0.3 g of catalyst at 300 °C and 30 bar of hydrogen with a residence time of 12 min. Catalyst deactivation occurred via aliphatic coke formation which resulted not only in a decrease in conversion but also significant selectivity changes with increasing time-on-stream. The apparent activation energy of dihydroeugenol hydrodeoxygenation in solventless isoeugenol hydrodeoxygenation was calculated to be 6.3 kJ mol−1 ascribed to both external mass transfer limitations of hydrogen dissolved in dihydroeugenol and by rapid catalyst deactivation in the initial isoeugenol hydrogenation. The spent catalyst was successfully regenerated by coke oxidation and subsequently reused.
1 Introduction
Substitution of petroleum with fuels obtained from renewable sources, such as bio-oil is an important step to prevent many environmental problems, including global warming. Because bio-oils have high acidity and oxygen content as well as low stability, they are not suitable to be used directly as fuels. Hydrodeoxygenation (HDO) is a cheap way to improve the quality of fuels from bio-oil by removing oxygen from the oxygen-containing compounds. HDO is the most commonly used method to upgrade bio-oil to deoxygenated hydrocarbon fuels, which have higher stability and energy density.1 Bio-oil consists of an aqueous and organic phase with the latter containing various acids, aldehydes, ketones and phenolic components, reflecting the complex nature of lignocellulosic biomass. Due to this complexity typically model compounds are used to study the reaction mechanism of HDO and its kinetics. Isoeugenol (IE) and its hydrogenated product, dihydroeugenol (DHE) are used in this work as model compounds, because isoeugenol containing hydroxyl, methoxy and allyl groups, also found in bio-oils derived from lignin, can be considered as a representative lignin model compound.1,2Batch-wise HDO of lignin-derived compounds was already performed with noble metals,1,3,4 noble metal-containing bimetallic catalysts5,6 and non-noble metal-based catalysts.2,3,7 As there is an increased interest in industry to use cheap non-noble metal materials as catalysts, abundant Fe and Ni were selected as active metals. It has been already reported that HDO of triolein was successfully performed over a bifunctional Ni–Fe/ZSM-5/SAPO-11 catalyst, which displayed higher catalytic activity than monometallic ones.7 Fe is oxophilic metal, which promotes oxygen adsorption and subsequently activity and selectivity in HDO of oxygen-containing compounds.8 Despite a few attempts reported in the literature, there is not much information about the application of non-noble bimetallic catalysts for HDO of lignin-derived compounds. Furthermore, it is known that acidity, structure and morphology of a support can influence catalytic activity,9 with strong acidity eventually resulting in cracking and catalyst deactivation. Subsequently in the current work, mildly acidic Beta-300 was chosen as a support.
The aim of the current work was to compare the performance of a bimetallic bifunctional 5–5 wt% FeNi/H-Beta-300 in batch and continuous reactors. According to our knowledge, continuous HDO of similar components has been previously made in the gas-phase,10 and the open literature is almost devoid of studies performed in the liquid-phase when IE and DHE are used as a feedstock under solvent-less conditions. A comprehensive product analysis was performed including the liquid, solid and gas phase analysis. One of the aims of the current research was to elucidate catalyst stability in the absence of any solvent, as the latter ones, especially long-chain hydrocarbons, are not inert under the reaction conditions.11 Furthermore, several physico-chemical methods were applied to characterize the fresh and spent catalysts, such as laser diffraction, scanning electron microscopy–energy-dispersive X-ray microanalysis, inductively coupled plasma – optical emission spectrometry, transmission electron microscopy, Fourier-transform infrared spectroscopy with pyridine, X-ray diffraction, Mössbauer spectroscopy, X-ray absorption spectroscopy, hydrogen temperature programmed reduction, nitrogen physisorption, thermogravimetric analysis, oxygen temperature-programmed oxidation, organic elemental analysis, soluble coke extraction with dichloromethane, and Raman spectroscopy.
2 Experimental
2.1 Preparation of the catalyst
In the current work, a low-cost bimetallic bifunctional 5–5 wt% FeNi/H-Beta-300 catalyst was synthesised by the subsequent incipient wetness impregnation method with two impregnation steps for each metal. The metal composition was selected based on the results of the previous work dealing with Fe–Ni metal ratios in the catalyst for co-processing of n-hexadecane with lignin-derived isoeugenol, resulting in complete deoxygenation over 5 wt% Fe–5 wt% Ni/H-Y-5.1.11 First, a commercial H-Beta-300 zeolitic support (CP811C-300, Zeolyst International) was pretreated in a muffle oven with a step calcination procedure: temperature ramp at 4 °C min−1 rate to 250 °C (held for 50 min), increased at 2 °C min−1 to 500 °C (held for 4 h). First iron and then nickel were subsequently introduced using a 1.6 M aqueous solution of Fe(NO3)3·9H2O (Sigma-Aldrich) and a 1.5 M Ni(NO3)2·6H2O (CJSC Souzchimprom) precursors, respectively. After each impregnation step, the sample was dried at 100 °C overnight, and the final sample with 10 wt% of the total metal nominal loading was calcined at 450 °C for 6 h under static air. Before the catalytic experiment, the catalyst was reduced in a flow of hydrogen, 40 mL min−1, with the heating rate of 2 °C min−1 to 250 °C (held for 2 h) and subsequently to 500 °C (held for 2 h).2.2 Characterization of the catalyst
Fresh and spent FeNi/H-Beta-300 catalysts were characterized by a range of physico-chemical methods. The fresh catalyst for characterization was pre-reduced ex situ with the same reduction program as used before the catalytic experiments. Most of the details of characterization methods and the corresponding equipment are given in the previous publications11,12 with the pertinent details presented below.The Fe valence state composition in the bulk was investigated by Mössbauer spectroscopy. The 57Fe Mössbauer spectra were acquired at 295 K using an 18-month-old 57Co:Rh source (Ritverc Co. 50 mCi June 2020) with a maximum Doppler velocity of 11.0 mm s−1.
X-ray absorption spectroscopy, in terms of X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), was used to study the bulk-averaged element-specific local structure around Fe and Ni atoms. Measurements were performed on the as-received (calcined) and reduced (in situ) fresh catalyst and also on the as-received (stored in air) and reduced (quasi in situ) spent catalyst, used in isoeugenol HDO in the batch reactor. For reduction, the catalyst samples were placed in a quartz capillary, 1.5 mm o.d., 0.02 mm wall thickness and sample bed length of 3 mm. Pure H2 was flowing through the samples at 20 mL min−1 flow rate which was heated by a hot air blower from 0 to 250 °C with a dwell time of 2 h and subsequently from 250 to 500 °C (2 h dwell time) with the temperature ramp of 2 °C min−1 (same temperature program as before the catalytic tests). After that, the fresh sample was directly measured (in situ) while the spent sample holder was sealed by means of two-way valves (Swagelok), packed in polyethylene bags using a vacuum food sealer, and transported for the measurements taking place approx. 48 hours later (quasi in situ). XAS spectra at Fe and Ni K absorption edges were recorded at the P65 beamline of the PETRA III synchrotron radiation source (DESY, Hamburg) in the transmission mode. Higher harmonics were rejected by a pair of Si plane mirrors installed in front of the monochromator. The energy of the X-ray photons was selected by a Si (111) double-crystal monochromator and the beam size was set by means of slits to 0.3 (vertical) × 1.5 (horizontal) mm2. X-ray absorption near edge spectra (XANES) were normalized and the extended X-ray absorption fine structure spectra (EXAFS) background was subtracted using the Athena program from the IFEFFIT software package.14 The k2-weighted EXAFS functions were Fourier transformed (FT) in the k range of 2–14 Å−1 and multiplied by a Hanning window with a sill size of 1 Å−1. The displayed FT EXAFS spectra were not corrected for the phase shift. Amplitude reduction factors S02 0.65 (Fe) and 0.81 (Ni) were obtained by fitting the Fe and Ni foil reference spectra. The fits of the EXAFS data were performed using Artemis software14 by the least squares method in R-space between 1.0 and 3.0 Å. Coordination numbers (CN), interatomic distances (r), energy shift (δE0) and the mean square deviation of interatomic distances (σ2) were refined during fitting. The absolute misfit between the theory and the experiment was expressed by ρ.
Microtrac Belcat II equipment was used to perform temperature-programmed oxidation (O2-TPO) measurements. Two different heating rates for TPO were used: 5 °C min−1 (up to 900 °C for 10 min) and 2 °C min−1 (up to 400 °C for 20 min) under 5 vol% oxygen in argon.
CHNS analysis was performed at 950 °C using A Thermo Fisher Scientific Flash 2000 organic elemental analyzer equipped with A TC detector. Cystine, 2,5-bis(5-tert-butyl-benzoxazol-2-yl)thiophene, sulphanilamide and methionine were used as standards.
The soluble coke in the spent catalyst was identified by extraction with CH2Cl2. Extracts (organic phase) were analyzed by GC-MS (Agilent GC/MS 6890N/5973) using the same column and the same temperature program (held for 10 extra minutes) as for the reaction product analysis.
Raman spectroscopy was carried out using a Renishaw inVia Qontor confocal Raman microscope system with Raman mapping and a focus track capability. The Raman microscope was equipped with four laser lines, ranging from near-IR (785 nm) to visible (633 and 532 nm) and near-UV (355 nm). In the current work, an external laser with wavelengths of 514 nm and 1064 nm was also employed.
2.3 Chemicals
The following chemicals were used in the current work: isoeugenol (mixture of cis and trans, 99% FG, As ≤ 3 ppm, Hg ≤ 1 ppm, Cd ≤ 1 ppm, Pb ≤ 10 ppm, Sigma-Aldrich), dihydroeugenol (≥99% FG, As ≤ 3 ppm, Hg ≤ 1 ppm, Cd ≤ 1 ppm, Pb ≤ 10 ppm, Sigma-Aldrich), hydrogen (N50, 99.999%, Woikoski Oy), argon (N50, 99.999%, Woikoski Oy), helium (N46, 99.996%, Woikoski Oy), oxygen in argon (5 vol%, Woikoski Oy), propylcyclohexane (99%, Sigma-Aldrich), hexane (≥99%, Fluka), cyclohexane (≥99.9%, Alfa Aesar), 2,5-dimethylhexane (99%, Sigma-Aldrich), ethylbenzene (≥99.5%, Merck), N,O-bis(trimethylsilyl)-trifluoroacetamide (BSTFA, ≥99%, Sigma-Aldrich), tetrahydrofuran (anhydrous, ≥99.9%, Merck), mixture of alkanes (Woikoski Oy): ethane (0.9985 mol%), methane (0.9965 mol%), isobutene (1.0053 mol%), n-butane (1.0036 mol%) in helium (96.00 mol%), mixture of alkenes (Woikoski Oy): 1-butene (1.0060 mol%), ethylene (0.9974 mol%), propylene (1.0035 mol%), isobutane (1.0060 mol%) in helium (95.99 mol%), and mixture of gases (Woikoski Oy): carbon monoxide (1.05 mol%), carbon dioxide (1.04 mol%), hydrogen (1.07 mol%) in helium (96.84 mol%).2.4 Catalytic tests
Solventless experiments of hydrodeoxygenation were performed with the lignin-derived model compound isoeugenol or dihydroeugenol over 5–5 wt% FeNi/H-Beta-300. The catalyst was sieved into a fraction of 150–180 μm and reduced in a flow of hydrogen (40 mL min−1), according to the TPR profile, in two steps: 25–250 °C (hold 2 h) and 250–500 °C (hold 2 h) with the temperature ramp of 2 °C min−1.Catalytic experiments in a semi-batch mode were performed in an autoclave reactor with a mechanical stirrer (300 mL, Parr Instruments, Fig. 1a and S1a†) under a continuous flow of hydrogen, 20 mL min−1. These experiments were carried out with and without injection of the reactant. In the former case, the following procedure was used: the pre-reduced catalyst (150–180 μ) was loaded into the reactor, the reactor was heated up to the reaction temperature at ca. half of the reaction pressure, and then the pre-heated reactant (100 °C) was injected; the reactor was pressurized and stirring was switched on. In the case of an experiment without injection of the reactant, the pre-reduced catalyst was loaded into the reactor together with the reactant and heated up to the reaction temperature with the heating ramp of 10 °C min−1. All batch experiments were carried out at 200 °C (for 1 h) – 250 °C (for 1 h) – 300 °C (for 1 h), 30 bar, 1000 rpm with the weight ratio of the reactant-to-catalyst of 25 (i.e. 55 g of the reactant and 2.2 g of the catalyst). The liquid samples were taken directly from the reactor at specific time intervals (1, 30, 60, 90, 120, 150, and 180 min). At the outlet line, a separator operating at 0 °C was placed to separate the liquid and gas phases.
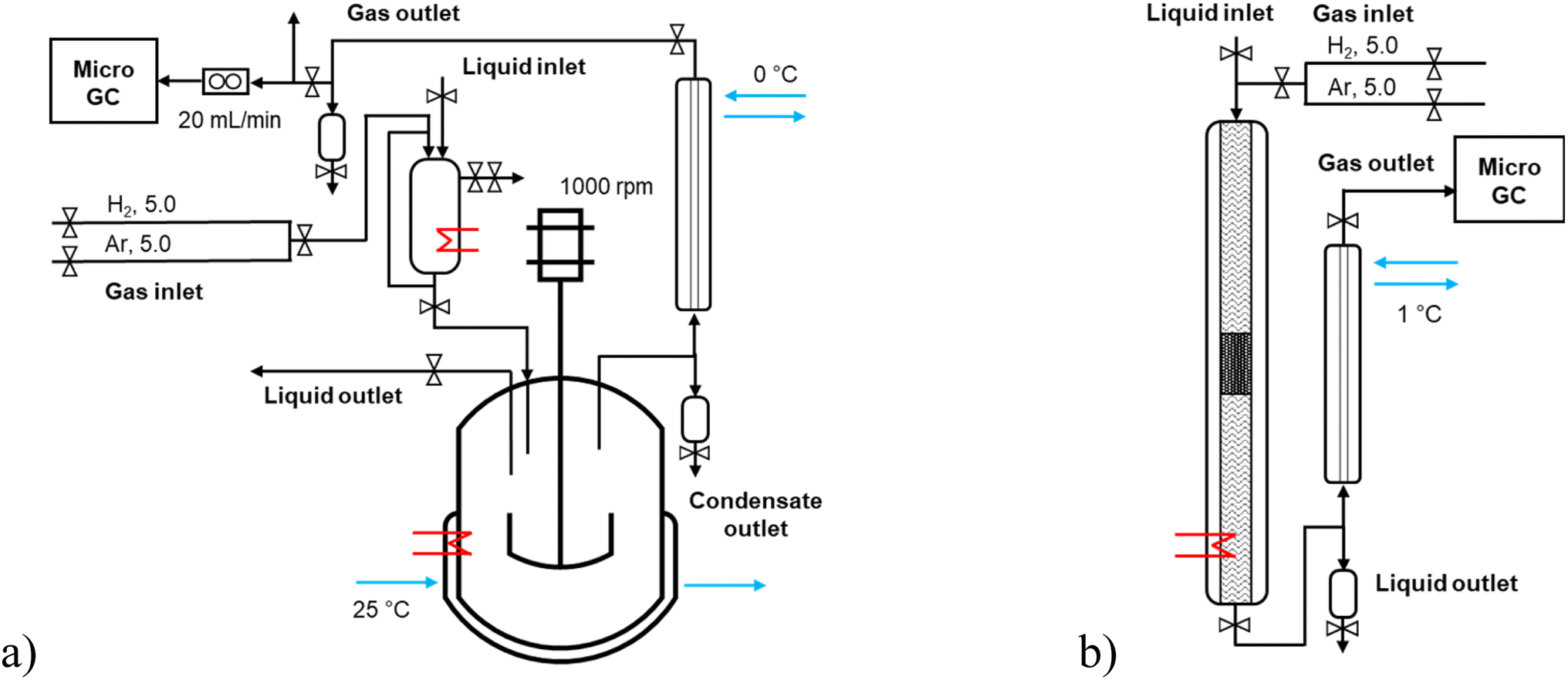 | ||
| Fig. 1 The scheme of the experimental setup: (a) batch and (b) continuous modes. | ||
Catalytic experiments in a continuous mode were performed in a stainless-steel tubular reactor (ID 4.3 mm, L 50 cm, Fig. 1b and S1b†) operating in a co-current down-flow regime. The catalyst (150–180 μm) particles were mixed in a one-to-one volume ratio with inert quartz of the size 250–350 μm, loaded into a reactor and reduced in situ. The catalytic bed was placed in the slightly lower part of the reactor to ensure that the upper layer (1–1.5 cm) was at the same level as the thermocouple measuring the reactor temperature. The empty space of the reactor upstream and downstream of the catalytic bed, separated by quartz wool, was filled with inert quartz of the size 250–350 μm. The liquid reactant was fed to the reactor by using a Fusion 6000-X high-pressure syringe pump (20 mL, 0.0001–51.22 mL min−1, up to 108 bar). All continuous experiments were carried out with 0.04 mL min−1 liquid flow rate of reactant, 15 molar excess of hydrogen, at 30 bar of total pressure. The temperature was 250, 270, and 300 °C, and the weight of the catalyst was 0.1, 0.2, and 0.3 g, respectively; the liquid residence time was 2, 5.5, and 11.9 min with the weight hour space velocity (WHSV) of 24.4, 12.4, 8.4 greactant/gcatalyst/h. At the outlet line, a cooler operating at 1 °C was placed to separate the liquid and gas phases. The liquid samples were taken from the separator at specific time intervals of time-on-stream (TOS 1, 15, 45, 60, 90, 120, 180, and 300 min, i.e. the first sample (TOS 1 min) was taken ca. one hour after the start of the liquid flow). A trickling flow regime in the tubular reactor was confirmed by analysis of the flow map considering low gas and liquid flow rates.15,16
The liquid samples were analysed using a gas chromatograph (GC) with an FID detector (Agilent 6890N) using a DB-1 column (30 m × 250 μm × 0.5 μm). The temperature programme consisted of 4 steps: 60 °C (5 min) – 138 °C (3 °C min−1) – 160 °C (1.5 °C min−1) – 300 °C (15 °C min−1, 1 min). The temperature of the detector was 280 °C. The following chemicals were used for the calibration of GC analysis: isoeugenol, dihydroeugenol, propylcyclohexane, cyclohexane, hexane, 2,5-dimethylhexane, and ethylbenzene. Other products were confirmed with an Agilent GC/MS 6890N/5973 using a DB-1 column and the same temperature program. Water content in the samples was analysed by Karl-Fischer titration (736 GP Titrino, Metrohm; Hydranal Composite 2, Fluka). A wide-bore short column GC-FID (PerkinElmer Clarus 500, Shelton, CT, USA) was used to analyze the phenolic dimers. The samples were silylated using N,O-bis(trimethylsilyl)-trifluoroacetamide (BSTFA) as a silylation agent at 60 °C for 1 h. The column parameters were: Agilent HP-1/SIMDIST, ∼6 m (length) × 0.530 mm (inner diameter), film thickness 0.15 μm. The flow rate of hydrogen serving as a carrier gas was 7 mL min−1. The following temperature program was used: 100 °C (after 0.5 min hold) at a rate of 12 °C min−1 to 340 °C (5 min hold), and with the following injector program: 80 °C (0.1 min hold) at a rate of 50 °C min−1 to 110 °C, and at a rate of 15 °C min−1 to 330 °C (7 min hold), while the temperature of the detector was maintained at 340 °C. The liquid phase samples were also analyzed by the size-exclusion chromatography–high-performance liquid chromatography (SEC – HPLC) technique to detect oligomeric compounds using tetrahydrofuran containing 1% acetic acid as eluent. The diluted samples were analyzed by high-performance size exclusion chromatography with an evaporative light scattering detector (HPSEC-ELSD, Shimadzu DGU-405 series modular HPLC, Shimadzu Corporation (Shimadzu, Japan)). The SEC–HPLC system was equipped with three different columns (2 x Jordi Gel DVB 500A (300 mm × 7.8 mm), guard column 50 × 7.8 mm). The flow rate of 0.8 mL min−1 was used with the column oven temperature of 40 °C. The components were detected with an ELSD detector (Sedex-100 LT-ELSD) having the following parameters: 40 °C, air pressure of 3.5 bar and gain of 1. The SEC – HPLC system contained the following parts: in-line degasser (Shimadzu DGU-405 degassing unit), the HPLC gradient pump (Shimadzu LG-40D solvent delivery module), an autosampler (Shimadzu SIL-20A HT), the column oven (Shimadzu CTO-10ACvp) and the system controller (Shimadzu CBM-40).
The gas samples were analysed online every 15 min by using a micro gas chromatograph with TCD detectors (Agilent 6890N) using 4 parallel channels: (1) and (2) 100 °C, MS 5A column (10 m × 320 μm × 30 μm) with Plot U pre-column (3 m × 320 μm × 30 μm); (3) 60 °C, Plot Q column (8 m × 320 μm × 10 μm); (4) 90 °C, OV-1 column (14 m × 150 μm × 2 μm). The injector temperature was the same for all channels, 100 °C. The following chemicals were used to calibrate the microGC: hydrogen, ethane, methane, isobutane, n-butane, 1-butene, ethylene, propylene, isobutene, carbon monoxide, and carbon dioxide.
2.5 Catalyst regeneration
The spent catalyst was regenerated directly in the reactor. After the experiment, the catalyst was kept inside and the reactor was flushed with Ar, 40 mL min−1, and cooled down overnight to 100 °C. The in situ regeneration was performed with a step-by-step procedure from 100 °C to 400 °C at the heating ramp 2 °C min−1, at 5 vol% O2 in an Ar atmosphere, 40 mL min−1. The outlet stream was analysed by microGC-TCD removing water upstream of the separator. After the regeneration procedure, the reactor was flushed with Ar, 40 mL min−1, overnight and then re-activated according to the reduction procedure described above.3 Results and discussion
3.1 Characterization results of the fresh and spent FeNi/H-Beta-300 catalysts
Fresh and spent FeNi/H-Beta-300 catalysts were characterized by a range of physico-chemical methods.| Entry | Catalyst | DHE/IE | B/C | Conditions | d FeNi | D FeNi | d Fe f | d Ni f | D Fe f | D Ni f | D FeNi f |
|---|---|---|---|---|---|---|---|---|---|---|---|
| nm | % | nm | nm | % | % | % | |||||
| a With injection of the reactant on the preheated reduced catalyst. b Two runs, f – fitting of kernel smooth distribution curves by the Gauss function in Origin. | |||||||||||
| 0a | Fresh, calcined | — | — | — | 7 | 16 | 5 | 16 | 24 | 6 | 15 |
| 0b | Fresh, reduced | — | — | — | 8 | 14 | 6 | 20 | 20 | 5 | 13 |
| 1 | Spent | IE | B | 250–300 °C, 2.2 g | 8 | 14 | 7 | — | 18 | — | — |
| 2 | Spent | DHE | B | 250–300 °C, 2.2 g | 15 | 7 | 6 | 20 | 18 | 5 | 12 |
| 3 | Spent | DHE | Ba | 250–300 °C, 2.2 g | 11 | 10 | 7 | 22 | 18 | 5 | 11 |
| 4 | Spent | IE | C | 300 °C, 0.2g | 9 | 12 | 5 | 21 | 22 | 5 | 13 |
| 5 | Spent | IE | C | 300 °C, 0.3 g | 8 | 14 | 6 | — | 18 | — | — |
| 6 | Spent | IE | C | 275 °C, 0.3 g | 6 | 18 | 5 | 18 | 26 | 5 | 16 |
| 7 | Spent | IE | C | 250 °C, 0.3 g | 8 | 14 | 5 | — | 23 | — | — |
| 8 | Spent | DHEb | C | 300 °C, 0.1 g | 7 | 16 | 6 | 15 | 20 | 7 | 13 |
| 9 | Spent | DHEb | C | 300 °C, 0.3 g | 15 | 7 | 6 | 18 | 20 | 6 | 13 |
Worth noting is that, for most catalysts, the histogram of the FeNi particle size clearly revealed two maxima. For this reason, the fitting of kernel smooth distribution curves by the Gauss function was also used (Fig. 2). Acknowledging all limitations of the applied approach, the results were compared with Fe2O3 (ref. 18) and the monometallic Fe (5–5.5 nm) and monometallic Ni (18.1–19.2 nm) catalysts on another zeolitic support (H-Y-5.1) used in the previous work.12 Both ranges are in line with the maxima for fresh and spent bimetallic FeNi/H-Beta-300 catalysts used in the current work. To be specific, the first maximum with the median particle size of 5–7 nm could hypothetically correspond to Fe and particles rich in Fe (dFef) assuming the same size of iron clusters formed after deposition on beta and Y zeolites, and the second maximum with a median particle size of 15–22 nm reflects subsequently Ni and particles rich in Ni (dNif).
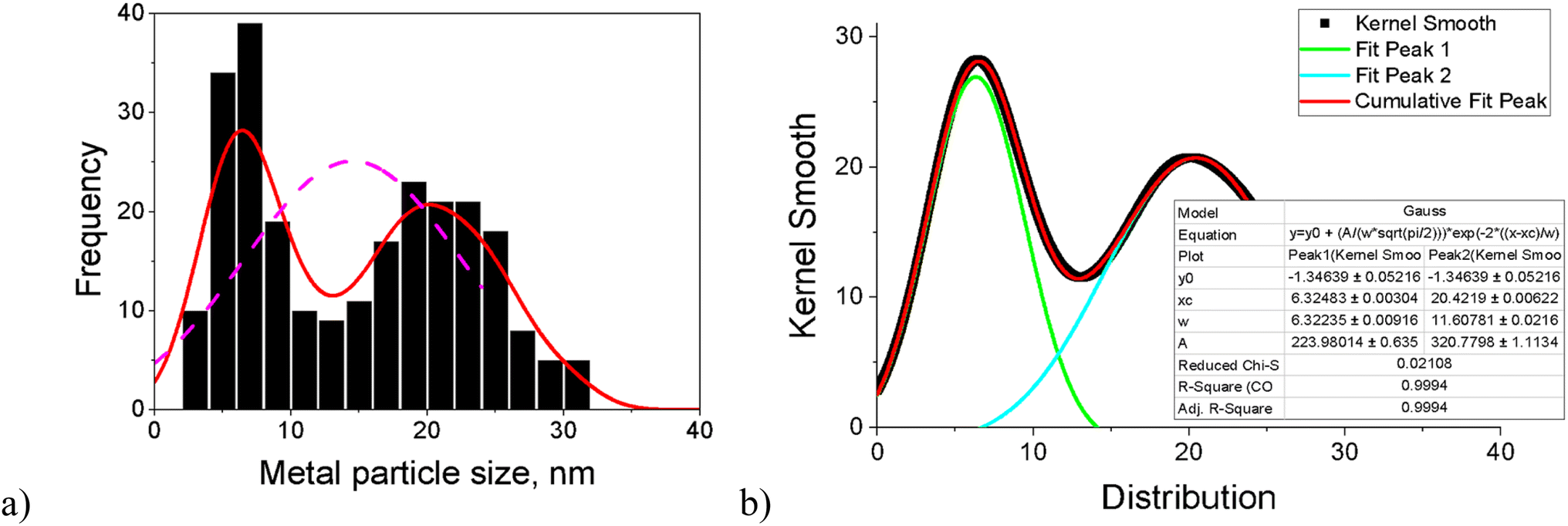 | ||
| Fig. 2 (a) Histogram of the FeNi particle size and (b) fitting curves of kernel smooth distribution for the spent catalyst after dihydrogenol hydrodeoxygenation in the batch reactor (Table 1, entry 2). | ||
Based on the comparison of the median particle size of both fresh and spent catalysts no sintering of the metal particles in hydrodeoxygenation of isoeugenol or dihydroeugenol in either batch (3 h) or continuous reactor (5 h) at 250–300 °C and 30 bar of total pressure could be confirmed. Moreover, neither a decrease in the particle size was observed (Table 1, entry 1), although slight leaching was detected by ICP-OES (Table S1†).
Metal dispersion in the fresh and spent catalysts calculated from the non-fitted and fitted median particle sizes of FeNi was relatively close to each other, DFeNi of 7–18% and DFeNif of 11–16%, respectively (Table 1, Fig. S4†). It should be also mentioned that the differences in metal particle size of catalysts could be related to the alloy formation. Metal dispersion was calculated according to:
| DFeNi = (xFe·116 + xNi·101)/dFeNi | (1) |
| DFeNif = xFe·DFef + xNi·DNif | (2) |
| Catalyst | TAS | BAS | LAS | BAS | LAS | B/L | n FeNi/nAS | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| — | μmol g−1 | w | m | s | w | m | s | — | mol mol−1 | ||
| H-Beta-300 | 77 | 66 | 11 | 16 | 28 | 23 | 5 | 2 | 3 | 6.0 | — |
| 5–5 wt% Fe–Ni/H-Beta-300 | 104 | 47 | 57 | 24 | 23 | 0 | 34 | 23 | 0 | 0.8 | 15.5 |
| Spent catalyst (batch, IE) | 77 | 46 | 31 | 18 | 28 | 0 | 16 | 15 | 0 | 1.5 | 17.3 |
After isoeugenol hydrodeoxygenation in the batch reactor, a slight decrease of weak and medium Lewis acid sites from 57 to 31 μmol g−1 was noticed which led to higher B/L. This decrease could be related to Fe and/or Ni leaching (Table S1†) as well as coke formation during the reaction (Table 4) which can adsorb onto the surface of the catalyst, potentially masking or blocking the Lewis acid sites. The metal-to-acid site ratio slightly increased after isoeugenol hydrodeoxygenation in the batch reactor from 15.5 to 17.3 (Table 2). While Fe and/or Ni leaching might cause a reduction in the total metal content on the catalyst surface, it does not necessarily imply a direct correlation with the decrease in Lewis acid sites. In fact, the increase in the metal-to-acid site ratio indicates that the metal content, relative to the remaining acid sites, has increased. This could be due to a preferential loss of acid sites or a redistribution of the metal species on the catalyst surface. FTIR-pyridine spectra are provided in the ESI in Fig. S5.†
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) lattice system is a common crystal structure for various metals (e.g., Cu, Co), which can lead to misinterpretation of the data.
m) lattice system is a common crystal structure for various metals (e.g., Cu, Co), which can lead to misinterpretation of the data.
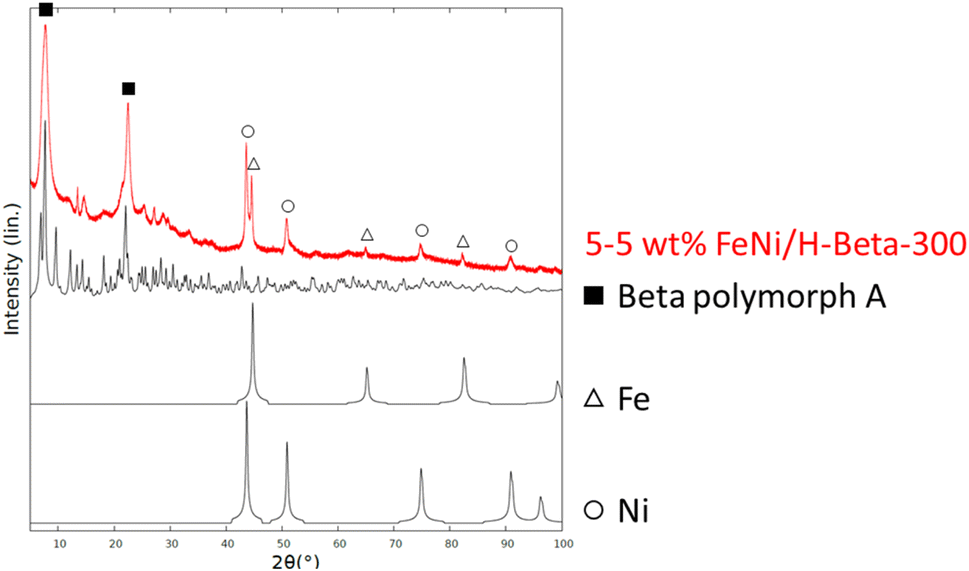 | ||
| Fig. 3 XRD pattern of the fresh catalyst. | ||
According to Mössbauer spectroscopy (Fig. 4a), the fresh 5–5 wt% Fe–Ni/H-Beta-300 catalyst at 22 °C is dominated by two sextets related to metallic Fe (red is more typical and blue is broadened and has a smaller field probably due to interactions with nickel). Cyan and brown doublets in Fig. 4a represent a small amount of Fe3+ or possible superparamagnetic Fe and marginal amounts of paramagnetic Fe2+, respectively. It should be mentioned that significant differences in results of the Fe valence state composition in the bulk and Mössbauer parameters (Table 3) were obtained for 5–5 wt% Fe–Ni/H-Y-5.1 synthesized and reduced by the same method (ref. 12Fig. 4b) as in the current work. While the metallic form of Fe dominated (93%) for the Fe–Ni catalyst supported on mildly acidic H-Beta-300, in the case of a more acidic H-Y-5.1 with the total acidity of the pristine support of 172 μmol g−1 comprising 154 μmol g−1 Brønsted acid sites,12 it was predominantly Fe3+ (52%). Such differences could be explained by different metal–metal and metal–support interactions in these catalysts.
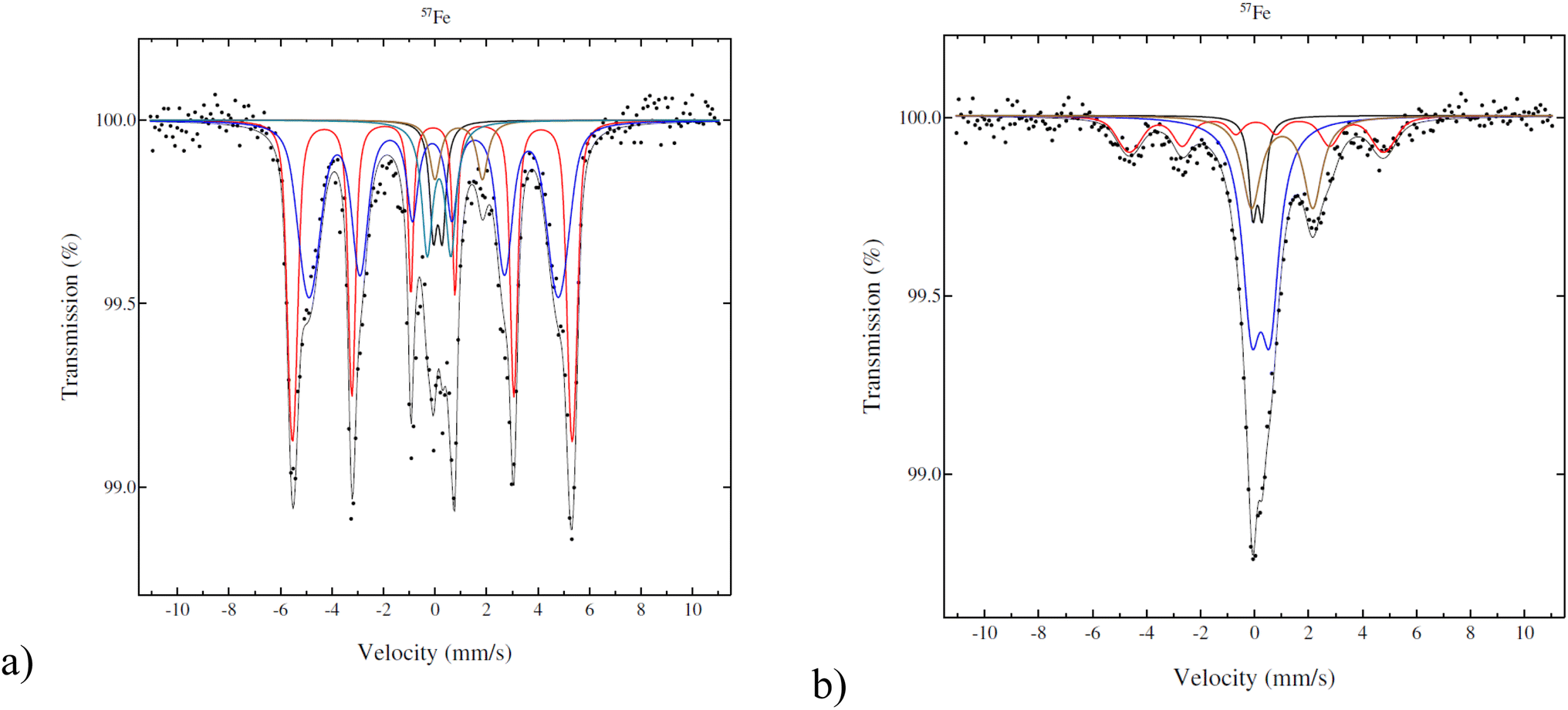 | ||
| Fig. 4 Mössbauer spectra of the fresh catalyst: (a) 5–5 wt% Fe–Ni/H-Beta-300, legend: metallic and magnetic Fe (red, blue), Fe2+ (brown), Fe3+ (cyan), Fe impurity in the detector (black); (b) 5–5 wt% Fe–Ni/H-Y-5.1,12 legend: metallic and magnetic Fe (red), Fe2+ (beige), Fe3+ (blue), Fe impurity in the detector (black). | ||
XAS was used to investigate the local structure around Fe and Ni atoms in the as-received (calcined) and reduced fresh and spent FeNi/H-Beta-300 (FeNi/β, batch, IE, Table S2†) catalysts. It is noteworthy that the catalysts were reduced directly in in situ cells with a plug-flow geometry according to the protocol used for catalytic testing, then cooled down and either directly measured in flowing H2 or sealed in the cell under H2 and measured later without a need for any passivation or exposure to air. XANES and Fourier transformed (FT) EXAFS spectra of the as-received catalysts (fresh after calcination, spent after exposure to air) are depicted in Fig. 5 alongside spectra of the most relevant oxidized reference compounds. The position and the shape of pre-edge and edge features in the Fe K XAS spectra (Fig. 5a and c) correspond to γ-Fe2O3 nanoparticles (nanosizing effect seen from lower scattering intensity on the 2nd and further shells compared to the reference spectrum, Fig. 5c). This is in line with the literature12 attributing the initial presence of Fe(III) to the used Fe(NO3)3·9H2O precursor. Qualitatively, the iron oxide nanoparticle size decreased after activation, the catalytic test and a subsequent exposure to air. Ni K XAS spectra (Fig. 5b and d) demonstrate the same trend: bulk-like NiO in the fresh calcined sample with a somewhat more dispersed oxide in the spent catalyst (Fig. 5d). This trend was not clearly demonstrated by TEM (Table 1, entry 1).
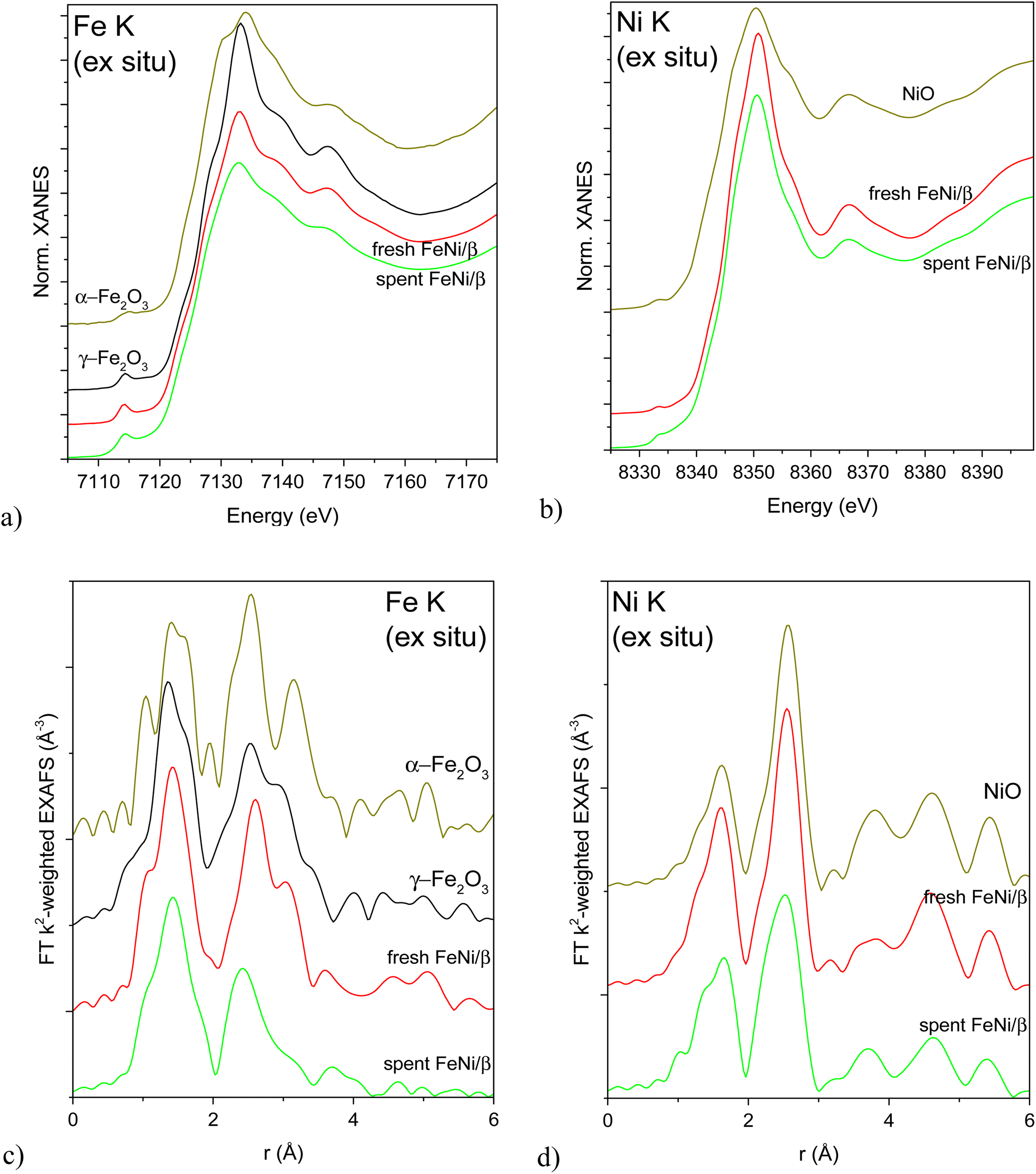 | ||
| Fig. 5 Normalized XANES (a and b) and Fourier transformed EXAFS (c and d) spectra of the calcined and exposed to air spent catalyst and relevant reference compounds measured at Fe K (a and c) and Ni K (b and d) edges. EXAFS spectra are not corrected for the phase shift. | ||
XAS spectra of the reduced catalysts are shown in Fig. 6. Edge positions in the XANES spectra confirm the reduced state of both Fe and Ni in all studied bimetallic catalysts (Fig. 6a and b). Bulk Fe and Ni metals crystallize in different structures under ambient conditions: bcc (Imm) in the case of Fe and fcc (Fmm) structure for Ni. The different crystal structures result in very different XANES spectra for the respective individual metals (Fig. 6a and b). The fcc structure around Ni atoms is preserved for both samples, although the Ni K XANES spectrum of the spent catalyst displays a white line with a higher intensity and shape, possibly due to partial oxidation (Fig. 6b). This partial oxidation may happen due to exposure of the sample to air during transportation to the synchrotron and therefore O-related features will not be used for drawing conclusions. On the other hand, the peaks at 8357 and 8382 eV are due to the metallic structure. These peaks are shifted to lower energies in the case of both bimetallic samples with the shift being less pronounced for the spent catalyst. This shift may indicate alloying with Fe, and subsequently different shifts reflect different degrees of alloying (i.e. partial dealloying after catalysis and exposure to air). All Ni K EXAFS spectra (Fig. 6d) unequivocally confirm the fcc Ni structure.
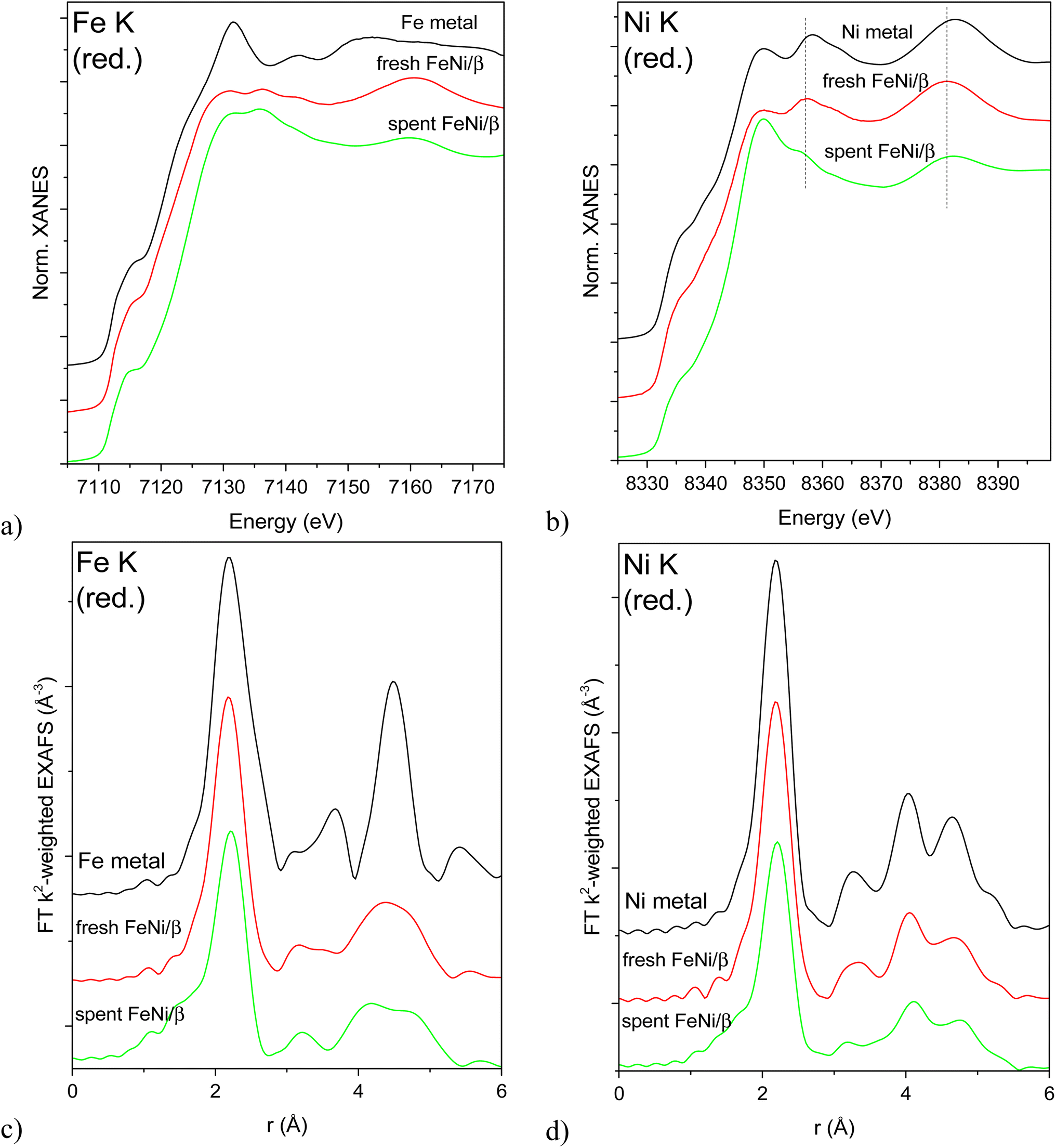 | ||
| Fig. 6 Normalized XANES (a and b) and Fourier transformed EXAFS (c and d) spectra of reduced fresh and re-reduced spent catalysts and the respective bulk metals (5 μm thick foils) measured at Fe K (a and c) and Ni K (b and d) edges. EXAFS spectra are not corrected for the phase shift. | ||
Fe K XAS spectra (Fig. 6a and c) of the reduced fresh and spent catalysts are markedly different from bcc Fe and rather suggest the fcc structure as in the case of Ni which signifies FeNi alloy formation.
The first shell analysis was performed on Fe K and Ni K EXAFS spectra of the reduced samples to identify structural parameters such as the coordination number and interatomic distances (Table S2, Fig. S6–S8†). Due to similar scattering factors of Fe and Ni, EXAFS cannot reliably distinguish between these two types of nearest neighbours. The obtained structural parameters point at fcc structures around both Fe and Ni atoms in both samples. Coordination numbers around 11 in the fresh reduced FeNi sample correspond to rather large, on the order of 3–10 nm (with large error bars due to uncertainty in CN determination and asymptotic behaviour of the model)24 fcc FeNi nanoparticles. The same CN around Ni and Fe suggests the random distribution of both metals in the alloy. Somewhat longer Fe-M means interatomic distance in the spectrum of the spent and rereduced FeNi/H-Beta-300 (FeNi/β) may signify a contribution from the bcc Fe structure (i.e. partial dealloying), while lower average coordination numbers around both Fe and Ni (ca. 9.3 at an average) stem from a smaller mean metal particle size of 1.5–2 nm. Significantly smaller particle sizes determined by EXAFS, compared to TEM, are probably related to the EXAFS sensitivity to single coherently scattering domains and TEM observing their aggregates and agglomerates.
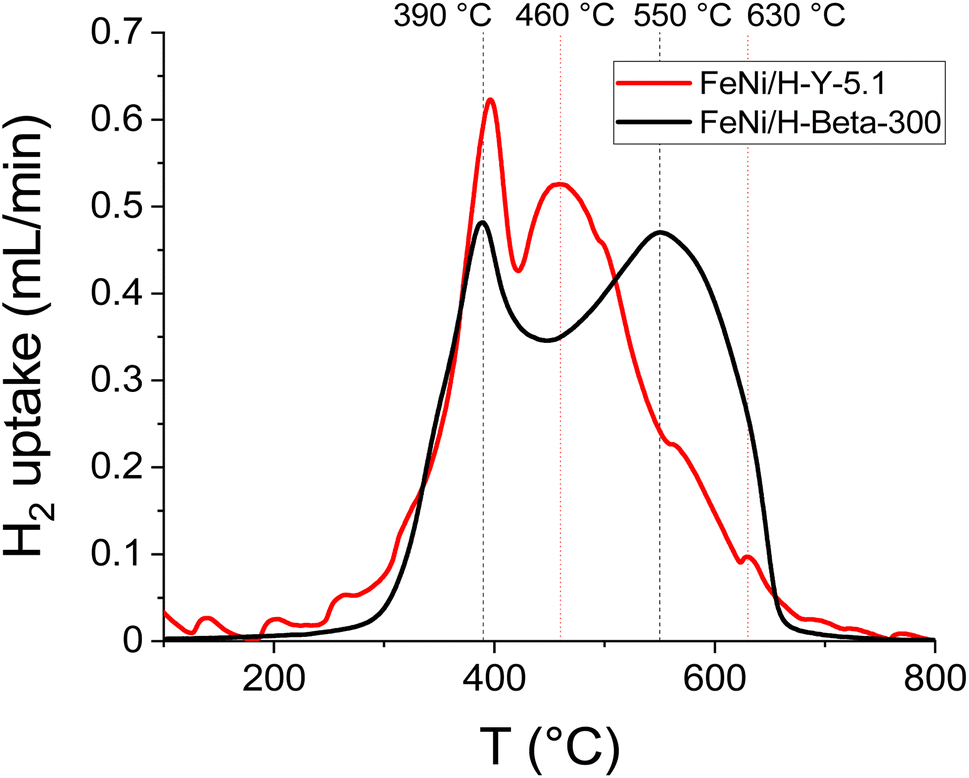 | ||
| Fig. 7 H2-TPR profiles of the fresh catalyst: 5–5 wt% Fe–Ni/H-Y-5.1 (data from ref. 12) and 5–5 wt% Fe–Ni/H-Beta-300. | ||
| Entry | Catalyst | B/C | DHE/IE | T | m cat | S D–R | V | V μ | V μ/Vm | R c | m c,TGA | m c,CHNS | m c,TPO |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| °C | g | m2 g−1 | cm3 g−1 | vol% | — | μg mg−1 h−1 | wt% | wt% | wt% | ||||
| a With injection of the reactant on the preheated reduced catalyst. b Two runs. | |||||||||||||
| 0 | Fresh | — | — | — | — | 516 | 0.26 | 81 | 4.2 | — | 0 | 0 | 0 |
| 1a | Spent | B | IE | 250–300 | 2.2 | 202 | 0.12 | 66 | 1.9 | 27.0 | 8.1 | 8.2 | 9.5 |
| 1b | Regenerated 1a cat. (TPO 100–400 °C) | 415 | 0.23 | 72 | 2.6 | — | 3.2 | — | — | ||||
| 1c | Regenerated 1a cat. (TPO 100–900 °C) | 413 | 0.21 | 77 | 3.3 | — | — | — | — | ||||
| 2 | Spent | B | DHE | 250–300 | 2.2 | 178 | 0.16 | 39 | 0.6 | 31.0 | 9.3 | — | — |
| 3 | Spent | Ba | DHE | 250–300 | 2.2 | 204 | 0.13 | 66 | 1.9 | 25.0 | 7.5 | — | — |
| 4 | Spent | C | IE | 300 | 0.2 | 4 | 0.01 | 14 | 0.2 | 36.2 | 18.1 | — | — |
| 5 | Spent | C | IE | 300 | 0.3 | 5 | 0.01 | 11 | 0.1 | 37.0 | 18.5 | — | — |
| 6 | Spent | C | IE | 275 | 0.3 | 19 | 0.17 | 1 | 0.01 | 26.2 | 13.1 | — | — |
| 7 | Spent | C | IE | 250 | 0.3 | 117 | 0.11 | 41 | 0.7 | 24.2 | 12.1 | — | — |
| 8 | Spent | C | DHEb | 300 | 0.1 | 163 | 0.09 | 75 | 2.9 | 12.2 | 6.1 | — | — |
| 9 | Spent | C | DHEb | 300 | 0.3 | 143 | 0.09 | 61 | 1.6 | 21.8 | 10.9 | — | 9.2 |
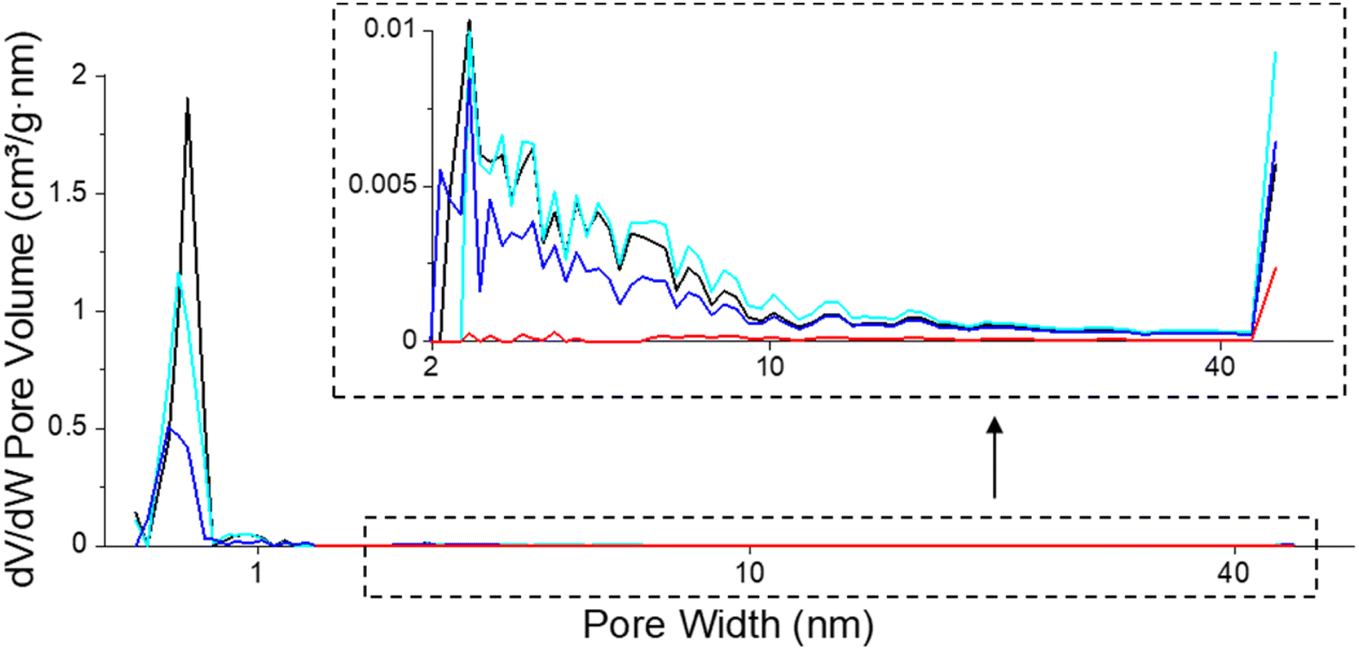 | ||
| Fig. 8 Pore size distribution of fresh (black), spent catalysts after isoeugenol hydrodeoxygenation in the batch reactor from (blue, Table 4, entry 1a) and in the continuous reactor (red, Table 4, entry 5) and regenerated catalyst (cyan, Table 4, entry 1b). | ||
In situ regeneration of the spent catalyst was simulated by an O2-TPO-TCD-MS measurement performed under the same conditions over the catalyst from the batch experiment of isoeugenol hydrodeoxygenation (Table 4, entry 1a). The textural properties of the regenerated catalyst obtained by this procedure clearly demonstrated the success of removing coke at 100–400 °C. The mesoporosity was fully regenerated while the specific surface area and microporosity of the regenerated catalyst achieved 80% of values compared to the fresh catalyst (Fig. 8, Table 4, entry 1b). In the case of the catalyst regeneration by burning coke up to 900 °C (O2-TPO-TCD-MS, 100–900 °C, 5 °C min−1), the comparable micropore volume was recovered compared to a similar treatment to a much lower temperature (O2-TPO-TCD-MS, 100–400 °C, 2 °C min−1). The specific surface area and total pore volume were comparable, i.e. ca. 80% of the original value for the fresh catalyst.
TEM images of the catalysts regenerated at 400 °C and 900 °C are shown in Fig. S9.† The metal particle size after regeneration at 400 °C varies in the range of 10–25 nm indicating that metal particles are not sintered due to strong interactions between the metal and acidic zeolite. However, sintering clearly occurred after catalyst regeneration at 900 °C, when the metal particles of 15–60 nm were observed in the images (Fig. S9c and d†). These results together with surface area measurements demonstrate that it is not possible to fully regenerate the catalyst even at a high temperature, because the surface area could not be fully recovered and metal sintering occurred.
It was also revealed that the specific surface area of the spent FeNi/H-Beta-300 catalysts, tested under different conditions, linearly decreased with the increasing amounts of coke determined by TGA analysis (6–19 wt%, Fig. 9, Table 4). This is valid for all catalysts including the in situ regenerated catalyst. In other words, the formation of one weight percent of coke in the catalyst led to a decrease in the specific surface area of approx. 12.7%; i.e. a 50% decrease in the specific surface area of the catalyst corresponds to ca. 7.7 wt% coke formation. This is the opposite trend that was obtained over the FeNi/H-Y-5.1 catalyst with different ratios of metals, tested in the co-processing of n-hexadecane with lignin-derived isoeugenol.11 It could be related to the catalyst composition but also to the different feedstock.
 | ||
| Fig. 9 Amount of coke determined by TGA as a function of the catalyst-specific surface area. Legend: fresh catalyst (diamond), spent catalyst from the batch experiment (triangle), spent catalyst from the continuous experiments (circle), spent catalyst from IE HDO (open symbols) and spent catalyst from DHE HDO (black symbols), regenerated catalyst (grey symbol, in situ regeneration of the spent catalyst simulated by O2-TPO). Notation is the same as in Table 4. | ||
According to the heat release, two maxima were obtained in 355–380 °C and 470–480 °C temperature regions for all the spent catalysts used in IE HDO (Fig. S10a†) and DHE HDO (Fig. S10b†). This is in line with filamentous type coke formation (400–550 °C) observed also over Ni-upgraded slug oxides,29 and FeNi/H-Y-5.1.12 Sánchez-Sánchez et al.30 reported that the oxidation of filamentous coke coupled with Ni particles occurs in the temperature range between 300 and 530 °C, while the oxidation of carbonaceous deposits with various degrees of graphitization occurs above 530 °C.
Similar results as from TGA were obtained from the TPO analysis (Fig. S11†), i.e. two maxima of carbon oxidation at 370 °C and 485 °C for the spent catalyst, used in IE HDO in the batch reactor. The weight ratio of CO-to-CO2 formation was 18/82 (wt%), i.e. 0.21. For the same catalyst (Table 4, entry 1a), 9.5 wt%, 8.1 wt%, and 8.2 wt% coke was determined by TPO (100–900 °C), TGA (100–800 °C), and CHNS (950 °C), respectively. Furthermore, CHNS analysis also revealed the molar H/C ratio of 1.8, pointing to the presence of aliphatic species with a mean value of molar H/C ratio ca. 1.7 (1.4 < H/C < 2.0), having higher H/C ratios than aromatic compounds (ca. 0.6).31
An analogous result was also obtained by identification of the extracted soluble coke species using GC-MS. The extracted soluble coke from the spent catalysts, used in the batch mode experiments without injection of the reactant on the preheated fresh catalyst, showed a broad range of predominantly aliphatic coke (primary compounds: C10–C18 straight-chain alkanes) and a low amount of aromatics (primary component: dimethylethyl benzene) (Fig. S12†). Analogous results were obtained from the co-processing of n-hexadecane with lignin-derived isoeugenol performed also in the batch mode.11 On the other hand, the results from the batch mode with injection (Fig. S12†) and the continuous mode (Fig. S13†), obtained in the current work in IE HDO and DHE HDO, revealed selective coke formation of n-C16 with minor amounts of n-C12 and i-C16 straight-chain alkanes. These results can be attributed to the specific reaction conditions in the applied experimental setups determining the selective formation and composition of the coke species. In the case of the batch experiment without the injection, the catalyst was exposed to the reactant during the heating period without stirring. The absence of stirring may have led to the strong adsorption of the reactant on the catalyst surface. The combination of strong adsorption and a sufficiently long contact time in the batch experiment could have been the reason for the formation of a wide range of coke species.
For the coke species analysis on the surface of the spent catalyst, also Raman spectroscopy with different wavelengths (355 nm, 514 nm, 532 nm, 633 nm, 785 nm and 1064 nm) has been employed. This technique was previously applied for metal-free ZSM-5, USY zeolites (UV Raman, ref. 32), Pt–Sn/Al2O3 (514 nm, ref. 33), Mn3O4 (785 nm, ref. 34), Pt-Re/γ-Al2O3 (785 nm, ref. 35), and Au nanoparticles (785 nm, ref. 36). In the current work, the Fe–Ni/H-Beta-300 spent catalyst from the batch experiment of isoeugenol hydrodeoxygenation (ca. 8 wt% of coke, Table 4) was analysed in both powder and thin pressed pellet form. However, all measurements performed under different conditions resulted in one big peak (Fig. S14†) attributed to the strong fluorescence interference.33,37
3.2 Activity and selectivity of the FeNi/H-Beta-300 catalyst in hydrodeoxygenation
A set of solventless experiments over the reference 5–5 wt% Fe–Ni/H-Beta-300 catalyst (sieved fraction 150–180 μm) has been performed in batch and continuous modes. Isoeugenol and its hydrogenated intermediate dihydroeugenol were used as feedstock. It should be also pointed out that in the case of isoeugenol as a reactant, the reaction proceeds via rapid hydrogenation to dihydroeugenol, even in the absence of any catalyst, which is in line with the literature.11 Therefore, catalytic results of IE HDO also comprise the conversion of the consecutive step, i.e. conversion of dihydroeugenol.In the case of dihydroeugenol (DHE) as a starting material, the initial reaction rate of dihydroeugenol HDO was ca. 3-fold lower for the experiment without the injection of the reactant on the preheated catalyst, which could be attributed to the strong adsorption of the reactant on the catalyst during the heating period of the reactor. When comparing the product distribution between DHE and IE HDO in a batch reactor in the current study, ca. two-fold yield of the desired oxygen-free compounds (6%) and a slightly higher water content (0.9%) were gained at the same DHE conversion level (19%) as for IE HDO performed under the same conditions. Except soluble coke species composition (Fig. S12†) and the initial reaction rate and turnover frequency (Table 5), the results of DHE HDO from both experiments, i.e. with and without an injection on the preheated catalyst, were comparable (Table 5). No heavy compounds were detected by GC-FID, short column analysis, or SEC analysis of the liquid phase. In the gas phase, only hydrogen was detected in all cases of the batch experiments.
For comparison with the literature,42 DHE conversion of 34% was observed in the batch reactor over a dual catalyst system, 0.1 g Ru/C and 0.2 g Nb2O5, at 250 °C and 6 bar of hydrogen in the mixture of DHE (0.2 mL), MeOH (0.8 mL) and distilled water (12 mL) giving 4% yield of the desired oxygen-free compounds and SOCC/SOFC equal to 90/10 in DHE HDO performed. It should also be noted that after 4 h DHE was fully converted giving the SOCC/SOFC ratio equal to 69/31.42 It can be concluded that more efficient deoxygenation of DHE was obtained in the presence of the solvent, as expected.
| Entry | DHE/IE | Conditions | in 1 min of time-on-stream | in 5 h of time-on-stream | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r DHE 0 | TOF0DHE | X 0 IE | X 0 DHE | S OCC(DHE) 0/SOFC0 | Y OCC 0 | Y OFC 0 | X IE | X DHE | CB(l) | S OCC(DHE)/SOFC | Y OCC | Y OFC | Y H2O | ΔXDHE | ΔSOFC | |||
| a 3 h TOS. b Two runs.2 | ||||||||||||||||||
| 4 | IE | 300 °C, 0.2 g | 0.11 | 70 | 96 | 72 | 61(41)/39 | 39.5 | 25.3 | 100 | 43 | 63 | 98(90)/2 | 62.2 | 1.5 | 1.5 | 0.53 | 0.64 |
| 5 | IE | 300 °C, 0.3 g | 0.08 | 49 | 100 | 99 | 24(1)/76 | 18.2 | 58.2 | 100 | 30 | 78 | 96(89)/4 | 75.3 | 3.2 | 1.3 | 1.09 | 1.20 |
| 6 | IE | 275 °C, 0.3 g | 0.07 | 46 | 100 | 91 | 19(11)/81 | 16.1 | 68.3 | 100 | 23 | 83 | 94(92)/6 | 79.2 | 4.7 | 0.8 | 1.01 | 1.33 |
| 7 | IE | 250 °C, 0.3 g | 0.07 | 43 | 100 | 90 | 14(11)/86 | 12.0 | 71.1 | 100 | 15 | 90 | 94(93)/6 | 85.5 | 5.6 | 1.3 | 1.24 | 1.47 |
| 8a | DHEb | 300 °C, 0.1 g | 0.06 | 36 | — | 47 | 38/62 | 9.3 | 15.1 | — | 9 | 95 | 45/55 | 2.2 | 2.7 | 0.7 | 0.69 | 0.07 |
| 8b | 0.02 | 12 | — | 15 | 66/34 | 5.3 | 2.7 | — | 7a | 98a | 77/23a | 1.3a | 4.4a | 0.2 | 0.00 | 0.03 | ||
| 9a | DHEb | 300 °C, 0.3 g | 0.06 | 35 | — | 78 | 20/80 | 12.4 | 50.2 | — | 23 | 93 | 62/38 | 11.3 | 6.9 | 1.8 | 0.72 | 0.78 |
| 9b | 0.03 | 20 | — | 45 | 71/29 | 11.9 | 4.7 | — | 17 | 96 | 65/35 | 8.7 | 4.7 | 0.7 | 0.33 | — | ||
Hydrodeoxygenation of isoeugenol (IE HDO) in the continuous mode revealed rapid catalyst deactivation during the first hour of time-on-stream (ΔXDHE 1–1.2% per minute over 0.3 g of catalyst, Fig. 10a). A slightly slower deactivation was observed in hydrodeoxygenation of dihydroeugenol (DHE HDO), the first intermediate of isoeugenol HDO (ΔXDHE 0.7% per minute over 0.3 g of catalyst, Fig. 10b). In both cases, the yield of the desired oxygen-free compounds (OFC, Fig. 10c and d) decreased during the first hour of time-on-stream too. The product distribution of the individual oxygen-free and oxygen-containing compounds detected in the liquid phase is presented in Fig. S19 and S20.†
 | ||
| Fig. 10 Dihydroeugenol conversion as a function of time-on-stream in (a) isoeugenol hydrodeoxygenation, (b) dihydroeugenol hydrodeoxygenation; yield of the oxygen-free compounds as a function of time-on-stream in (c) isoeugenol hydrodeoxygenation, (d) dihydroeugenol hydrodeoxygenation. Conditions: 250–300 °C, 30 bar, 0.1–0.3 g of catalyst with 0.04 mL min−1 of liquid flow and a 15-fold excess of hydrogen. | ||
In the gas phase (Fig. S21†), mainly methane, one unknown product, and methanol were detected. While methane and methanol decreased with increasing catalyst deactivation and water formation, the concentration of the unknown product had the opposite trend. Ethane, propane, butane, isobutene, and carbon oxides were detected in negligible amounts. Overall, the total concentration of the gas-phase products decreased with decreasing DHE conversion in all the cases of continuous experiments.
IE HDO performed at different temperatures demonstrated a higher initial selectivity to OFC at a lower temperature. The apparent activation energy of dihydroeugenol hydrodeoxygenation over the FeNi/H-Beta-300 catalyst (150–180 μm) in solventless isoeugenol HDO was calculated to be 6.3 kJ mol−1 (Fig. S22†). This value is 2.4-fold lower than that reported in ref. 41, i.e. 15 kJ mol−1, for 0.3 g granulated PtRe (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/Sibunit catalyst (1 mm) at 75–200 °C, 30 bar of H2 and 0.5 mL min−1 liquid flow of 0.012 M isoeugenol in dodecane. The activation energy in the kinetic study of dihydroeugenol hydrodeoxygenation, without the hydrogenation step of isoeugenol, in the packed-bed microreactor over the sulfided NiO/MoO3/Al2O3 catalyst (75–150 μm) at 200–450 °C and 20.7 bar was calculated to be 34 kJ mol−1.43 Note that the lower activation energy of DHE obtained in the current work from IE HDO in practice can depend not only on the catalyst type but also on the catalyst deactivation, commonly modeled as an exponential decay function of active sites with TOS,44 appearing in the first step of isoeugenol hydrogenation. A low value of apparent activation energy in the current work could also be attributed to the presence of a mass transport limitation regime in a non-diluted reaction system with a significantly lower ratio of reactant-to-catalyst.
:1)/Sibunit catalyst (1 mm) at 75–200 °C, 30 bar of H2 and 0.5 mL min−1 liquid flow of 0.012 M isoeugenol in dodecane. The activation energy in the kinetic study of dihydroeugenol hydrodeoxygenation, without the hydrogenation step of isoeugenol, in the packed-bed microreactor over the sulfided NiO/MoO3/Al2O3 catalyst (75–150 μm) at 200–450 °C and 20.7 bar was calculated to be 34 kJ mol−1.43 Note that the lower activation energy of DHE obtained in the current work from IE HDO in practice can depend not only on the catalyst type but also on the catalyst deactivation, commonly modeled as an exponential decay function of active sites with TOS,44 appearing in the first step of isoeugenol hydrogenation. A low value of apparent activation energy in the current work could also be attributed to the presence of a mass transport limitation regime in a non-diluted reaction system with a significantly lower ratio of reactant-to-catalyst.
The mass transport limitations were estimated for DHE HDO in the continuous mode (Table 6, entry 8a) assuming the spherical catalyst particles and the first-order reaction.45–47 The Mears criterion of external mass transfer limitation (CMears < 0.15) for H2 dissolved in DHE and for DHE was calculated to be 4423 and 2.3 × 10−4, respectively. Low values of the Damköhler number (Da = 7.8 × 10−4) and the Thiele modulus (ϕ = 2.8 × 10−5) giving subsequently the catalyst effectiveness factor equal to one also pointed out the absence of external or internal mass transfer limitations of the liquid compounds. It can be concluded that the low apparent activation energy obtained in the current work for DHE HDO was affected by both external mass transfer limitation of hydrogen dissolved in dihydroeugenol and by the rapid catalyst deactivation in the initial isoeugenol hydrogenation.
To elucidate the catalyst deactivation, the catalyst was consecutively reused in dihydroeugenol hydrodeoxygenation after in situ regeneration, which was performed by simply flushing the catalyst with hydrogen flow of 40 mL min−1 overnight at the reaction temperature (Table 6, entry 8) or by coke oxidation increasing temperature step-by-step from 200 °C to 400 °C, with the heating ramp of 2 °C min−1 with 40 mL min−1 of 5 vol% oxygen in argon after flushing with argon (Table 6, entry 9). In the latter case, the outlet gas stream was monitored by microGC-TCD (Fig. S17†) and after regeneration, the catalyst was flushed with argon again and reduced by the same procedure as described above.
In the first case, when the spent catalyst was flushed out in the hydrogen flow, the initial conversion of dihydroeugenol in the second run was only ca. 30% of the first run. After 3 h with time-on-stream, the conversion level was just by ca. 20% lower than with the fresh catalyst, while selectivity to the desired oxygen-free compounds was lower by ca. 60% and YOCC/YOFC ratio by ca. 70% (Fig. S18†). On the other hand, in the second case, when the coke in the spent catalyst was oxidized in 5 vol% of oxygen in argon flow at up to 400 °C, the initial conversion of dihydroeugenol during the second run was two-fold higher than in the case of flushing in hydrogen, i.e. ca. 60% of the initial value compared to the first run. The initial reaction rate dropped only by 1.7-fold for the second run. After 5 h of time-on-stream, the conversion level was lowered by ca. 25% compared to the fresh catalyst, while selectivity to the desired oxygen-free compounds was lower only by ca. 8% and YOCC/YOFC ratio by ca. 10% (Fig. S18†). Overall, it can be concluded that in situ regeneration at up to 400 °C in the presence of oxygen was successful resulting in slightly lower activity and similar selectivity of the consecutively reused catalyst compared to the results obtained from the fresh one. A slightly lower reaction rate of DHE, 0.4 mol g−1 h−1, was obtained from HDO of DHE over the sulfided NiO/MoO3/Al2O3 catalyst at 300 °C and 14.3 bar with 2.1 mol L−1 DHE concentration in hexane.43
As mentioned above, catalyst deactivation was directly related to the formation of aliphatic coke on the spent catalyst, which led to a decrease in its specific surface area (Fig. 9). Furthermore, it was observed that the rate of coke formation (Rc, μg of coke/mg of catalyst/h) increased with increasing dihydroeugenol conversion (Fig. 11a) and with increasing yield of water (Fig. 11b) as a side product. This applies to all experiments independently of the reaction system, temperature, or reactant. Simultaneously, the liquid phase carbon balance closure decreased with the increasing amount of coke in line with the strong adsorption of heavy compounds on the catalyst. Analogously, the coke formation was also considered as the reason for catalyst deactivation in bio-oil hydrodeoxygenation on Ni/H-ZSM-5 and Ni–Cu/H-ZSM-5 catalysts.48 The rate of coke formation of 10 μg of coke/mg of catalyst/h reported over Ni/HZSM-5 catalyst in bio-oil hydrodeoxygenation at 300 °C (ref. 47) was comparable with the lowest rate of coke formation of 12 μg of coke/mg of catalyst/h obtained in the current work at the lowest DHE conversion. A similar range of the rate of coke formation as in the current work (12–37 μg of coke/mg of catalyst/h) was also obtained over the 10 wt% Fe/SiO2 catalyst in guaiacol HDO at 400 °C in the presence of H2, CH4 or H2O (13–36 μg of coke/mg of catalyst/h).49 A lower rate of coke formation 8 μg of coke/mg of catalyst/h was observed in HDO of the bio-oil in a continuous-flow two-stage catalytic reactor system that contained a mild hydrogenation zone at 130 °C over Ru/C as the catalyst followed by a more severe HDO zone between 300 and 400 °C over a Pt/ZrP catalyst.50
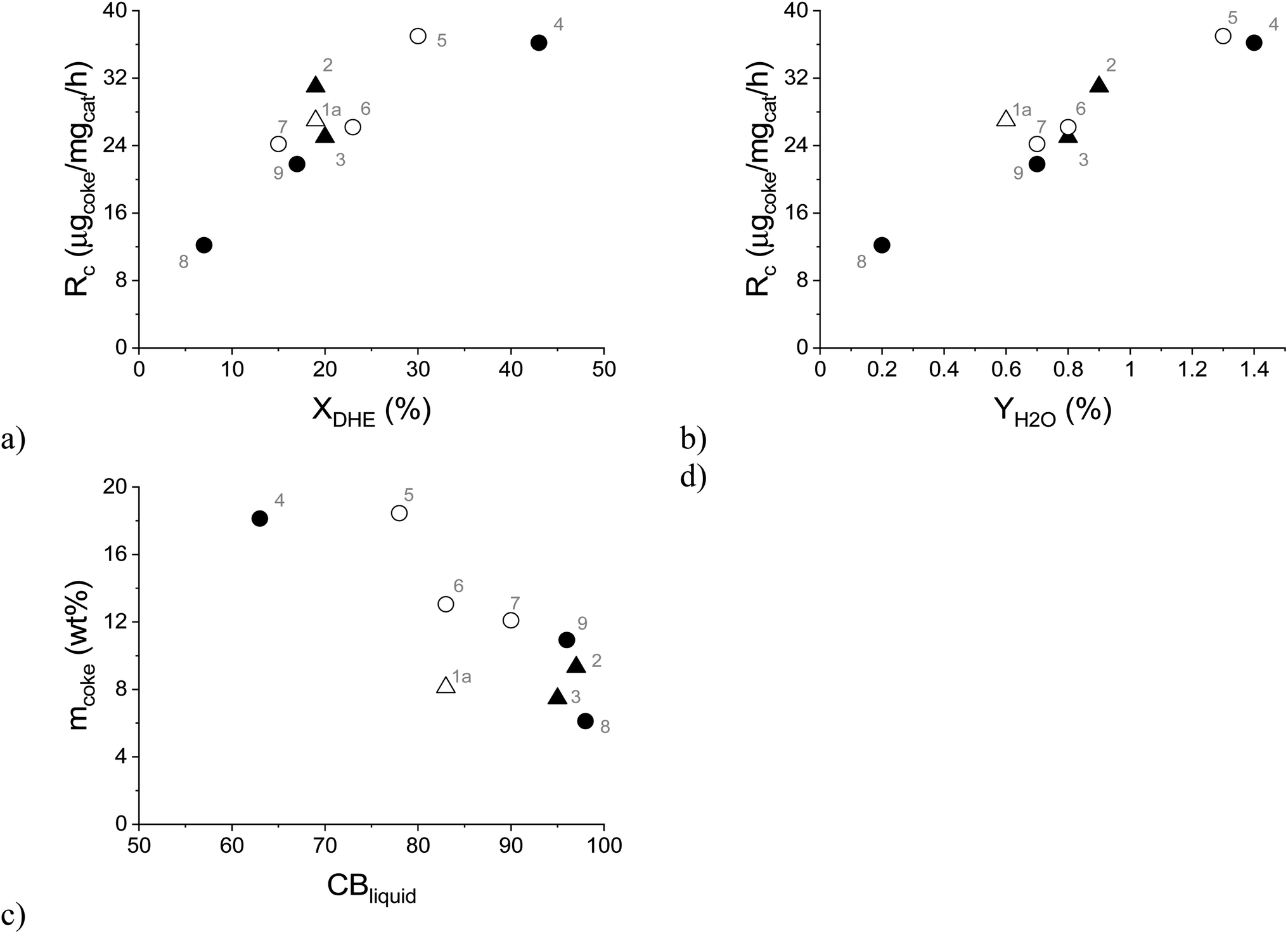 | ||
| Fig. 11 Formation rate of coke as a function of: (a) dihydroeugenol conversion, (b) yield of water; (c) amount of coke as a function of the liquid phase carbon balance closure. Conditions: solventless hydrodeoxygenation over 0.1–0.3 g of FeNi/H-Beta-300 (150–180 μm) catalyst at 250–300 °C, 30 bar of H2, 0.04 mL min−1 of liquid flow and a 15-fold excess of hydrogen. Legend: spent catalyst from the batch experiment (triangle), the spent catalyst from the continuous experiments (circle), the spent catalyst from IE HDO (open symbols) and the spent catalyst from DHE HDO (black symbols). Notation is the same as in Table 4. | ||
4 Conclusions
A low-cost bimetallic bifunctional 5–5 wt% FeNi/H-Beta-300 catalyst synthesized by the subsequent incipient wetness impregnation was investigated in hydrodeoxygenation in the batch and continuous modes. Solventless experiments were performed with lignin-derived model compounds isoeugenol or dihydroeugenol. The liquid phase of the reaction mixture was analysed by GC-FID, GC-MS and Karl-Fischer titration while the gas phase was analysed by microGC-TCD. Characterization of the fresh and spent catalysts was made by several physico-chemical methods. The spent catalyst was regenerated in situ and consecutively reused.After metal impregnation, the total acidity of the catalyst remained mild, 104 μmol g−1, while the ratio of the Brønsted and Lewis acid sites significantly decreased from 6 to 0.8. The nanoparticle size of Fe and Ni metals of 6 and 20 nm, respectively, was determined by fitting of kernel smooth distribution curves from TEM. The results from Mössbauer spectroscopy, XAS, SEM-EDX, and TPR confirmed metal–metal and metal–upport interactions. After the reduction, 93% of iron was in the metallic form. In the FeNi alloy, the distribution of both metals was random. The specific surface area for the fresh catalyst was 516 m2 g−1 comprising 81 vol% micropores. After the reaction, a significant drop in both the specific surface area and pore volume of the catalyst was observed due to the formation of filamentous-type coke. The specific surface area of the spent catalysts, tested under different conditions, linearly decreased with the increasing amounts of coke determined by TGA analysis (6–19 wt%). The extracted soluble coke species were predominantly identified as the aliphatic ones. After in situ regeneration by coke oxidation, the mesoporosity was fully regenerated while 80% of the specific surface area and microporosity was restored in the regenerated catalyst compared to the fresh one.
The batch experiments resulted in low activity and poor selectivity to the desired oxygen-free compounds accompanied by rapid catalyst deactivation. The experiments in the continuous mode demonstrated slower catalyst deactivation showing not only a decrease in conversion but also significant selectivity changes with increasing time-on-stream. A slightly slower catalyst deactivation was observed in the hydrodeoxygenation of dihydroeugenol (ΔXDHE 0.7% per minute) compared to isoeugenol hydrodeoxygenation in the continuous mode. The apparent activation energy of dihydroeugenol hydrodeoxygenation over the FeNi/H-Beta-300 catalyst (150–180 μm) in solventless isoeugenol hydrodeoxygenation was calculated to be 6.3 kJ mol−1, which can be ascribed to external mass transfer limitations and the catalyst deactivation in isoeugenol hydrogenation. Conversion of 78% of the initial dihydroeugenol with 80% of selectivity to the desired oxygen-free compounds was obtained over 0.3 g of catalyst at 300 °C and 30 bar of hydrogen with a residence time of 12 min. Oxidative regeneration at up to 400 °C was successfully done resulting in a slightly lower catalyst activity and similar selectivity of the regenerated catalyst compared to the fresh one.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Business Finland for funding through the project: Catalytic Slurry Hydrotreatment. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to thank Dr Edmund Welter for his assistance in using the beamline P65. We thank the Electron Microscopy Laboratory, Institute of Biomedicine, University of Turku, and Biocenter Finland. The authors are grateful to Dr Irina Simakova for the discussion regarding catalyst preparation, and Dr Rose-Marie Latonen and MSc. Minette Kvikant for help with Raman spectroscopy.References
- A. A. Dwiatmoko, J. Seo, J. W. Choi, D. J. Suh, J. M. Jae and J. M. Ha, Improved activity of a CaCO3-supported Ru catalyst for the hydrodeoxygenation of eugenol as a model lignin-derived phenolic compound, Catal. Commun., 2019, 127, 45–50, DOI:10.1016/j.catcom.2019.04.024.
- M. X. Zhao, J. Hu, P. Lu, S. L. Wu, C. Liu and Y. H. Sun, Efficient hydrodeoxygenation of lignin-derived bio-oil to hydrocarbon fuels over bifunctional RuCoWx/NC catalysts, Fuel, 2022, 326, DOI:10.1016/j.fuel.2022.125020.
- A. Bjelic, M. Grilc and B. Likozar, Bifunctional metallic-acidic mechanisms of hydrodeoxygenation of eugenol as lignin model compound over supported Cu, Ni, Pd, Pt, Rh and Ru catalyst materials, Chem. Eng. J., 2020, 394, 14, DOI:10.1016/j.cej.2020.124914.
- X. K. Yue, S. Zhang, N. Z. Shang, S. T. Gao, Z. Wang and C. Wang, Porous organic polymer supported PdAg bimetallic catalyst for the hydrodeoxygenation of lignin-derived species, Renewable Energy, 2020, 149, 600–608, DOI:10.1016/j.renene.2019.12.066.
- M. X. Zhao, J. Hu, S. L. Wu, L. Yang, X. An, P. Yuan and P. Lu, Hydrodeoxygenation of lignin-derived phenolics over facile prepared bimetallic RuCoNx/NC, Fuel, 2022, 308, 121979, DOI:10.1016/j.fuel.2021.121979.
- T. Liu, Z. Tian, W. Zhang, B. Luo, L. Lei, C. Wang, J. Liu, R. Shu and Y. Chen, Selective hydrodeoxygenation of lignin-derived phenols to alkyl cyclohexanols over highly dispersed RuFe bimetallic catalysts, Fuel, 2023, 339, 12916, DOI:10.1016/j.fuel.2022.126916.
- S. Khan, K. M. Qureshi, A. N. K. Lup, M. F. A. Patah and W. Daud, Role of Ni-Fe/ZSM-5/SAPO-11 bifunctional catalyst on hydrodeoxygenation of palm oil and triolein for alternative jet fuel production, Biomass Bioenergy, 2022, 164, 106563, DOI:10.1016/j.biombioe.2022.106563.
- J. W. Zhou and W. An, Unravelling the role of oxophilic metal in promoting the deoxygenation of catechol on Ni-based alloy catalysts, Catal. Sci. Technol., 2020, 10, 6849–6859, 10.1039/d0cy01361g.
- L. Bomont, M. Alda-Onggar, V. Fedorov, A. Aho, J. Peltonen, K. Eranen, M. Peurla, N. Kumar, J. Warna, V. Russo, P. Mäki-Arvela, H. Grénman, M. Lindblad and D. Yu. Murzin, Production of cycloalkanes in hydrodeoxygenation of isoeugenol over Pt- and Ir-modified bifunctional catalysts, Eur. J. Inorg. Chem., 2018, 24, 2841–2854, DOI:10.1002/ejic.201800391.
- N. K. G. Silva, R. A. R. Ferreira, R. M. Ribas, R. S. Monteiro, M. A. S. Barrozo and R. R. Soares, Gas-phase hydrodeoxygenation (HDO) of guaiacol over Pt/Al2O3 catalyst promoted by Nb2O5, Fuel, 2021, 287, 119509, DOI:10.1016/j.fuel.2020.119509.
- Z. Vajglová, B. Gauli, P. Mäki-Arvela, I. L. Simakova, N. Kumar, K. Eränen, T. Tirri, R. Lassfolk, M. Peurla, D. E. Doronkin and D. Yu. Murzin, Co-Processing of fossil feedstock with lignin-derived model compound isoeugenol over Fe-Ni/H-Y-5.1 catalysts, J. Catal., 2023, 421, 101–116, DOI:10.1016/j.jcat.2023.03.016.
- Z. Vajglová, B. Gauli, P. Mäki-Arvela, N. Kumar, K. Eränen, J. Warna, R. Lassfolk, I. L. Simakova, I. Provirin, M. Peurla, J. Lindén, H. Huhtinen, P. Petriina, D. E. Doronkin and D. Yu. Murzin, Interactions between iron and nickel in Fe-Ni nanoparticles on Y zeolite for co-processing of fossil feedstock with lignin-derived isoeugenol, ACS Appl. Nano Mater., 2023, 6, 10064–10077, DOI:10.1021/acsanm.3c00620.
- ImageJ program, https://imagej.net/Welcome, accessed March 2023 Search PubMed.
- B. Ravel and M. Newville, Athena, Artemis, Hephaestus: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541, DOI:10.1107/s0909049505012719.
- M. I. Urseanu, J. G. Boelhouwer, H. J. M. Bosman, J. C. Schroijen and G. Kwant, Estimation of trickle-to-pulse flow regime transition and pressure drop in high-pressure trickle bed reactors with organic liquids, Chem. Eng. J., 2005, 111, 5–11, DOI:10.1016/j.cej.2005.04.015.
- G. Mary, A. Esmaeili and J. Chaouki, Simulation of the selective hydrogenation of C-3-cut in the liquid phase, Int. J. Chem. React. Eng., 2016, 14, 859–874, DOI:10.1515/ijcre-2015-0095.
- Z. Vajglová, N. Kumar, M. Peurla, J. Peltonen, I. Heinmaa and D. Yu. Murzin, Synthesis and physicochemical characterization of beta zeolite-bentonite composite materials for shaped catalysts, Catal. Sci. Technol., 2018, 8, 6150–6162, 10.1039/c8cy01951g.
- X. Y. Yang, X. W. Cheng, A. A. Elzatahry, J. Y. Chen, A. Alghamdi and Y. H. Deng, Recyclable Fenton-like catalyst based on zeolite Y supported ultrafine, highly-dispersed Fe2O3 nanoparticles for removal of organics under mild conditions, Chin. Chem. Lett., 2019, 30, 324–330, DOI:10.1016/j.cclet.2018.06.026.
- J. J. F. Scholten, A. P. Pijpers and A. M. L. Hustings, Surface characterization of supported and nonsupported hydrogenation catalysts, Catal. Rev. Sci. Eng., 1985, 27, 151–206, DOI:10.1080/01614948509342359.
- A. J. Plomp, H. Vuori, A. O. I. Krause, K. P. de Jong and J. H. Bitter, Particle size effects for carbon nanofiber supported platinum and ruthenium catalysts for the selective hydrogenation of cinnamaldehyde, Appl. Catal., A, 2008, 351, 9–15, DOI:10.1016/j.apcata.2008.08.018.
- G. Bergeret, and P. Gallezot, Particle size and dispersion measurements, Handbook of Heterogenous Catalysis, ed. Ertl, G., Knözinger, H., Schüth, F. and Weitkamp, J., Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- D. Kubička, N. Kumar, T. Venalainen, H. Karhu, I. Kubičkova, H. Osterholm and D. Yu. Murzin, Metal-support interactions in zeolite-supported noble metals: influence of metal crystallites on the support acidity, J. Phys. Chem. B, 2006, 110, 4937–4946, DOI:10.1021/jp055754k.
- H. Metiu, S. Chretien, Z. P. Hu, B. Li and X. Y. Sun, Chemistry of Lewis acid-base pairs on oxide surfaces, J. Phys. Chem. C, 2012, 116, 10439–10450, DOI:10.1021/jp301341t.
- A. Puig-Molina, F. M. Cano and T. V. W. Janssens, The Cu promoter in an iron-chromium-oxide based water-gas shift catalyst under industrial conditions studied by in-situ XAFS, J. Phys. Chem. C, 2010, 114, 15410–15416, DOI:10.1021/jp1035103.
- M. Nagai, O. Uchino, J. Okubo and S. Omi, TPR and XPS studies of iron-exchanged Y zeolites and their activity during dibenzothiophene hydrodesulfurization, Stud. Surf. Sci. Catal., 1997, 105, 989–995, DOI:10.1016/S0167-2991(97)80731-8.
- N. Czuma, K. Zarebska, M. Motak, M. E. Galvez and P. Da Costa, Ni/zeolite X derived from fly ash as catalysts for CO2 methanation, Fuel, 2020, 267, 117139, DOI:10.1016/j.fuel.2020.117139.
- S. E. Eliana-Misi, A. Ramli and F. H. Rahman, Characterization of the structure feature of bimetallic Fe-Ni catalysts, J. Appl. Sci., 2011, 11, 1297–1302, DOI:10.3923/jas.2011.1297.1302.
- Z. X. Cheng, X. G. Zhao, J. L. Li and Q. M. Zhu, Role of support in CO2 reforming of CH4 over a Ni/gamma-Al2O3 catalyst, Appl. Catal., A, 2001, 205, 31–36, DOI:10.1016/s0926-860x(00)00560-3.
- O. A. Sahraei, A. Desgagnes, F. Larachi and M. C. Iliuta, Ni-Fe catalyst derived from mixed oxides Fe/Mg-bearing metallurgical waste for hydrogen production by steam reforming of biodiesel by-product: investigation of catalyst synthesis parameters and temperature dependency of the reaction network, Appl. Catal., B, 2020, 279, 119330, DOI:10.1016/j.apcatb.2020.119330.
- M. C. Sánchez-Sánchez, R. M. Navarro and J. L. G. Frierro, Ethanol stam reforming over Ni/La-Al2O3 catalysts: influence of lanthanum loading, Catal. Today, 2007, 129, 336–345, DOI:10.1016/j.cattod.2006.10.013.
- J. Cain, A. Laskin, M. R. Kholghy, M. J. Thomson and H. Wang, Molecular characterization of organic content of soot along the centerline of a coflow diffusion flame, Phys. Chem. Chem. Phys., 2014, 16, 25862–25875, 10.1039/c4cp03330b.
- C. Li and P. C. Stair, Coke formation in zeolites studied by a new technique: ultraviolet resonance Raman spectroscopy, Stud. Surf. Sci. Catal., 1997, 105, 599–606, DOI:10.1016/S0167-2991(97)80606-4.
- H. Z. Wang, L. L. Sun, Z. J. Sui, Y. A. Zhu, G. H. Ye, D. Chen, X. G. Zhou and W. K. Yuan, Coke formation on Pt-Sn/Al2O3 catalyst for propane dehydrogenation, Ind. Eng. Chem. Res., 2018, 57, 8647–8654, DOI:10.1021/acs.iecr.8b01313.
- C. C. Zhang, S. Hartlaub, I. Petrovic and B. Yilmaz, Raman Spectroscopy characterization of amorphous coke generated in industrial processes, ACS Omega, 2022, 7, 2565–2570, DOI:10.1021/acsomega.1c03456.
- S. R. Bare, F. D. Vila, M. E. Charochak, S. Prabhakar, W. J. Bradley, C. Jaye, D. A. Fischer, S. T. Hayashi, S. A. Bradley and J. J. Rehr, Characterization of coke on a Pt-Re/gamma-Al2O3 Re-forming catalyst: experimental and theoretical study, ACS Catal., 2017, 7, 1452–1461, DOI:10.1021/acscatal.6b02785.
- P. J. Wang, D. Zhang, L. S. Zhang and Y. Fang, The SERS study of graphene deposited by gold nanoparticles with 785 nm excitation, Chem. Phys. Lett., 2013, 556, 146–150, DOI:10.1016/j.cplett.2012.11.018.
- P. D. Green, C. A. Johnson and K. M. Thomas, Applications of laser Raman microprobe spectroscopy to the characterization of coals and cokes, Fuel, 1983, 62, 1013–1023, DOI:10.1016/0016-2361(83)90134-5.
- M. Alda-Onggar, P. Mäki-Arvela, A. Aho, I. L. Simakova and D. Yu. Murzin, Hydrodeoxygenation of phenolic model compounds over zirconia supported Ir and Ni-catalysts, React. Kinet., Mech. Catal., 2019, 126, 737–759, DOI:10.1007/s11144-018-1502-1.
- S. Tieuli, P. Mäki-Arvela, M. Peurla, K. Eränen, J. Warna, G. Cruciani, F. Menegazzo, D. Yu. Murzin and M. Signoretto, Hydrodeoxygenation of isoeugenol over Ni-SBA-15: Kinetics and modelling, Appl. Catal., A, 2019, 580, 1–10, DOI:10.1016/j.apcata.2019.04.028.
- C. Lindfors, P. Mäki-Arvela, P. Paturi, A. Aho, K. Eränen, J. Hemming, M. Peurla, D. Kubička, I. L. Simakova and D. Yu. Murzin, Hydrodeoxygenation of isoeugenol over Ni- and Co-supported catalysts, ACS Sustainable Chem. Eng., 2019, 7, 14545–14560, DOI:10.1021/acssuschemeng.9b02108.
- M. E. Martinez-Klimov, P. Mäki-Arvela, Z. Vajglová, M. Alda-Onggar, I. Angervo, N. Kumar, K. Eränen, M. Peurla, M. H. Calimli, J. Muller, A. Shchukarev, I. L. Simakova and D. Yu. Murzin, Hydrodeoxygenation of isoeugenol over carbon-supported Pt and Pt-Re catalysts for production of renewable jet fuel, Energy Fuels, 2021, 35, 17755–17768, DOI:10.1021/acs.energyfuels.1c02656.
- S. M. Li, B. Y. Liu, J. Truong, Z. Y. Luo, P. C. Ford and M. M. Abu-Omar, One-pot hydrodeoxygenation (HDO) of lignin monomers to C9 hydrocarbons co-catalysed by Ru/C and Nb2O5, Green Chem., 2020, 22, 7406–7416, 10.1039/d0gc01692f.
- N. Joshi and A. Lawal, Hydrodeoxygenation of 4-Propylguaiacol (2-Methoxy-4-propylphenol) in a microreactor: performance and kinetic studies, Ind. Eng. Chem. Res., 2013, 52, 4049–4058, DOI:10.1021/ie400037y.
- I. L. Simakova, Z. Vajglová, P. Mäki-Arvela, K. Eränen, L. Hupa, M. Peurla, E. M. Makila, J. Warna and D. Yu. Murzin, Citral-to-menthol transformations in a continuous reactor over Ni/mesoporous aluminosilicate extrudates containing a sepiolite clay binder, Org. Process Res. Dev., 2022, 26, 387–403, DOI:10.1021/acs.oprd.1c00435.
- G. Ertl, H. Knözinger, F. Schüth and J. Weikamp, Handbook of Heterogeneous Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, DOI: DOI:10.1002/9783527610044.hetcat0001.
- D. Yu. Murzin, Chemical Reaction Technology, De Gruyter, 2022 Search PubMed.
- S. H. Fogler, Elements of Chemical Reaction Engineering, Pearson, 2020 Search PubMed.
- Y. Li, C. S. Zhang, Y. G. Liu, S. S. Tang, G. H. Chen, R. Q. Zhang and X. Q. Tang, Coke formation on the surface of Ni/HZSM-5 and Ni-Cu/HZSM-5 catalysts during bio-oil hydrodeoxygenation, Fuel, 2017, 189, 23–31, DOI:10.1016/j.fuel.2016.10.047.
- R. Olcese, M. M. Bettahar, B. Malaman, J. Ghanbaja, L. Tibavizco, D. Petitjean and A. Dufour, Gas-phase hydrodeoxygenation of guaiacol over iron-based catalysts. Effect of gases composition, iron load and supports (silica and activated carbon), Appl. Catal., B, 2013, 129, 528–538, DOI:10.1016/j.apcatb.2012.09.043.
- K. Routray, K. J. Barnett and G. W. Huber, Hydrodeoxygenation of pyrolysis oils, Energy Technol., 2016, 5, 80–93, DOI:10.1002/ente.201600084.
Footnote |
| † Electronic supplementary information (ESI) available: Definitions, catalyst characterization results: SEM, BSE, EDX, ICP-OES, TEM, EXAS, O2-TPO, GC-MS analysis of the extracted soluble coke species, Raman, and catalytic results. See DOI: https://doi.org/10.1039/d3se00371j |
| This journal is © The Royal Society of Chemistry 2023 |