
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRejuvenation of dearomative cycloaddition reactions via visible light energy transfer catalysis
Angshuman
Palai†
,
Pramod
Rai†
and
Biplab
Maji
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, West Bengal, India. E-mail: bm@iiserkol.ac.in
First published on 13th October 2023
Abstract
Dearomative cycloaddition is a powerful technique to access sp3-rich three-dimensional structural motifs from simple flat, aromatic feedstock. The building-up of unprecedentedly diverse polycyclic scaffolds with increased saturation and stereochemical information having various applications ranging from pharmaceutical to material sciences, is an essential goal in organic chemistry. However, the requirement of large energy inputs to disrupt the aromaticity of an arene moiety necessitates harsh reaction conditions for ground state dearomative cycloaddition. The photochemical requirement encompasses use of ultraviolet (UV) light to enable the reaction on an excited potential energy surface. The microscopic reversibility under thermal conditions and the use of high energy harmful UV irradiation in photochemical manoeuvres, however, constrain their widespread use from a synthetic point of view. In this context, the recent renaissance of visible light energy transfer (EnT) catalysis has become a powerful tool to initiate dearomative cycloaddition as a greener and more sustainable approach. The excited triplet state population is achieved by triplet energy transfer from the appropriate photosensitizer to the substrate. While employing mild visible light energy as fuel, the process leverages an enormous potential of excited state reactivity. The discovery of an impressive portfolio of organic and inorganic photosensitizers with a range of triplet energies facilitates visible light photosensitized dearomative cycloaddition of various substrates to form sp3-rich fused polycyclic architectures with diverse applications. The tutorial review comprehensively surveys the reawakening of dearomative cycloadditions via visible light-mediated energy transfer catalysis in the past five years. The progress ranges from intra- and intermolecular [2π + 2π] to [4π + 2π], and ends at intermolecular [2π + 2σ] cycloadditions. Furthermore, the review provides potential possibilities for future growth in the growing field of visible light energy transfer catalysis.
1. Introduction
Rapid generation of molecular complexity from readily available starting materials is one of the trickiest approaches in synthetic chemistry. The cycloaddition reactions bring exciting opportunities in this regard due to their perfect atom economy, stereospecificity, and single-step operation.1,2 Aromatic compounds could, in principle, be ideal reaction partners for cycloaddition reactions, as they, at least from a topological point of view, contain requisite ene-components. Furthermore, the dearomative cycloaddition reactions could be considered an advanced synthetic and practically rewarding tactic for producing polycyclic three-dimensional architectures from widely abundant flat molecules.3,4 However, the aromatic stability of these conjugated systems must be broken during these reactions, which is translated as kinetic and thermodynamic barriers for these processes.5–7 Even if the kinetic barrier can be overcome, the endothermicity of these reactions makes these reactions reversible and favors the starting materials under thermal conditions (Fig. 1A).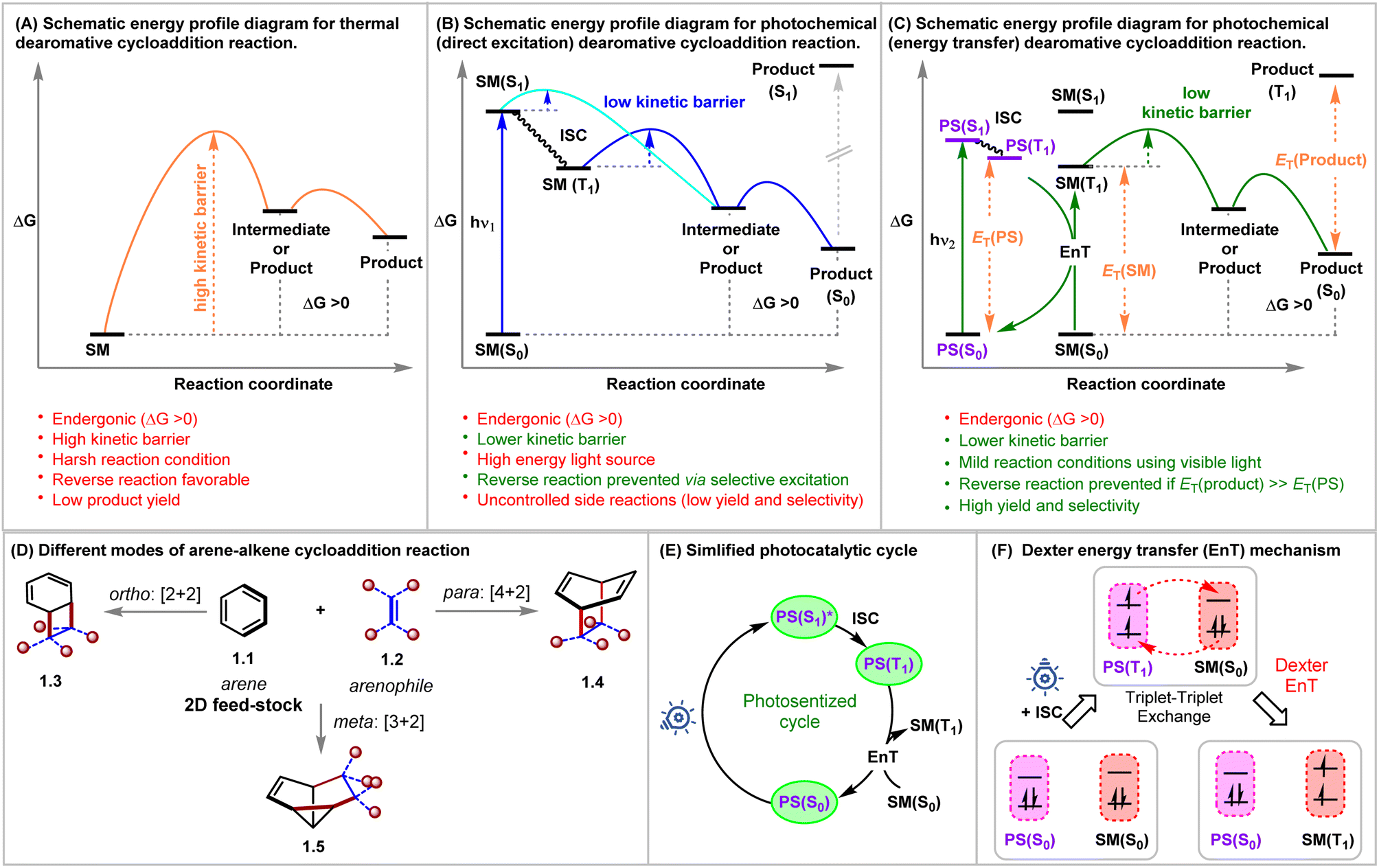 | ||
| Fig. 1 Schematic energy profile diagram for (A) thermal, (B) UV light-mediated photochemical, and (C) visible light-energy transfer mediated dearomative cycloaddition reactions. (D) Different modes of the arene–alkene cycloaddition reaction. (E) Simplified photocatalytic cycle. (F) Schematic mechanism of Dexter energy transfer (EnT). SM = starting material; PS = photosensitizer; ISC = intersystem crossing; ET = ΔG(T1) − ΔG(S0). | ||
Decades of rigorous research have unraveled the panorama of prospective cycloadditions involving fascinating substrate activations.8 As illustrated in Fig. 1A, the thermal pathway for achieving dearomative cycloadditions is endergonic. This can be attributed to the loss of molecular stability from the loss of aromatic stabilization. The high potential barrier requires the employment of harsher reaction conditions, which, however, favors reversibility, thus giving low yields of the desired cycloadduct. Shifting to photochemical routes (Fig. 1B and C) makes it possible to incite the reactant selectively by judicious choice of light sources of appropriate wavelengths while leaving the product intact.9 As illustrated in Fig. 1B, a starting material can be selectively excited when the light energy (hν1) is matched with the [ΔG(S1) − ΔG(S0)]SM. Thus, the endergonic cycloadduct ([ΔG(S1) − ΔG(S0)]product > hν1) would be trapped kinetically, and the backward thermal retro-cycloaddition reaction can be prevented effectively by negating the principle of microscopic reversibility, keeping the much lower kinetic barrier in view. In this regard, the extent of photochemical reactivity for aromatic compounds ranges from isomerizations, additions, and substitutions to cycloadditions via direct excitation of the aromatic chromophore.3,10 Based on the orbital symmetry and mode of connection, the arene–arenophile cycloaddition can be considered to be of three types: ortho [2 + 2], meta [3 + 2], and para [4 + 2] (Fig. 1D).3
The foundation for photochemical dearomative cycloadditions was laid in 1959 with the commencement of the pioneering work on classical ortho-cycloaddition by Angus and Bryce-Smith.11Meta-photocycloaddition was discovered in 1966, independently by Wilzbach and Kaplan.12 Five years after this unearthing of the meta-variant, the same duo realized the para-cycloaddition of benzene with olefins forming bicyclo[2.2.2]-octa-2,5-dienes upon irradiation of their reaction mixture at 254 nm.13 Bryce-Smith later conducted a detailed investigation on the orbital symmetry relationships requisite for ortho-, meta-, and para-cycloadditions of benzene under thermal and photochemical conditions.14 In 1982, Houk interpreted frontier orbital overlap between benzene and ethylene, revealing that the ortho- and meta-cycloadditions are photochemically allowed, whereas para-cycloaddition is photochemically forbidden.15 This was in total correlation with the well-established Woodward–Hoffman rule.16 Bryce-Smith, Gilbert, and Orger raised an open question about the involvement of singlet or triplet intermediates in the existing reports as molecular orbital symmetry arguments only allow meta-cycloadditions to occur in a concerted fashion if benzene's first excited singlet state, i.e., from S1 benzene (1B2u = particular concerted process occurring from the lowest excited singlet state in the system; that is from S1) is involved. Bryce-Smith showed the interaction between the first excited singlet state, i.e., from S1 of arene (1B2u) and an alkene to be symmetry allowed, whereas the same reaction from the triplet excited state, i.e., T1 of arene (3B1u = particular concerted process occurring from the triplet state in the system; that is from T1) state was symmetry forbidden.14 Ferree et al. provided evidence for the occurrence of meta-cycloaddition via the singlet excited state only under the influence of direct excitation.17 The para-cycloadduct formation was found to be feasible if either the reaction took place in a non-concerted manner (independent of the Woodward–Hoffmann rule) or via the involvement of the second singlet excited state (1B1u = particular concerted process occurring from the second excited-state of benzene) of benzene.18 The selectivity of the type of cycloaddition depends on the electronic properties of coupling partners engaged in the process. The Rehm–Weller equation is utilized to draw inferences regarding the reaction mode from the parameters showcasing the possibilities of electron and charge transfer in the excited state (eqn (1)).19 The free energies (ΔGET) corresponding to the charge transfer could be calculated from the given equation simply by using the oxidation potential of the donor Eox(D), the reduction potential of the acceptor Ered(A), and the excitation energy of the electronically excited species. According to Mattay, electron-rich and electron-deficient olefin partners preferentially produce the meta-cycloadduct with benzene given that ΔGET > 1.4–1.6 eV and in other cases, give rise to the ortho-cycloaddition product. The Weller equation shows that the meta- to ortho-cycloadduct transition increases upon decreasing the value of ΔGET below 1.4 eV.20
| Weller equation: ΔGET = Eox1/2(D) − Ered1/2(A) − ΔEexcit + ΔEcoul | (1) |
In addition to product selectivity issues discussed above, the use of high-energy UV irradiation is a severe concern for the classic photochemical dearomative cycloaddition reaction.21,22 Low product yields, low functional group tolerance, and unpredictable side reactions are common characteristics of these methods, posing a limitation on their widespread applicability. As a result, developing mild visible-light-mediated catalytic techniques can serve as an important tool for addressing the problems mentioned above.23,24
In this regard, an external photosensitizer (PS) allows aromatic hydrocarbons (ArH) to get photochemically excited by incident visible light energy (Fig. 1C). The PS absorbs the visible light energy to reach its triplet state (T1) and transfers it to the ArH, a process known as triplet sensitization or energy transfer (EnT), which excites ArH indirectly to its triplet state. A simplified photosensitized cycle is depicted in Fig. 1E. Mechanistically, EnT is classified into three types: primitive EnT, Förster resonance EnT (a radiation-free transmitter-receiver mechanism),25 and Dexter EnT (Fig. 1F).26 Dexter described the concurrent two-electron transfer mechanism in 1953 (Fig. 1F).26 In the context of dearomative cycloaddition reactions in organic synthesis, the photosensitized pathway generally follows the Dexter EnT mechanism, providing mild conditions and establishing the requirement of low activation energy over a thermodynamically unfavorable thermal route. In Dexter EnT, a photochemically excited PS transfers an electron to the lowest unoccupied molecular orbital (LUMO) of the ArH substrate while simultaneously receiving another electron from its highest occupied molecular orbital (HOMO). Excited state energy transfer and spin multiplicity reversal are observed synchronously. The proximity between the PS and the ArH is essential for efficient Dexter EnT, and the rate decays exponentially with the increasing separation between them.25 The triplet state energy differences ΔET = ET(PS) − ET(ArH) can be used to predict the efficiency of Dexter EnT, and the overall efficiency of EnT is co-related with the ΔET.27 When ΔET > 0 (exergonic), many coupled transitions are able to populate the substrate's triplet state. When ΔET < (endergonic), coupled transitions only exist from excited vibrational and rotational levels of the PS's triplet state, leading to inefficient triplet-state excitation of the substrates.28 The overall rate of EnT is determined by using the diffusion rate and the spectral overlap integral, which can be calculated from the energies of the triplet excited states.25,29 The product could be kinetically trapped if the ET(PS) < ET(product), and the product selectivity can be determined.
An efficient energy transfer catalyst or a PS should have the following characteristics: (1) a large absorption cross-section, (2) an effective intersystem crossing (ISC) to its triplet state, (3) a long excited state lifetime, and (4) high triplet state energy compared to that of the substrate. In 1950, the sensitization-induced synthetic technique was first introduced, in which ketones, with the lowest π* excited state, acted as the PS.30 However, the application of said ketones as photosensitizers is quite limited owing to their low absorption at longer wavelengths. Besides, the n–π* transition produces an unpaired electron on the oxygen atom and promotes hydrogen atom transfer (HAT), resulting in unwanted products. Later, ruthenium and iridium-based photocatalysts emerged, demonstrating high visible-light absorption and longer excited state lifetimes.31
The triplet state energy transfer (EnT) catalysis has been recently employed for diverse organic transformations and reviewed as well.23,32–40 The last review focused on arene–alkene cycloaddition was reported in 2016.3 The modern visible-light mediated EnT dearomative cycloaddition has been advanced since 2018 and is rapidly developing en route to becoming the cornerstone of synthetic methodology. This tutorial article is envisioned to summarize the recent developments to educate the readers and demonstrate the exciting opportunities ahead. The following sections provide an in-depth study and critical analysis of the dearomative cycloaddition of different aromatic compounds and their applications in various sustainable chemical syntheses. The literature is classified based on the mode of connection and sub-categorized based on the intra- and intermolecularity of the cycloadditions involved.
2. Dearomative [2 + 2] ortho-cycloaddition
Substantial research has been conducted on arene–alkene [2π + 2π] cycloaddition reactions (Fig. 1D), often referred to as ortho-cycloaddition reactions employing direct excitation of the arene by UV irradiation.3 As discussed, the first ortho photo-cycloaddition of benzene with maleic anhydride was reported by Angus and Bryce-Smith.11 Since then, this field has received significant contributions from many groups, including Faggiani,41 Hanzawa and Panqutte,42 Wagner and Nahm,43 Zupancic and Sket,44 and Hoffmann and Pete.45 The visible-light-induced variant was discovered only recently. Triplet sensitization is employed to excite the arene moiety for a stepwise reaction with the olefin counterpart to produce the cyclobutane fragment, widely present in several natural products, pharmaceuticals, and agrochemicals.46,47 The following section summarizes the recent developments.2.1. Intramolecular dearomative [2π + 2π] ortho-cycloaddition
In 2018, Glorius et al. reported the first visible light-mediated EnT dearomative [2 + 2] cycloaddition reaction (Fig. 2).48 When the iridium-based photosensitizer PS-1 (ET = 49.2 kcal mol−1) was irradiated with 455 nm LEDs in MeOH in the presence of the substrates 2.1, alkene tethered 1-naphthols bearing an adjacent electron-withdrawing group, the [2 + 2] cycloadduct 2.2a was isolated in 93% yield. However, when PS-2, having a higher ET (=61.8 kcal mol−1), capable of exciting 2.2, was used, a different cascade product 2.3 with a 6/4/6/5 fused ring structure was isolated. The ET of 2.2a was computed to be 55.9 kcal mol−1 (B3LYP/6-311+G(2d,p) level of density functional theory (DFT)), and PS-1 could not productively interact with 2.2a since it has a low triplet energy of 49.2 kcal mol−1. The consequent experimental studies demonstrated the selective synthesis of [2 + 2] cycloadduct 2.2 using PS-1 (1 mol%) in 0.02 M MeOH (condition A) and cascade product 2.3 using PS-2 (1 mol%) in 0.04 M 1,4-dioxane (condition B). A detailed study on the photophysical properties of the PSs confirmed that photoinduced single electron transfer (SET) was not responsible for the generation of 2.2 and 2.3. Instead, the authors correlated the differing product identities with the different ETs of the respective PSs. Both PSs absorbed visible light and became excited to their triplet state, thus activating naphthols 2.1via EnT to their triplet state 2.1(T1), followed by the commencement of intramolecular stepwise [2 + 2] cycloaddition to generate cyclobutanes 2.2. The selectivity mentioned above was found to be controlled by the relative rates of EnT between the PS and substrate. Stern–Volmer quenching studies showed that 2.2 effectively quenched the luminescence of PS-2 only. PS-1 having a lower ET does not interact with 2.2. Thus, it was proposed that rapid conversion of 2.1 to 2.2 initially occurred. 2.3 was formed subsequently from 2.2via EnT-mediated vinyl cyclobutane rearrangement. It should be noted that no reaction was observed in the absence of the electron-withdrawing carbonyl group, which is essential to lower the ET of the naphthol moiety to undergo EnT with desired efficiency.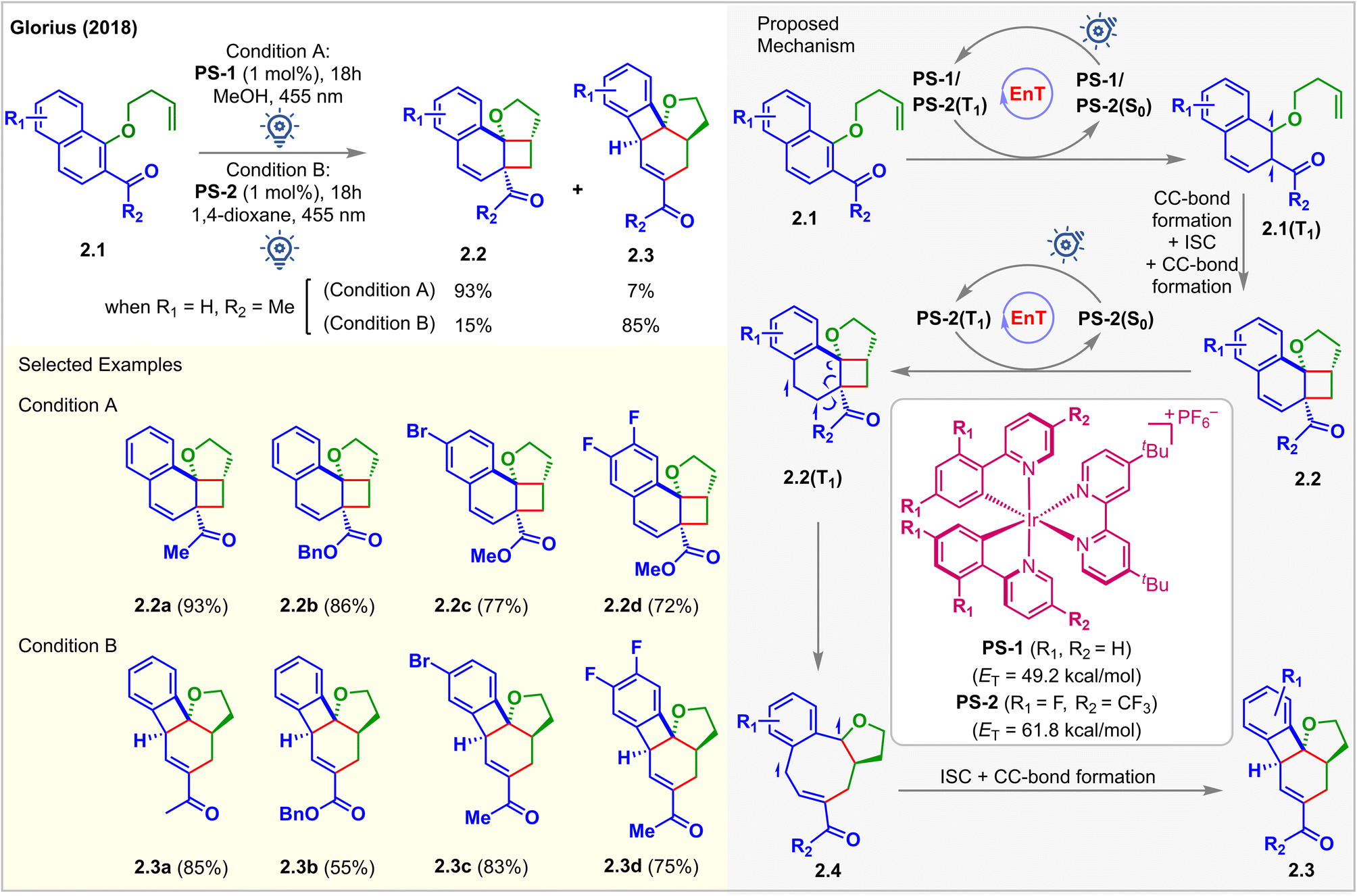 | ||
| Fig. 2 Visible light mediated EnT dearomative intramolecular [2π + 2π] cycloaddition of alkene-tethered 2-acetyl naphthalene derivatives. | ||
The You group was the first to report visible light-mediated EnT intramolecular partial dearomative [2 + 2] cycloaddition of indole derivatives 3.1 bearing a tethered alkene (Fig. 3, condition A).49 The ETs of C2-unsubstituted and unprotected indole derivatives (3.1, R1 = R2 = H) was calculated to be 65 kcal mol−1 (B3LYP/6-311+G(2d,p) level of DFT), which bore no parity with the ETs of PS-1 (ET = 49.2 kcal mol−1) and PS-2 (ET = 61.8 kcal mol−1), and thus does not react (yield of 3.2a = 0%) under the irradiation of visible light. Extending the conjugation by incorporating a Ph group at the C2 position (3.1, R2 = Ph) lowered the ET to 55.9 kcal mol−1. It smoothly underwent the [2 + 2] cycloaddition in the presence of PS-2 (4 mol%) in a CH2Cl2/CH3CN solvent mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to produce cycloadduct 3.2b with an excellent 95% yield. Notably, the reactivity also increased significantly for the N-electron withdrawing protecting group. The N-trifluoroacetyl indole derivative produced 3.2d in 98% yield. The proposed mechanism involved the excitation of PS-2 under blue light irradiation and EnT to yield a diradical species 3.1(T1) (Fig. 3). Subsequent 5-exo-trig cyclization forms diradical species 3.3, which undergoes ISC followed by radical recombination to give the desired [2 + 2] cycloadduct 3.2. Although this method tolerates substituted pendant olefin derivatives well, the cationic nature of iridium complexes and their low solubility limit the widespread application of this reaction. Nevertheless, a successful strategy was initiated for developing such highly strained molecules with contiguous quaternary centers. In 2022, Nolan et al. modified the above approach by using the carbene–metal–amido complex (Fig. 3, condition B).50 The gold carbazolyl complex [Au(SIPr)(Cbz)] PS-3 (ET = 66.6 kcal mol−1; uB3LYP/6-311+G(2d,p) level of DFT) acted as the sensitizer. It allowed the more facile intramolecular cycloaddition to occur in a shorter reaction time using greener solvents (MeOH, EtOAc, and i-PrOAc). However, 365 nm UV light irradiation was required. The authors successfully probed the cycloaddition of challenging unprotected indole, yielding a single diastereomer of the desired product 3.2a in 80% yield. It was also confirmed that diester-substitution is unnecessary to form the cycloadduct 3.2f. Indoles with electron-donating substituents were also well tolerated for this reaction forming 3.2g with 91% yield. Quenching studies, light on/off experiments, cyclic voltammograms, and quantum yield determination provided evidence to support the mechanism via the EnT pathway.
:1) to produce cycloadduct 3.2b with an excellent 95% yield. Notably, the reactivity also increased significantly for the N-electron withdrawing protecting group. The N-trifluoroacetyl indole derivative produced 3.2d in 98% yield. The proposed mechanism involved the excitation of PS-2 under blue light irradiation and EnT to yield a diradical species 3.1(T1) (Fig. 3). Subsequent 5-exo-trig cyclization forms diradical species 3.3, which undergoes ISC followed by radical recombination to give the desired [2 + 2] cycloadduct 3.2. Although this method tolerates substituted pendant olefin derivatives well, the cationic nature of iridium complexes and their low solubility limit the widespread application of this reaction. Nevertheless, a successful strategy was initiated for developing such highly strained molecules with contiguous quaternary centers. In 2022, Nolan et al. modified the above approach by using the carbene–metal–amido complex (Fig. 3, condition B).50 The gold carbazolyl complex [Au(SIPr)(Cbz)] PS-3 (ET = 66.6 kcal mol−1; uB3LYP/6-311+G(2d,p) level of DFT) acted as the sensitizer. It allowed the more facile intramolecular cycloaddition to occur in a shorter reaction time using greener solvents (MeOH, EtOAc, and i-PrOAc). However, 365 nm UV light irradiation was required. The authors successfully probed the cycloaddition of challenging unprotected indole, yielding a single diastereomer of the desired product 3.2a in 80% yield. It was also confirmed that diester-substitution is unnecessary to form the cycloadduct 3.2f. Indoles with electron-donating substituents were also well tolerated for this reaction forming 3.2g with 91% yield. Quenching studies, light on/off experiments, cyclic voltammograms, and quantum yield determination provided evidence to support the mechanism via the EnT pathway.
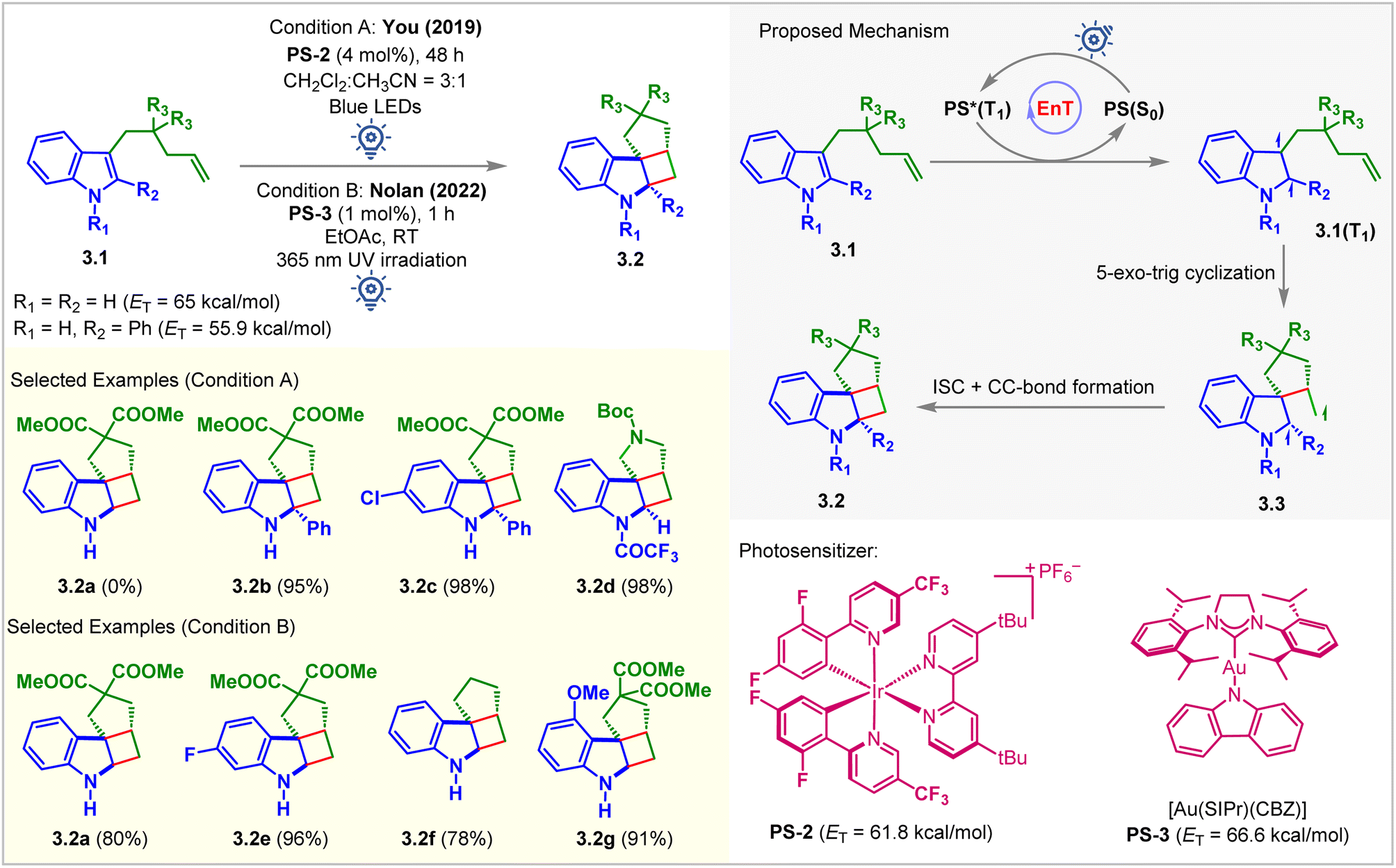 | ||
| Fig. 3 Visible light-mediated EnT dearomative intramolecular [2π + 2π] cycloaddition of C3-alkene-tethered indole derivatives. | ||
In 2020, the Fu group fostered a novel and practical photocatalytic partial dearomative intramolecular [2 + 2] cycloaddition of indole derivatives 4.1via hydrogen bond-influenced EnT (Fig. 4A).51 Substrate 4.1 (ET = 62.5 kcal mol−1) has a relatively higher ET than PS-2 (ET = 61.8 kcal mol−1) and PS-4 (ET = 55.2 kcal mol−1), thus making the energy transfer step thermodynamically unfeasible. The reaction produced lower yields when PS-2 and aprotic solvents, including CH2Cl2, MeCN, and DMF, were employed. Switching the nature of the solvent from aprotic to protic by introducing trifluoroethanol resulted in a dramatically improved yield of 4.2 up to 96%. The aggregation-induced and cooperatively enhanced H-bonds between the oxygen atom of the amide and trifluoroethanol were reasoned to be responsible for lowering the ET of the substrate below that of the PS-2. In particular, when the electron-withdrawing group (COOMe, formyl, and phenyl) was substituted at the C2 or C3 position, an excellent, up to 97%, yield of 4.2d was obtained.
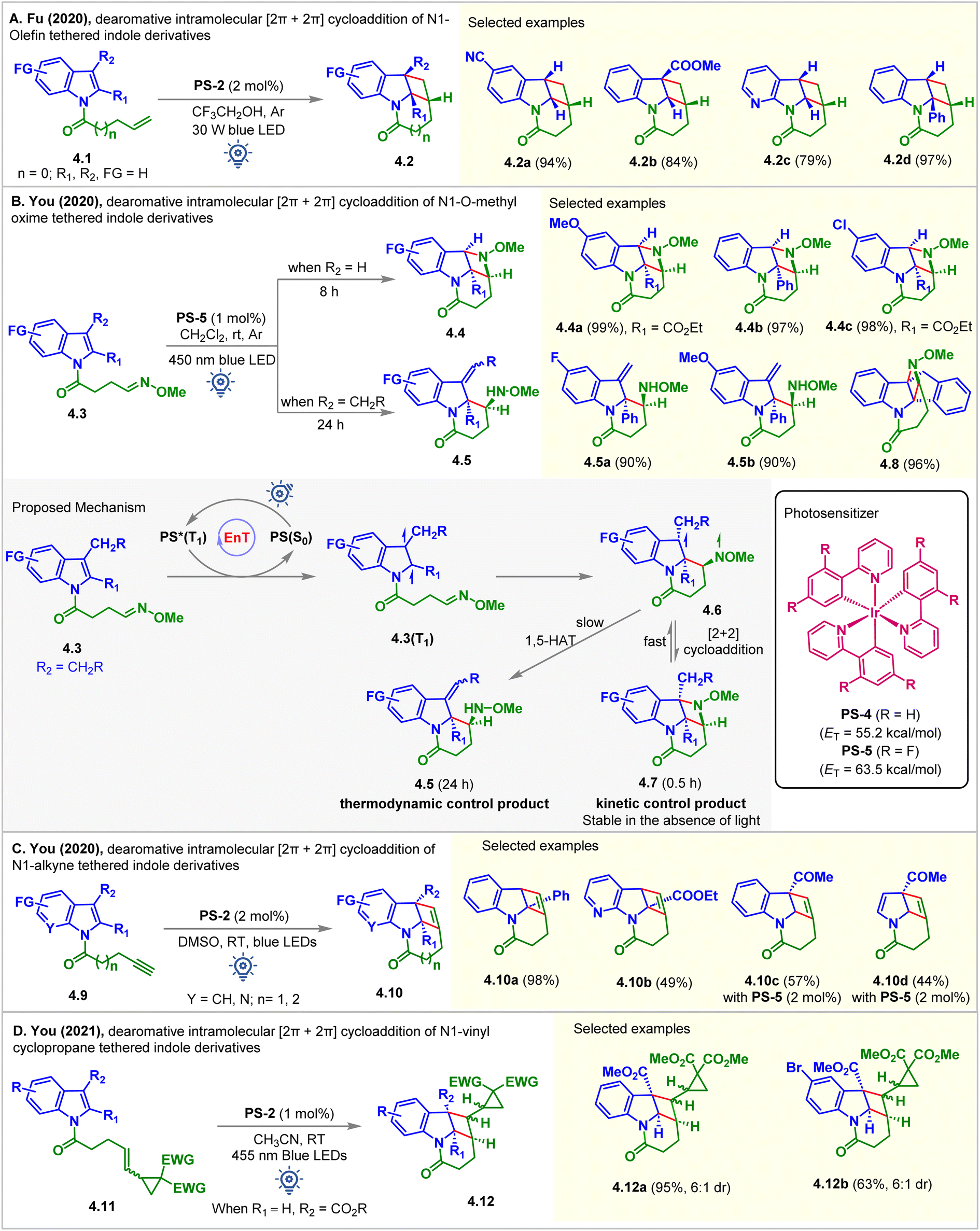 | ||
| Fig. 4 Visible light mediated EnT dearomative intramolecular [2π + 2π] cycloaddition of (A) N1-olefin, (B) N1-O-methyl oxime, (C) N1-alkyne, and (D) N1-vinyl cyclopropane tethered indole derivatives. | ||
In the same year, the You group disclosed the visible light-mediated EnT divergent partial dearomative cycloaddition of indole-tethered O-methyl oximes 4.3 (Fig. 4B).52 The reaction was conducted in CH2Cl2 (0.01 M) in the presence of PS-5. The [2 + 2] cycloadduct 4.4b was isolated as a single diastereomer in 97% yield. The presence of an EWG or an aryl group at the C2 position of indole achieved excellent results. However, the [2 + 2] cycloaddition of the 3-alkyl substituted indole moiety was interrupted after the first C–C bond formation. 1,5-hydrogen atom transfer (HAT) took place faster, delivering exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonded products 4.5 (Fig. 4B). The computation studies (B3LYP/6-31G(d,p) level of DFT) suggested that formation of early stage [2 + 2] intramolecular dearomative cycloadducts 4.7 was kinetically controlled. When the reaction was allowed to continue, the consumption of 4.7 caused the regeneration of 4.6, which then underwent 1,5-HAT to yield the thermodynamically controlled products 4.5. Due to conformational restrictions preventing a 1,5-HAT process, an indene-fused substrate underwent dearomative [2 + 2] cycloaddition, providing product 4.8 in 96% yield.
C bonded products 4.5 (Fig. 4B). The computation studies (B3LYP/6-31G(d,p) level of DFT) suggested that formation of early stage [2 + 2] intramolecular dearomative cycloadducts 4.7 was kinetically controlled. When the reaction was allowed to continue, the consumption of 4.7 caused the regeneration of 4.6, which then underwent 1,5-HAT to yield the thermodynamically controlled products 4.5. Due to conformational restrictions preventing a 1,5-HAT process, an indene-fused substrate underwent dearomative [2 + 2] cycloaddition, providing product 4.8 in 96% yield.
At the same time, the You group also registered the intramolecular partial dearomative [2 + 2] cycloaddition of indole-tethered terminal alkynes 4.9 in the presence of PS-2 under blue LED irradiation (Fig. 4C).53 Tetracyclic cyclobutene-fused benzindolizidines 4.10 were formed in good to excellent yields and >20:1 dr. An EWG (CO2Et, CO2Me) at the C2 position was necessary. Pyrrolo[2,3-b]pyridine derivative 4.10b also underwent the reaction, albeit in moderate yields. However, due to the high ET (up to 59.9 kcal mol−1) for C3-EWG substituted substrates (4.9, R1 = H, R2 = CO2Me, COMe, CN), PS-2 furnishes the corresponding products in low yields (up to 57%). Interestingly, PS-5 having higher ET = 63.5 kcal mol−1 enabled their [2 + 2] cycloaddition to produce 4.10c in moderate yields. Due to the lack of resonance stabilization for the photo-generated unpaired electron, a pyrrole substrate underwent cycloaddition reactions to produce 4.10d in a moderate 44% yield in the presence of PS-5.
Vinylcyclopropanes have demonstrated their applicability as an essential building block in several organic syntheses and frequent recurrence in many natural products.54 The tendency to undergo easy ring-opening and capacity to generate diradicals make them a well-known reagent in radical clock experiments.55 In 2021, You studied the partial dearomative cyclization of indoles and pyrroles with these units (Fig. 4D).56 Upon irradiation with blue LEDs (24 W, λmax = 455 nm), PS-2 (1 mol%) catalyzed the dearomative [2 + 2] cycloaddition of C3-carboalkoxy substituted substrates 4.11 in MeCN. The cyclobutene-fused indolines 4.12 were produced in high yields with 6:1 dr. However, without a C3-carbomethoxy substituent, the same reaction produced [5 + 2] cycloadducts, not discussed in this manuscript. The authors attributed this alteration of the reaction pathway to the increase in steric bulk at the C3-position of the indole ring, which leads to alkene-generated diradical addition directly to the indole fragment in a facile [2 + 2] manner, thus circumventing the [5 + 2] cycloadduct formation.
In 2020, Dhar et al. reported the visible light-induced EnT partial dearomative intramolecular [2 + 2] cycloaddition reaction of C2-substituted heteroarenes 5.1 for the synthesis of cyclobutane–pyrrolidinone-fused tetracyclic scaffolds 5.2 (Fig. 5A, condition A).57 There are two potential routes of ring closure leading to the formation of the product: (a) 5-exo-trig; and (b) 7-endo-trig, as demonstrated in the proposed mechanism (Fig. 5A). A computation study (B3LYP/6-311+G(2d,p) level of DFT) revealed that the former pathway was energetically more favorable than the latter. Tethering of olefin in the carboxamide substituent at the C2 position lowers the ET to <60 kcal mol−1 when compared to the olefin tethered at the C3 position of carboxamide, which has ET > 60 kcal mol−1. This was due to the enhanced stability via extended conjugation of the diradical intermediates 5.1(T1), which eliminated the requirement for the phenyl and ester group at the C2 position, as shown in Fig. 3 (condition A). The scope of the substrates went well beyond the indole-based moiety where C2 substituted indenes (5.1, X = CH2), benzothiophenes (5.1, X = S), and benzofurans (5.1, X = O) with ETs less than 60 kcal mol−1 were also capable of undergoing intramolecular dearomative cycloaddition with excellent 93–95% yield to produce 5.2b, 5.2c, and 5.2d, respectively. With the irradiation of the photosensitizer (PS-5), substrates 5.1 undergo 57% conversion in 24 h, and for the complete transformation, 48 h was required. With 400 nm purple light, the reaction was accelerated, and 5.1 was fully reacted within 18–24 h. However, for the 6-bromo-functionalized indole derivative, a 5% proto-debromination side product was observed in the presence of higher energy 400 nm light. An excellent yield of 97% was also observed for cycloadduct 5.2e with an unprotected indole. However, only small quantities of cycloadduct 5.2f were observed for substrates bearing an unprotected carboxamide group.
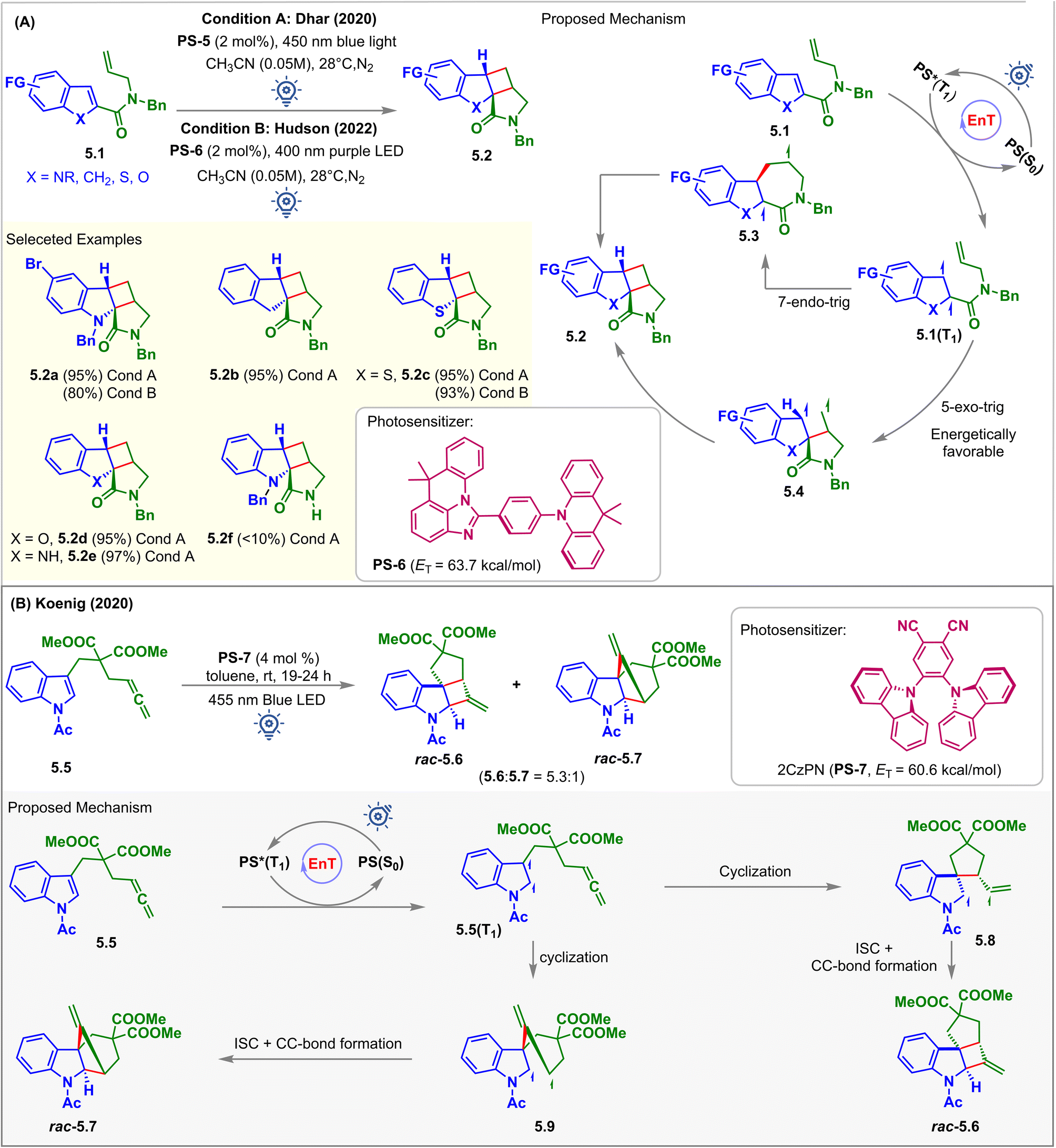 | ||
| Fig. 5 Visible light mediated EnT dearomative intramolecular [2π + 2π] cycloaddition of (A) alkene tethered carboxamide of the indole/benzofuran/benzothiophene derivative, and (B) allene tethered indole derivatives. | ||
In 2022, Hudson et al. demonstrated imidazoacridine-based thermally activated delayed fluorescence (TADF) material (ACR-IMAC, PS-6) to be an affordable, highly effective organic alternative to the commonly used iridium-based PSs for visible light EnT catalysis (Fig. 5A, condition B).58 The twisted donor–acceptor architecture of these materials accelerates rapid ISC and widens the HOMO–LUMO energy gap, thus minimizing electron transfer phenomena. Besides, high ETs (63.7 kcal mol−1), extended excited state lifetimes (64 μs), and poor oxidizing properties make them effective PSs in visible light-mediated EnT reactions. It has been demonstrated that the TADF material PS-6 completely converts 5.1 to cycloadducts 5.2 under purple LED irradiation. However, the efficiency dropped to a yield of 51% when exposed to blue light. The derivatives of benzo-thiophene, chromene, and indene also experience a similar dearomative [2 + 2] cycloaddition with excellent yield and high diastereocontrol (dr > 99:1). However, the cycloaddition of indole with an alkyne did not proceed successfully.
In 2020, Koenig investigated the potential of low-cost 2CzPN (PS-7, ET = 60.6 kcal mol−1) as a PS over well-known iridium-catalysts to carry out the partial dearomative cycloaddition of allene-tethered indoles 5.5 in the presence of blue LEDs (Fig. 5B).59 Ohkuma previously attempted the same reaction using a high catalyst loading of 3′,4′-dimethoxyacetophenone (50 mol%) as the PS. However, this involved irradiation from a high-energy mercury lamp through Pyrex glass.60 In this report, the desired cis-fused methylenecyclobutane-containing products were obtained in 80% yields with high stereoselectivity along with a trace amount of undesired alkyne product via 1,5-HAT. Also, incorporating –OMe and –COOMe groups at the C5 position reduced the reaction rates, producing undesirable deacylated or photo-Fries rearranged side products. However, the employment of PS-7 by Koenig et al. overcame the difficulties mentioned above. With the irradiation of PS-7 in the presence of a 455 nm light source, substrates 5.5 produced a mixture of rac-5.6 and rac-5.7 in a 5.3:1 ratio in toluene. The solubility of uncharged PS-7 in toluene allows its easy recovery and reuse over several cycles. Non-polar solvents inhibited electron transfer from indole derivatives 5.5 to PS-7, indicating EnT to be the operative mechanistic pathway. The presence of two ester groups facilitates ring closure via the Thrope–Ingold effect.
In 2022, Bach accessed the low-lying triplet states of aryl iminium ions 6.1via EnT (Fig. 6).61 Olefin-tethered aryl iminium ions 6.1 produced the [2 + 2] cycloadducts 6.2 in the presence of 2.5 mol% thioxanthone (TXT, PS-8, ET = 63.1 kcal mol−1) under 420 nm blue light irradiation for 18 h. Cycloadducts 6.2 undergo iminium ion-triggered rearrangement to produce cyclized products 6.3. The rearranged products 6.4 were isolated after treating the reaction mixture with aq. NaOH. Carbon-tethered olefinic and alkyne-tethered aryl iminium ions were found to be unreactive, proving the necessity of the oxygen substituent at the 2-position of iminium ions 6.1 to carry out the cycloaddition. Notably, under standard conditions, methoxy-substituted arenes and those with a substituent at the 5-position failed to give expected products which the authors attributed to inaccessibility of the respective triplet states due to change in electronic properties.
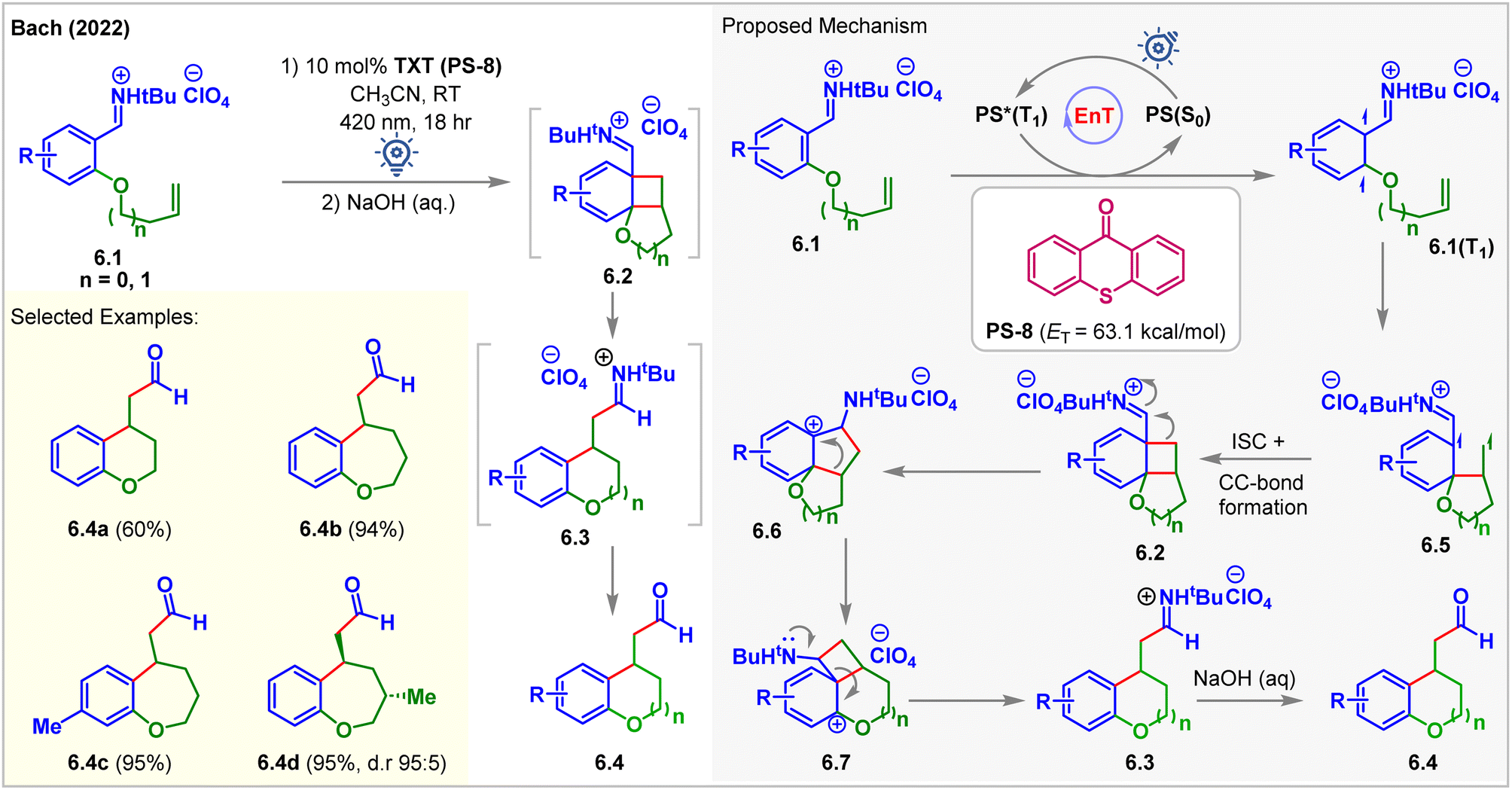 | ||
| Fig. 6 Visible light-mediated EnT dearomative intramolecular [2π + 2π] cycloaddition of an olefin tethered aryl-iminium ion. | ||
2.2. Intermolecular [2π + 2π] ortho-cycloaddition
Due to entropic challenges, the intermolecular [2 + 2] cycloaddition reactions are more demanding than the intramolecular ones. Besides, the EnT-generated excited state of the substrate must be long-lived to be intercepted by the coupling partner. In 2018, Meggers et al. reported the first visible-light-induced intermolecular dearomative [2 + 2] cycloaddition of 2-N-acylpyrazole-substituted benzofurans 7.1 with styrenes 7.2 using a chiral-at-rhodium complex PS-9 (2 mol%) as the catalyst (Fig. 7).62 The cycloadducts 7.3 were isolated in high yields with up to 98% ee on exposure to blue LED irradiation with PS-9 for 18 h. The reaction also yielded minor diastereomers 7.4 and regioisomers 7.5. Due to the lability of the N-acyl pyrazole moiety, the initially formed cycloadduct was treated with methanol in the presence of a base to isolate methyl ester 7.3 as the product. The diastereo- and regioisomeric ratios depended on the nature of substituents on styrenes. The reaction with parent styrene produced 7.3a in 78% yield with 98% ee, 6.2:1 dr, and 5.3:1 rr. Computation studies (B3LYP/6-31G(d,p) level of DFT) supported this observation. The (Z)-β-methyl styrene produced 7.3b in 90% yield with 97% ee and very high (>20:1) dr and (>20:1) rr when irradiated at −30 °C. The dr was found to be temperature dependent, as the same reaction produced a poor 1.8:1 dr under room temperature conditions. This observation was consistent with the formation of a biradical intermediate where temperature plays a crucial role in the rotation of the C–C single bond, vide infra. When (E)-β-methyl styrene took part in the reaction, the opposite diastereomer 7.4a was formed as the major product (>20:1 dr, >20:1 rr) in 90% yield with 99% ee. The presence of electron-donating, electron-withdrawing, and sterically hindered groups at the meta and para positions of the styrene aromatic ring was well tolerated while safeguarding the dr and rr. As an exception, an ortho-substituted styrene gave lower rr and dr of the product 7.3c. Interestingly, a chiral-at-iridium analog of PS-9 showed no conversion at all. Mechanistically, the substrate 7.1 was initially associated with PS-9 to form the [7.1-Rh] complex, as shown in Fig. 7. After the EnT from excited photosensitizer PS*(T1), the substrate-sensitizer complex [7.1-Rh] got excited to its triplet state [7.1-Rh](T1). The latter then reacted with 7.2b to form the biradical intermediate 7.6, and the stability of the 1,4-benzyl radical 7.6 determined the regioselectivity. Then, ISC, followed by C–C bond formation, generated the 7.7 complex, followed by the ligand exchange between 7.1 and 7.7 to liberate the product 7.8 and close the catalytic cycle. As a post-synthetic transformation, the methanolysis of 7.8 afforded 7.3b in 90% yield and 97% ee.
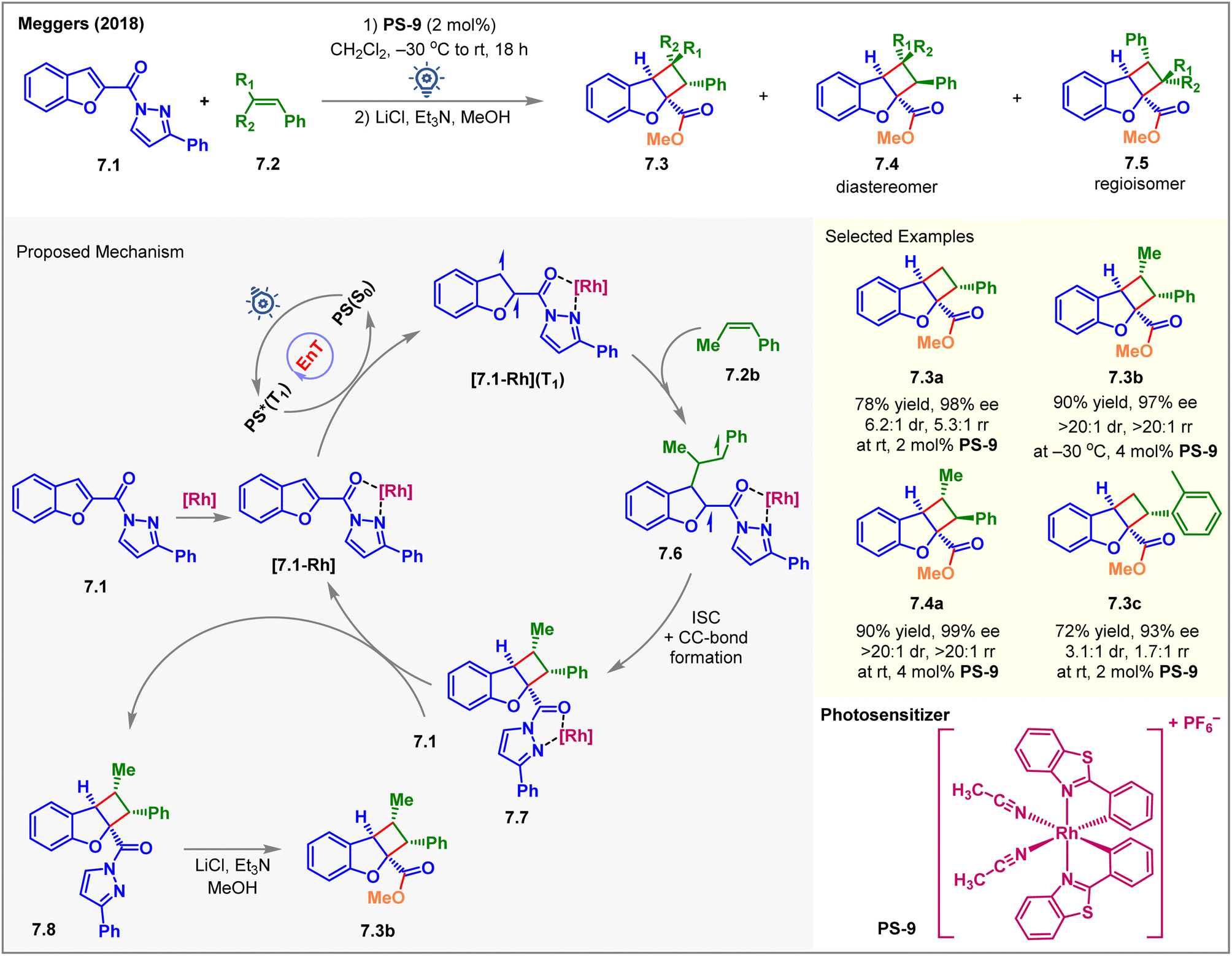 | ||
| Fig. 7 Visible light-mediated EnT asymmetric dearomative intermolecular [2π + 2π] cycloaddition of 2-N-acylpyrazole-substituted benzofuran derivatives with a chiral-at-rhodium photosensitizer. | ||
In 2021, the Oderinde group employed a substrate-based screening approach to examine indole derivatives 8.1a–8.1g for partial dearomative intermolecular [2 + 2] cycloaddition reaction (Fig. 8).63 Thermodynamically, accessing the indole triplet-states (ET = 68–70 kcal mol−1) from most PSs (ET = 40–64 kcal mol−1) is improbable which is also supported by the B3LYP/6-311+G(2d,p) level of DFT calculations. Installing an EWG at the C2 position lowers the ET of the indole moiety and should ideally make the desired cycloadditions more facile. However, with the irradiation of [Ir(dF(Me)ppy2)dtbbpy]PF6 (PS-10, ET = 62.9 kcal mol−1) under 450 nm blue LEDs, the indole derivatives 8.1d–8.1g did not undergo intermolecular [2 + 2] cycloadditions with vinylsilane 8.2, despite having a lower ETs than PS-10. It was hypothesized that the excited triplet diradical species derived from 8.1d–8.1g were too short-lived to undergo intermolecular cycloaddition reactions. While a 76% head-to-head homo-dimer product along with 24% minor cycloadduct 8.3a was produced from 8.1a, the indole derivative 8.1b experienced cycloadditions with vinylsilane 8.2 to produce cycloadduct 8.3b in 50% yield, along with head-to-tail homo-dimers 8.4. Notably, N-Boc-indole 8.1c reacted with vinylsilane 8.2 to produce the cycloadduct 8.3c in an excellent 95% yield, >20:1 rr, and 16:1 dr. Without vinylsilane, 8.1c underwent homodimerization to produce 8.4c in 98% isolated yield, implying that productive cross-cycloaddition occurred significantly faster than homodimerization. Additionally, the reactions could also be carried out in higher yields using purple LEDs (=400 nm) in the presence of poorly oxidizing sensitized PS-5 (Ir(III)*/Ir(II) = 0.36 V vs. SCE). While exploring the alkene scope, the authors observed that bulky substituents on the alkenes enhanced the dr. Modest yields were obtained while extending the methodology for the reactions with various alkynes. Homodimerized products are the major side products. It is implied that the cross-cycloaddition reaction with alkynes was less efficient than that with alkenes.
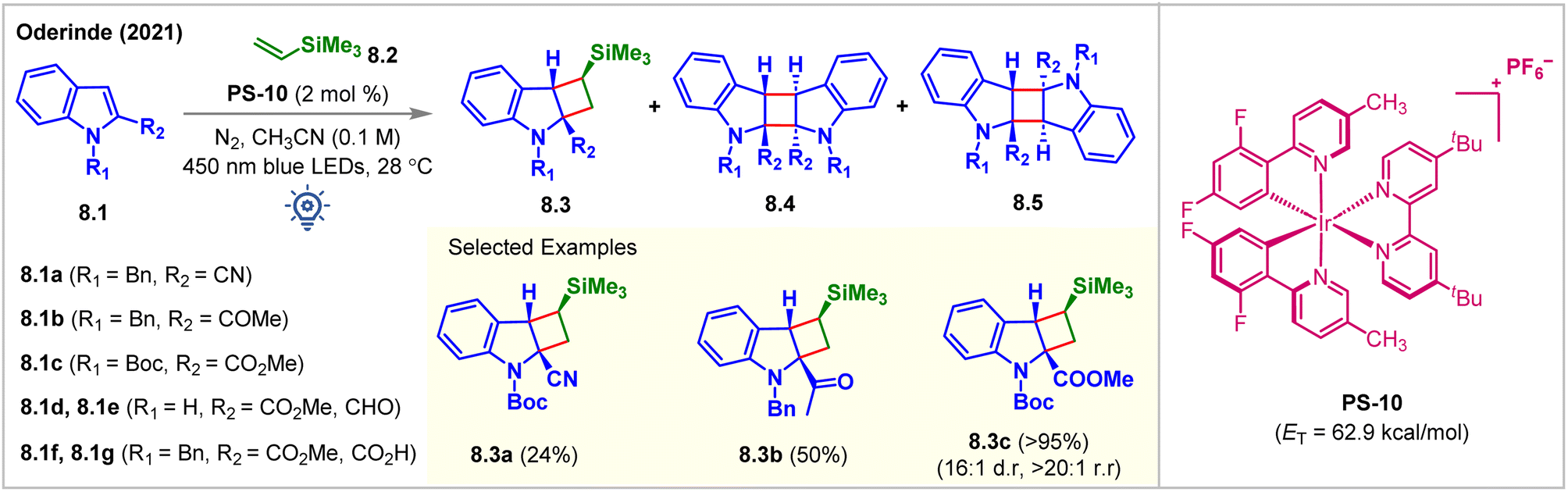 | ||
| Fig. 8 Visible light mediated EnT dearomative intermolecular [2π + 2π] cycloaddition of various indole derivatives with vinylsilane. | ||
2.3. Intermolecular [2π + 2σ] ortho-cycloaddition
Rigid saturated cyclic scaffolds with complex three-dimensionality, such as bicylo[1.1.0]butanes (BCBs), have become increasingly relevant bioisosteres and valuable medicinal chemistry motifs.64,65 The distortions of the ring in BCBs forced them to flip the ring between different configurations at high temperatures.66 They have different bond angles distorting from a planar conformation with high p-character (96%) in the central C–C σ-bond (Fig. 9A). This central C–C σ-bond has been shown to interact with a wide variety of electrophiles and nucleophiles, in processes ranging across a variety of thermal67 and photochemical cycloadditions (Fig. 9A).68 Recent studies showed that the BCBs could undergo [2π + 2σ] cycloadditions with alkenes by cleaving this bridge σ-bond.69–72 Glorius and Houk utilized this strategy for the intermolecular dearomative [2π + 2σ] cycloaddition of quinolines 9.1 with BCBs 9.2 (Fig. 9B).73PS-2 (2 mol%) catalyzed the reaction with the aid of a Lewis acid Sc(OTf)3 (0.5 equiv.). The reaction produced C(sp3)-rich bicyclo[2.1.1]hexanes (BCHs, 9.3) in CH2Cl2 under 450 nm blue LED irradiation of PS-2 for 16 h at room temperature. The BCH 9.3a was isolated from 7-methoxyquinoline 9.1a in 89% yield. It was observed that both the reaction partners could not absorb 450 nm light. However, in the presence of Sc(OTf)3, a bathochromic absorbance shift for both substrates was noticed. The product 9.3a was formed in 43% yield even in the absence of PS-2 with Sc(OTf)3 (1 equiv.) via direct excitation. Sulfone-substituted BCB also underwent the [2π + 2σ] cycloaddition reaction to produce 9.3b in 67% yield. 1,3-Disubstituted BCBs also participated, and the product 9.3f bearing a quaternary carbon center at the bridge-head position was formed without Lewis acid. 4-Substituted quinolines were also tolerated. However, a 7-methoxy substituent was necessary. Stern–Volmer quenching studies revealed significant quenching of the excited PS by 7-methoxy quinoline 9.1a. The DFT study at the UωB97XD/def2TZVPP/SMD (solvent = DCM)//UM06-2X/def2SVP level of theory showed that the Lewis acid Sc(OTf)3 lowers the ET of 9.1a (ET = 62.6 kcal mol−1) by 10.5 kcal mol−1 such that the EnT from PS-2 (ET = 61.8 kcal mol−1) to the quinoline–scandium adduct 9.4 is highly exergonic. The resulting 9.4(T1) biradical species attacked the BCBs 9.2. The ISC is followed by C–C bond formation to give the desired [2π + 2σ] cycloadduct. The authors also proposed an alternative biphotonic radical chain propagation mechanism where the radical cations 9.7 were hypothesized to be formed via the SET mechanism in two different reaction pathways. The intermolecular cyclization of 9.7 forms another radical cation, 9.8, which then transforms into product 9.3 after undergoing a SET from quinolines 9.1, propagating the chain. Recently, Glorius reported the insertion of BCB to the thiophene moiety towards dearomative ring expansion via a photocatalyzed SET mechanism.74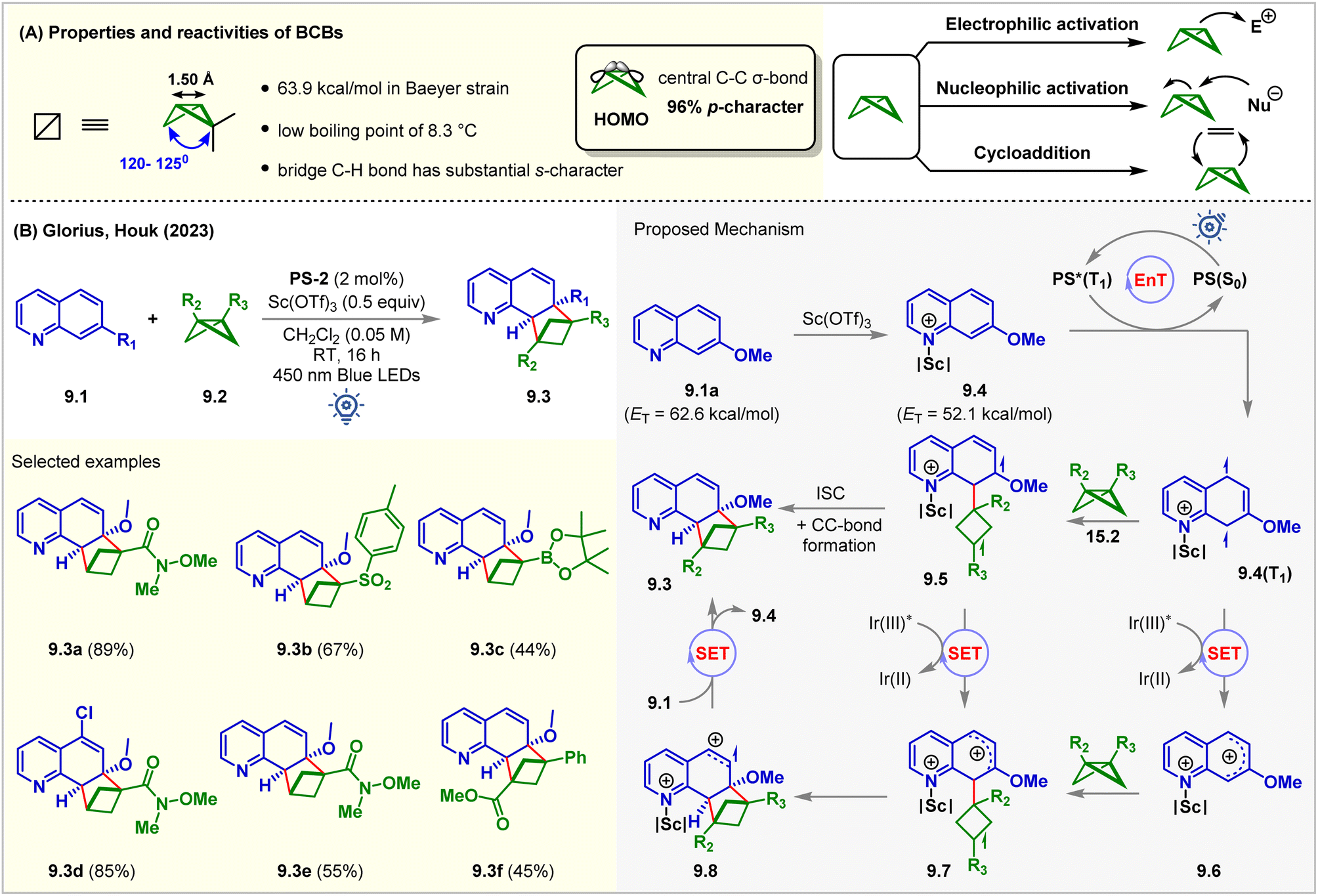 | ||
| Fig. 9 (A) Properties and reactivity of BCBs and (B) visible light mediated EnT dearomative intermolecular [2π + 2σ] cycloaddition of 7-methoxy quinolines with BCBs. | ||
3. Dearomative [4π + 2π] para-cycloaddition
The Diels–Alder reaction represents one of the powerful methods for six-membered ring synthesis.75–79 Electron-rich dienes and electron-deficient dienophile partners or vice versa assure high yields and reactivity explained by using the frontier molecular orbital (Fig. 10A).80 In 1989, Hamrock and Sheridan reported selective [4 + 2] or para-cycloaddition of benzene with 4-methyl-1,2,4-triazoline-3,5-dione (MTAD) under visible light irradiation at −60 °C (Fig. 10B).81 Low-temperature requirement was necessary for generating an arene–alkene cycloadduct as retro-cycloaddition happens at a temperature greater than −10 °C. The cycloaddition mechanism was not appropriately understood, indicating that either electron transfer or charge transfer or both become plausible pathways of cycloaddition between photo-activated MTAD and benzene. Subsequently, para-cycloaddition appeared in the literature through the work of Wilzbach and Kaplan,13 Gilbert and Taylor,82 DÖpp,83 Bochet,84 and others under high energy UV irradiation. Recently, Sarlah studied arene-MTAD cycloaddition in detail and coupled the photocycloaddition reaction with transition metal-catalyzed amino-functionalization (Fig. 10C),85–87 and dihydoxylation reactions.88 It was found that MTAD got excited for the cycloaddition process. The application of dearomatization has become a revolutionary method in addressing the significant challenges synthetic chemists face. The isolation and detailed characterization of MTAD-benzene cycloadducts were difficult due to their lower thermodynamic stability. This becomes a significant drawback that restricts understanding reactivity and further applications. Recently, Matsunaga and Yoshino89 demonstrated that the [4 + 2] cycloadduct of 1,2-dihydro-1,2,4,5-tetrazine-3,6-diones (TETRADs) and benzene could be isolated by the chromatographic technique and characterized by NMR and XRD (Fig. 10D). Interestingly, the further transformation of the TETRAD-benzene cycloadduct occurs above room temperature.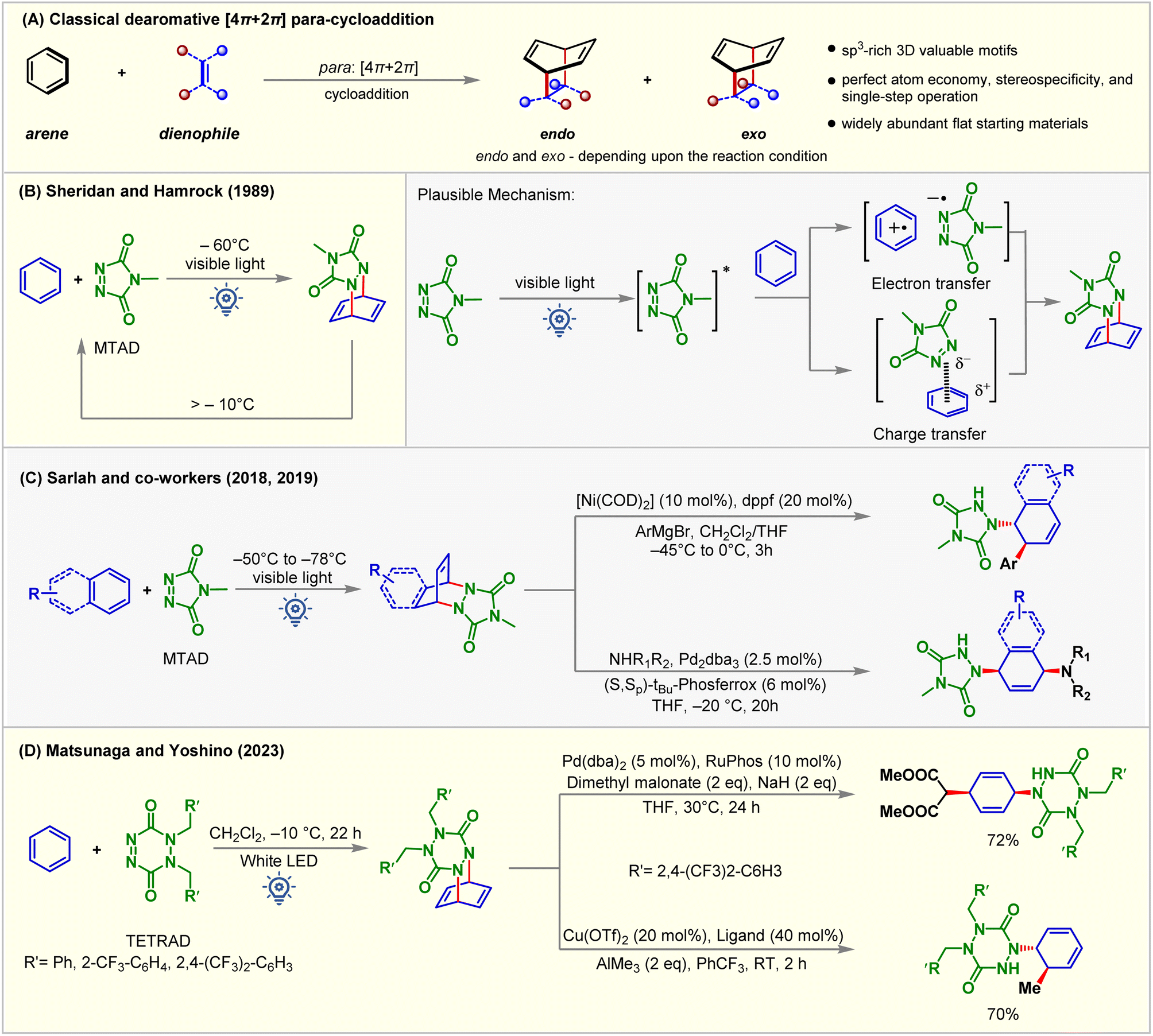 | ||
| Fig. 10 (A) Classical [4 + 2] para-cycloaddition reaction, (B) para [4 + 2] cycloaddition of benzene with MTAD under visible light irradiation, (C) transition metal-catalyzed amino-functionalization of simple arenes, and (D) [4 + 2] para-cycloaddition of TETRAD and benzene and further applications. | ||
When electronically mismatching cycloaddition partners are used, the requirement of high temperature, pressure, and UV irradiation strongly limits their widespread application in chemical and pharmaceutical industries.90 Electronically mismatched cycloaddition can be enabled by photoinduced SET or chemical oxidation using stoichiometric oxidants.91 Since the high-energy UV-light and stoichiometric redox reagents negatively impacted the sustainability of the reaction, the visible-light-induced [4 + 2] cycloaddition reaction using catalytic PSs is a promising alternative.89 As discussed earlier, the reluctance of arenes to lose aromaticity is a crucial challenge that needs to be overcome. It is relatively easy for heteroaromatic compounds and phenol derivatives with less aromaticity to undergo dearomative transformations, whereas it is highly challenging for benzene derivatives due to their enhanced aromaticity.92 Dearomative [4 + 2] para-cycloaddition employing light mainly focuses on sensitizing alkene partners instead of the arene.93 Visible-light-EnT-mediated direct sensitization of the arene derivatives is still developing. The main focus of the next section is to summarize the recent development of EnT dearomative [4 + 2] cycloaddition reactions under the influence of visible light. Both arene and arenophile excitation are discussed depending on the accessibility of their triplet states.
3.1. Intramolecular [4π +2π] para-cycloadditions
Pyridine derivatives are incompatible with photocatalytic reactions due to their redox inactivity and high ET > 70 kcal mol−1, making it challenging for them to undergo SET and EnT processes.94 However, the triplet states of the alkenes could be accessed to enable their cycloaddition with pyridines. In 2019, Glorius reported an intramolecular dearomative [4 + 2] cycloaddition of N-cinnamoyl picolinamide derivatives 11.1via EnT catalysis (Fig. 11).95 A polymer immobilized [Ir-F]@polymer PS-11 (1.5 mol%, ET = 60.8 kcal mol−1) was used as a catalyst. The EnT from photoexcited PS-11 to the pyridine substrates 11.1 (ET = 46 kcal mol−1) was thermodynamically feasible, which is also supported by computational studies at the (U)CAM-B3LYP/def2-TZVPP/CPCM (MeCN) level of theory. The [4 + 2] cycloadducts 11.2 were isolated in excellent yield when irradiated with 455 nm blue 6 W LEDs in acetone. Interestingly, the cycloaddition was highly tolerant to changes in reaction conditions. While optimizing the reaction conditions, it was observed that diverse solvents, including CH2Cl2, THF, CH3CN, and even water, could be used without affecting the yield of the products. 0.75 mol% of PS-11 loading also reproduced the same outcome. In addition, isoquinoline, naphthyl, and phenyl derivatives also underwent the dearomatization reaction smoothly with excellent dr under the same conditions. Ease of separation from the reaction mixture and a high degree of recyclability brings added benefits for PS-11. The catalyst was recyclized up to 10 times without any drop in its efficiency. Kinetic monitoring of the reaction showed the formation of only [4 + 2] cycloadducts 11.2. E to Z isomerization of the substrate was also noticed. This implied that E-11.1 and Z-11.1 converted to the same 1,2-biradical intermediates 11.1(T1) that produced 11.2. The selectivity for the [4 + 2] cycloadduct over [2 + 2] and [3 + 2] is justified by the thermodynamic stability of the products. Stern–Volmer analysis revealed the quenching of excited PS by N-cinnamoyl picolinamides 11.1, whereas no quenching was observed for the reduced substrates 11.3. This inferred that the EnT took place from the PS to the styrene pendant of the substrate to form 1,2-biradical intermediates 11.1(T1) that then produced 11.2via CC-bond formation, ISC, and the radical coupling sequence.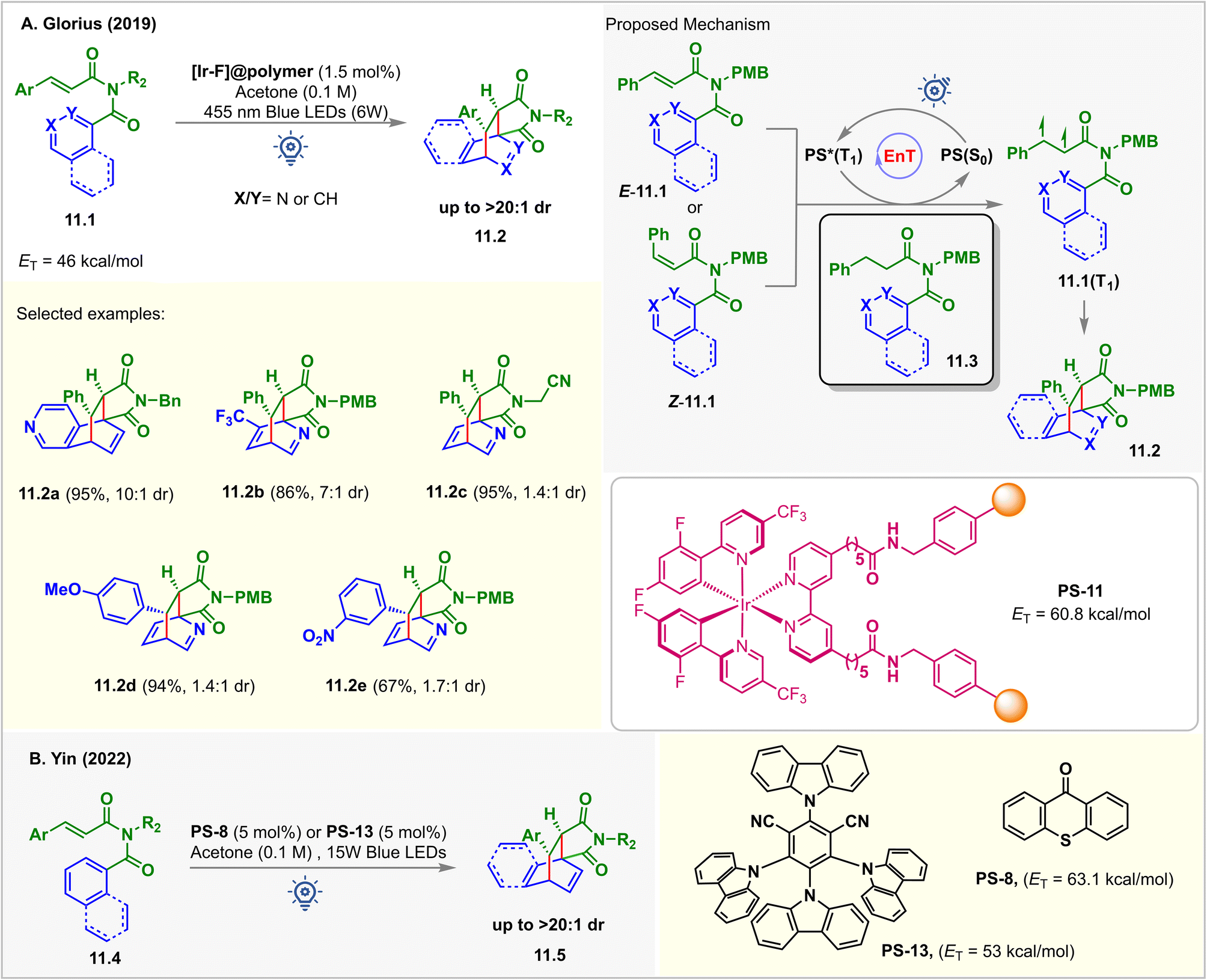 | ||
| Fig. 11 Visible light mediated EnT dearomative intramolecular [4π + 2π] cycloaddition of (A) pyridine tethered N-cinnamoyl picolinamide derivatives using [Ir-F]@polymer PS-11 as a photosensitizer and (B) arene tethered N-cinnamoyl picolinamide derivatives. | ||
In 2022, the Yin group demonstrated a similar dearomative intermolecular [4 + 2] para-cycloaddition of arene-tethered N-cinnamoyl picolinamide derivatives 11.4 (Fig. 11B).96PS-8 (1 mol%) or PS-13 (5 mol%) catalyzed the reaction in acetone solvent under irradiation of 15 W purple or blue LEDs. A series of bridged bicyclo scaffolds 11.5 were prepared with excellent diastereoselectivity (>20:1 dr).
In 2021, the You group developed a double dearomative cycloaddition of indole derivatives bearing a pendant 1-naphthyl ring 12.1 (Fig. 12).97 Bridged cyclic-indoline 12.2a was produced in 97% isolated yield with >20:1 dr in the presence of 1 mol% of PS-5 in CH2Cl2 at rt under the irradiation of blue LEDs for 5 h. With the increasing steric bulk at the C2- and C3-positions of the indole moiety, a longer time (up to 3 days) was required to reach high yields. Also, indoles with both electron-donating and withdrawing groups reacted smoothly to produce excellent 93–98% yields. The pendant 2-naphthyl ring and 9-anthracene rings led to a [4 + 2] cycloaddition product. In comparison, 9-phenanthrene gave [2 + 2] cycloadduct 12.3 with 96% yield. The [2 + 2] cycloaddition products were also observed for pendant 2-furyl (12.4), 3-furyl, and 2-benzofuryl derivates of indole. The kinetic analysis of the reaction revealed that dearomative [2 + 2] cycloadducts 12.5 were produced initially. As the reaction progressed, 12.5 were consumed, and 12.2 were formed. During this process, 12.5 is in equilibrium with the 1,4-biradical 12.6, which is itself removed from equilibrium by radial recombination from its resonance form 12.7. The desired double dearomative [4 + 2] cycloadducts 12.2 were formed after radical addition.
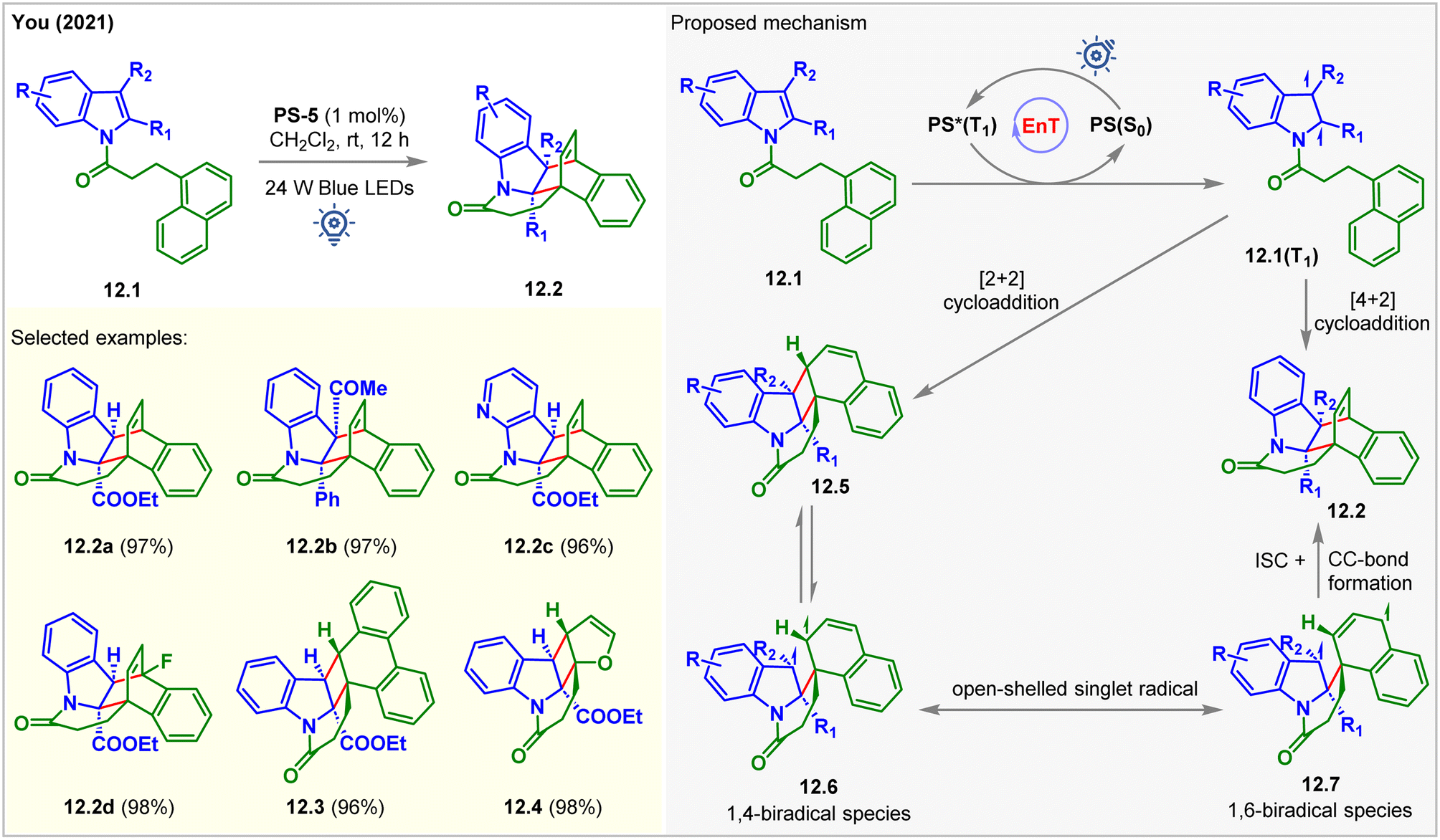 | ||
| Fig. 12 Visible light mediated EnT double dearomative intramolecular [4π + 2π] cycloaddition of an N1-naphthalene tethered indole derivative. | ||
Recently, the Maestri group photosensitized allenamides for their intramolecular dearomative [4 + 2] cycloaddition with tethered arenes (Fig. 13).98PS-4 was used as the catalyst under visible light irradiation in DMF. The author found that naphthyl derivatives 13.1 reacted twice faster than the derivative of simple arenes 13.2. The products 13.3 and 13.4 were isolated in 18–99% yields in 16–96 h. Allenamides having naphthyl side arms 13.1 were found to quench the excited PS-4 at a higher rate KSV = 404 M−1. The formation of [4 + 2] cycloadducts 13.3 from 13.1 could possibly occur via two reaction pathways involving two different biradical intermediates, 13.5 and 13.6. The spin density of the triplet biradical intermediates 13.5 were found to be localized on the allenyl fragment. It was suggested that the intramolecular attack of the excited allene onto the naphthyl moiety of 13.5 proceeds via a low-energy transition state to generate spiro triplet biradical intermediates 13.7. On the other hand, the spin density of 13.6 was localized on the naphthyl moiety. Since the formation of the intermediate 13.7 from 13.6 involved a high-energy transition state, this pathway was considered to be less favorable. However, in the case of allenamide containing a phenyl arm 13.2, the involvement of a more energy-costly TS and less stable intermediate 13.9 was responsible for the slower reaction. Mechanistically, the EnT-generated triplet vinyl radical intermediates 13.8 attacked the benzyl arm, broke the aromaticity, and produced the triplet biradical intermediates 13.9. The ISC to the singlet intermediates 13.10, followed by CC-bond formation, yields the [4 + 2] cycloadducts 13.4. An alternative pathway where the cleavage of a CC-bond of 13.10 occurs regenerating the starting material. Interestingly, this pathway is kinetically favored by 0.2 kcal mol−1 relative to the one derived for the radical addition. Computational studies at the M06/def2/TZVP level of theory were carried out for the mechanistic investigations. As a result of this unwanted side reaction, lower yields were obtained. Notably, the authors employed an excess of naphthalene as an additive to stabilize the biradical intermediate and the productive transition state via π-cloud interaction with the substrate. The yields were significantly improved under these conditions.
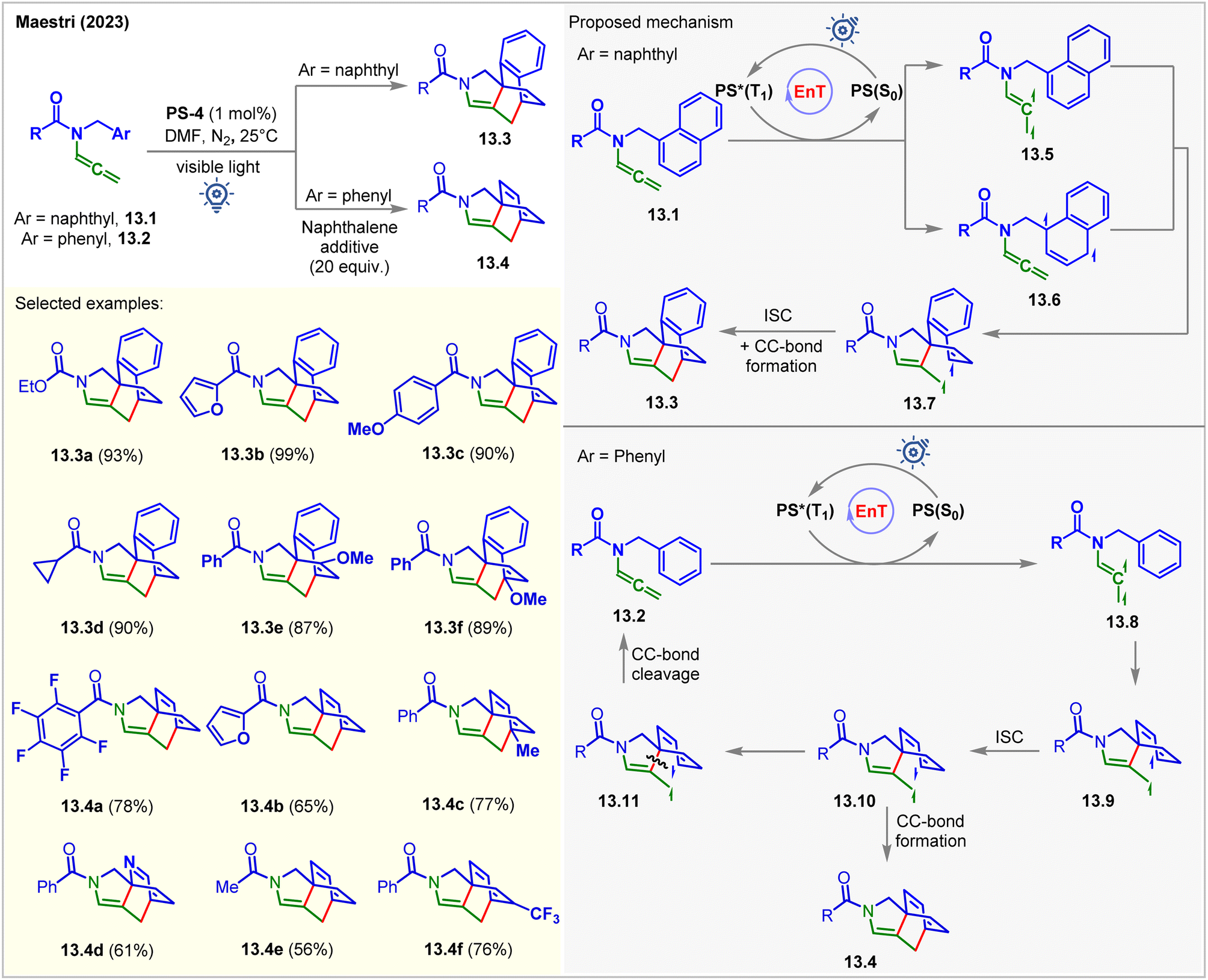 | ||
| Fig. 13 Visible light mediated EnT dearomative intramolecular [4π + 2π] cycloaddition of allenamide tethered arenes. | ||
3.2. Intermolecular [4π + 2π] para-cycloaddition
In 2021, in collaboration, Glorius, Brown, and Houk established Brønsted and Lewis acid-assisted dearomative [4 + 2] cycloaddition of quinolines 14.1 with olefins via visible-light EnT catalysis (Fig. 14A).99 The authors introduced two different conditions for differently substituted quinoline derivatives: condition A: PS-2 (2 mol%) in HFIP, and condition B: PS-2 (1 mol%) in CH2Cl2 with BF3‧OEt2 (1 equiv.). The dearomative cycloaddition of 6-methyl quinoline with 1-hexene under condition A forms the cycloadduct 14.3a in 88% yield with >95:5 dr as the only regioisomer. Whereas the reaction of the unsubstituted quinoline under condition B yields the cycloadduct 14.4a in 78% yield with 83:17 dr as the major regioisomer with 18% of other regioisomers 14.3. Condition A performed well for 5-, 6-, 7-, and 8-substituted quinolines. Anti-regioselectivity was observed for 2-, 3-, 4-, 5-, and 7-substituted quinolines, whereas 6-, and 8-substituted quinolines gave syn regioselectivity. No conversion occurred without HFIP and BF3‧OEt2 in two separate reaction setups, indicating the requirement of either the Brønsted acid or the Lewis acid in achieving the desired cycloaddition reaction. Based on these findings, the authors proposed that the HFIP solvent and the BF3‧OEt2 additive were adhering to the quinoline via hydrogen bonding and Lewis acid–base interaction, which lowers ET of 6-methyl quinoline from 61.7 kcal mol−1 to 61.2 kcal mol−1 (in HFIP), and to 58.9 kcal mol−1 (with BF3‧OEt2), respectively, which was supported by the ωB97X-D/6-311++G(d,p) level of computational study.
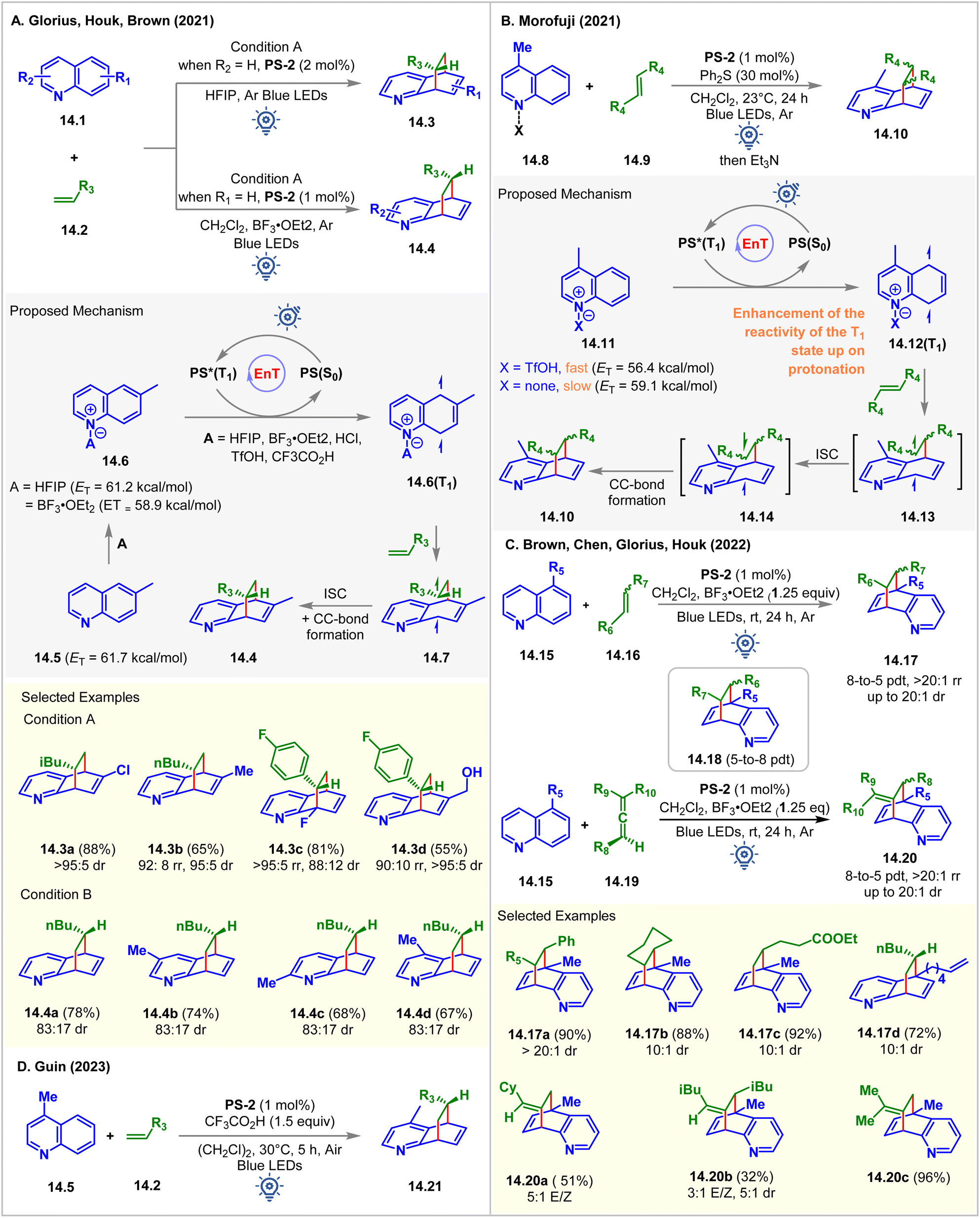 | ||
| Fig. 14 Visible light mediated EnT dearomative intermolecular [4π + 2π] cycloaddition of (A) quinolines with olefins using Lewis acid and HFIP solvent, (B) quinoline with disubstituted olefins using TfOH, (C) quinolines with disubstituted olefins and allene usings lewis acid, and (D) lepidine with olefins using CF3CO2H. | ||
Independently, Morofuji reported the same reaction under similar conditions (Fig. 14B).100 It was shown that adding triflic acid sped up the [4 + 2] cycloaddition reaction for the less reactive 5- or 8- substituted quinolines. The addition of acid increases the electrophilic nature of quinoline and helps to accelerate the reaction. Besides, upon protonation, the ETs of lepidine decreased (to 56.4 from 59.1 kcal mol−1 of free lepidine). However, by performing a laser flash photolysis experiment, the authors found that the transfer of the PS's triplet energy to both neutral and cationic quinoline substrates produced the triplet state of quinoline 14.12(T1). The Stern–Volmer experiments portrayed the quenching of PS-2 by both the neutral quinoline derivative and cationic quinoline derivative 14.11. It was suspected that the acceleration of EnT might not be the exact function of acids but to enhance the reactivity of the triplet state of quinolines toward olefins. The authors concluded that the highly electron-rich nature of the biradical intermediate produced from a neutral quinoline moiety was the reason for its slow reaction with its olefin partner. The DFT calculations at the (U)-B3LYP/6-311G+(d,p) level of theory suggested that the cycloaddition pathway of neutral lepidine involves a high Gibbs energy barrier, which is responsible for no product formation.
In 2022, Brown, Glorius, Chen, and Houk expanded the substrate scope to disubstituted alkenes 14.16 and allenes 14.19 (Fig. 14C) for the Lewis acid-mediated [4 + 2] cycloaddition of quinolines via visible light EnT catalysis.101 5-Alkene-tethered quinoline also underwent [4 + 2] cycloaddition effortlessly to produce 14.17d, leaving the tethered olefin intact. This indication was sufficient to realize the faster intermolecular reaction over the intramolecular reaction due to the slow radical addition step between C8 and the terminal carbon center of the tethered alkene forming an 8- or 9-membered transition state. The reaction of 5-alkyl quinolines 14.15 with styrene derivatives were observed to form an endo product 14.17 with high diastereoselectivity (up to >10:1 dr) due to the London dispersion force. The regioselectivity to form 8-to-5 products 14.17 were decided by the relative stability of the long-lived biradical intermediates. In comparison, a more polar solvent favored 5-to-8 products 14.18. Allenes 14.19 also underwent cycloaddition with 5-alkyl quinolines 14.15 to exclusively produce 8-to-5 [4 + 2] cycloadducts 14.20.
In 2023, the Guin group demonstrated a similar dearomative intermolecular [4 + 2] cycloaddition of lepidines 14.5 with alkenes 14.2 in the presence of 1.5 equiv. CF3CO2H as an additive (Fig. 14D).102PS-2 (1 mol%) catalyzed the reaction in dichloroethane solvent under irradiation of blue LEDs. Notably, the reaction was moisture- and air-tolerant, giving the product 14.21 with up to 91% isolated yield and up to 19:1 dr.
In 2022, the Maji group commenced the intermolecular dearomative [4 + 2] cycloaddition of aromatic hydrocarbon 2-acetyl naphthalenes 15.1 with styrenes 15.2 employing PS-2 (1 mol%) as the photocatalyst in acetonitrile at room temperature (Fig. 15A).103 The bicyclo[2.2.2]octa-2,5-diene derivative 15.3a was isolated as a single regioisomer in 98% yield with 2:1 dr favoring the endo diastereomer. The EnT from the excited photocatalyst PS-2 (ET = 61.8 kcal mol−1) to naphthalene derivatives 15.1 (ET = 55.1 kcal mol−1) is exergonic and more favorable compared to the energy transfer to the styrene (ET = 57.9 kcal mol−1) moiety. The triplet energy values of 15.1 and 15.2 were calculated at the B3LYP/6-311+G(2d,p) level of DFT. The Stern–Volmer quenching study validated this fact (kq(15.1) = 1.60 × 104 M−1 s−1vs. kq(15.2) = 0.41 × 104 M−1 s−1). A styrene moiety with both EDG and EWG substitutions at the aryl group underwent cycloaddition with the same productivity.
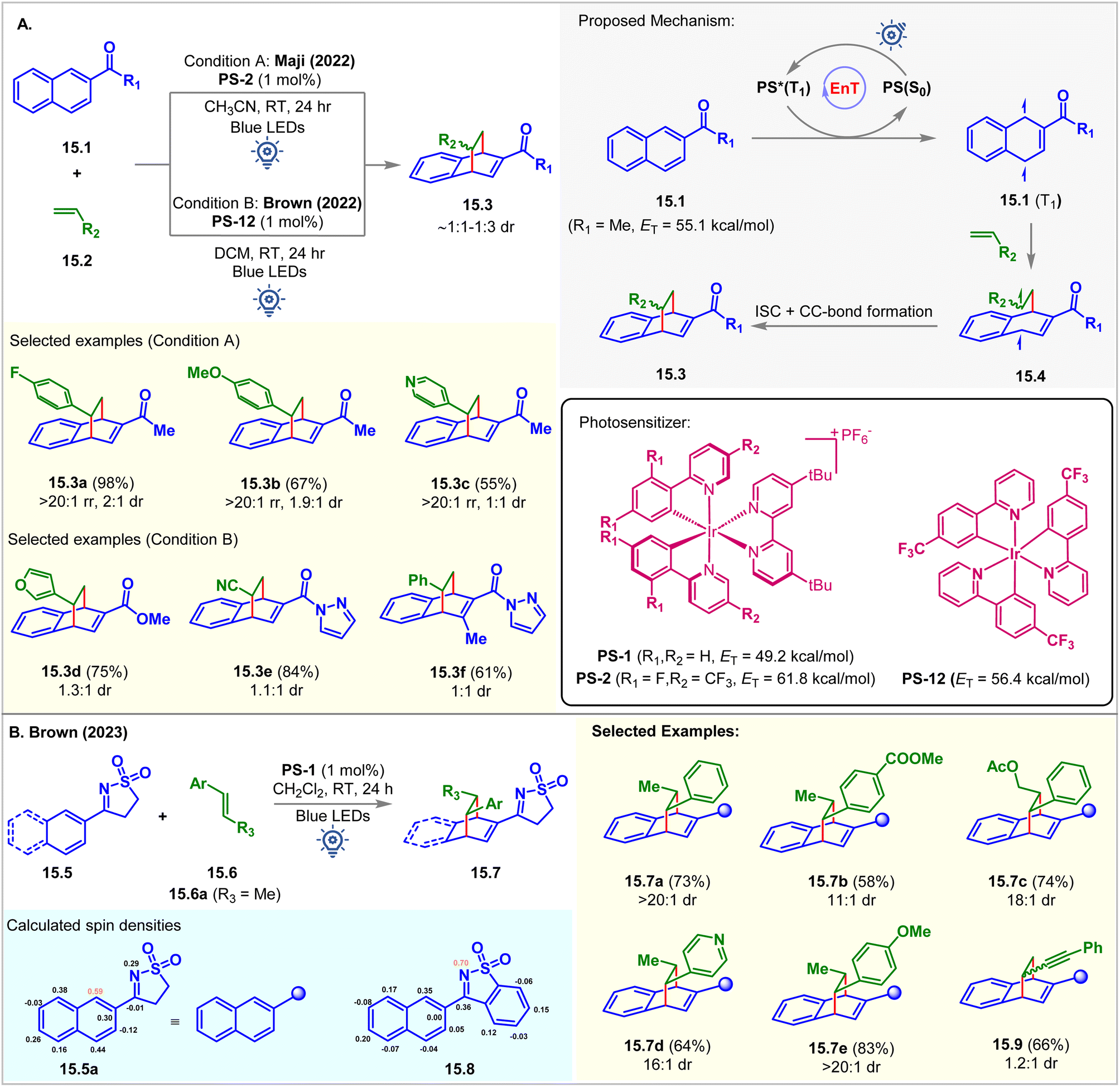 | ||
| Fig. 15 Visible light mediated EnT dearomative intermolecular [4π + 2π] cycloaddition of (A) 2-COR-naphthalenes and (B) naphthyl tethered rigid sulfonylimine derivatives. | ||
Independently, Brown reported a similar reaction using [Ir(p-CF3-ppy)3] (PS-12, ET = 56.4 kcal mol−1) as the PS (Fig. 15A).104 It was observed that the ester and amide-substituted naphthalenes did not undergo cycloaddition using PS-2. However, a promising result was shown when employing PS-12. This reaction was also extended to activated olefins bearing EWG groups, including cyano, ester, and ketones. The authors also found that the reaction proceeded well in the absence of PS upon irradiation with 365 nm LEDs, suggesting that the reaction proceeded via excited state triplet intermediates. Although good regioselectivity was obtained, the diastereoselectivity was poor (∼1:1 dr). Recently, the Brown group carried out further study to overcome the low diastereoselectivity challenge using a sulfonyl amine activating group (Fig. 15B).105 Introduction of a larger activating group could distinguish two diastereomeric transition states to achieve high diastereoselectivity. Rigid sulfonylimines 15.5 underwent dearomative [4 + 2] cycloaddition with β-methyl styrene 15.6a under blue LED irradiation of PS-12 to produce cycloadduct 15.7a with 73% yield and >20:1 dr. Electron donating and withdrawing groups at the aryl ring of 15.6 provided excellent diastereoselectivity of for the [4 + 2] cycloadducts 15.7e and 15.7b, respectively. Alkenyl heterocycle also undergoes cycloaddition to achieve 15.7d with excellent 16:1 dr. Interestingly, the derivatives of benzoisothiazole 15.8 undergo [2 + 2] cycloaddition with styrene to produce azetidine in a single diastereomer under 450 nm blue LED irradiation of PS-12. Spin density calculation of the excited triplet state of 15.5a and 15.8 revealed the highest spin density at C1 of N-aryl sulfonylimine 15.5a and the N-atom of benzoisothiazole derivative 15.8, which correlates with the bond-formation sites and supports the behavior of the cycloaddition reaction.
4. Conclusion and outlook
In conclusion, this review covers the applications of visible light EnT catalysis in dearomative cycloaddition reactions. Appropriate choice of photosensitizer and reaction conditions enables direct and selective activation of the substrate molecules employing visible light sources (Fig. 16). The mildness of the protocol allowed the isolation of endergonic products in high yield and selectivities. Initial work in this field employed intramolecular reactions to compensate for entropic issues and the high reactivity and low lifetime of the visible light-activated substrates. Subsequent development allowed the discovery of intermolecular reactions, even the enantioselective variants. This advancement in the field of triplet-energy photosensitized dearomative cycloadditions is not only restricted to the π-based systems, but the recent inclusion of the σ-frameworks (for instance, BCBs) has also ensured the expansion of its horizon. The synthetic work parallels experimental and computational mechanistic understanding of reactivity and selectivity.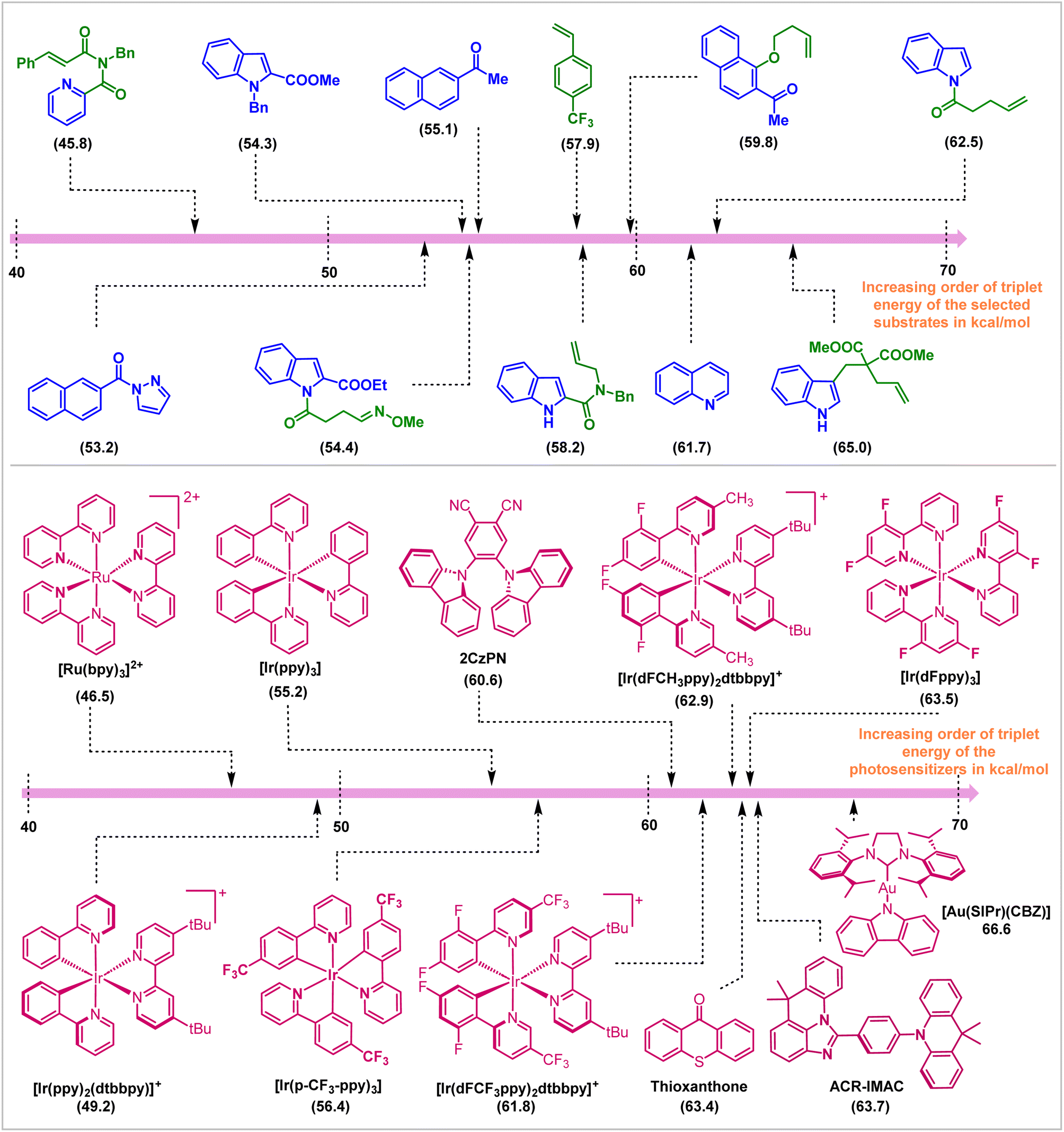 | ||
| Fig. 16 Photosensitizers and substrates and their ETs (in kcal mol−1, shown in parentheses) employed in visible-light mediated dearomative cycloaddition reactions. | ||
Despite significant advances in this field, visible light induced EnT dearomatization reactions are still in their early stages. There are still substantial challenges and underexplored directions that continue to arouse more interest in the field. For instance, most EnT-induced dearomative cycloaddition reactions are involved in racemic product formations, and, except for one example, the asymmetric variants of such reactions are yet to be explored. Moreover, some of the reactions are less diastereoselective. Hence, modulation of the reaction parameters and improving the catalysis to address these selectivity issues would be one of the subjects of future research. Additionally, only a limited section of arenes are considered thus far. The challenges are not only accessing their excited states but also enabling their productive reactions with the coupling partners. A more interesting and widely applicable strategy is to design photosensitizers with higher triplet excited state energies that will allow activation of these recalcitrant molecules for functionalization reactions, as well as to redesign the existing substrates to reduce the triplet energy. Another important direction of such an EnT reaction should also be towards [3 + 2] cycloaddition reactions, which are yet to be performed under visible-light irradiation. Late-stage modification of drug molecules and their application in the total synthesis is a worthwhile future direction. We expect that the insights gained in this field to date, elaborated in this tutorial review, will provide an informative tool to aid the development of this emerging triplet energy transfer catalysis for selective transformative innovation.
Author contributions
All authors have given approval to the final version of the paper.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank DST-SERB, GoI (grant no. CRG/2019/001232) for financial support. A. P. and P. R. thank CSIR and INSPIRE for fellowships.References
-
W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Elsevier Science, 2013 Search PubMed
.
-
S. Kobayashi and K. A. Jorgenson, in Cycloaddition Reactions in Organic Synthesis, Wiley-VCH Verlag GmbH, 2001, pp. 1–329 Search PubMed
- R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816–9849 CrossRef CAS PubMed
- U. Streit and C. G. Bochet, Chimia, 2008, 62, 962–966 CrossRef CAS
- D. Chappell, A. T. Russell and B. Chemistry, Org. Biomol. Chem., 2006, 4, 4409–4430 RSC
- J. Mattay, Angew. Chem., Int. Ed., 2007, 46, 663–665 CrossRef CAS PubMed
- C. W. Bird, Tetrahedron, 1996, 52, 9945–9952 CrossRef CAS
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed
- L. D. Elliott, S. Kayal, M. W. George and K. Booker-Milburn, J. Am. Chem. Soc., 2020, 142, 14947–14956 CrossRef CAS
- D. Bryce-Smith, Pure Appl. Chem., 1968, 16, 47–64 CrossRef CAS
- H. J. F. Angus and D. Bryce-Smith, Proc. Chem. Soc., 1959, 326–327 CAS
- K. E. Wilzbach and L. Kaplan, J. Am. Chem. Soc., 1966, 88, 2066–2067 CrossRef CAS
- K. E. Wilzbach and L. Kaplan, J. Am. Chem. Soc., 1971, 93, 2073–2074 CrossRef CAS
- D. Bryce-Smith, J. Chem. Soc. D, 1969, 806–808 RSC
- K. N. Houk, Pure Appl. Chem., 1982, 54, 1633–1650 CrossRef CAS
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed., 1969, 8, 781–853 CrossRef CAS
- W. Ferree Jr, J. B. Grutzner and H. Morrison, J. Am. Chem. Soc., 1971, 93, 5502–5512 CrossRef
- U. Streit and C. G. Bochet, Beilstein J. Org. Chem., 2011, 7, 525–542 CrossRef CAS PubMed
- F. Mueller and J. Mattay, Chem. Rev., 1993, 93, 99–117 CrossRef CAS
- J. Mattay, Tetrahedron, 1985, 41, 2405–2417 CrossRef CAS
- I. Yoshihisa, N. Katsuyuki, I. Koichi, H. Tadao and N. J. Turro, Chem. Lett., 1982, 11, 471–474 CrossRef
- D. C. Blakemore and A. Gilbert, J. Chem. Soc., Perkin Trans. 1, 1992, 2265–2270 RSC
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed
- F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183–2194 CAS
-
N. J. Turro, Modern Molecular Photochemistry, University Science Books, 1991 Search PubMed
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS
-
P. Klán and J. Wirz, in Photochemistry of Organic Compounds, Wiley, Chichester, 2009, pp. 44–75 Search PubMed
-
S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, New York, 1993 Search PubMed
-
V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, John Wiley & Sons, 2014 Search PubMed
- P. J. Wagner, Topics in current chemistry, Top. Curr. Chem., 1976, 1–52 CAS
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS
- D. S. Lee, V. K. Soni and E. J. Cho, Acc. Chem. Res., 2022, 55, 2526–2541 CrossRef CAS PubMed
- Y. Zhang, M. Xia, M. Li, Q. Ping, Z. Yuan, X. Liu, H. Yin, S. Huang and Y. Rao, J. Org. Chem., 2021, 86, 15284–15297 CrossRef CAS PubMed
- Y. Matsushita, R. Ochi, Y. Tanaka, T. Koike and M. Akita, Org. Chem. Front., 2020, 7, 1243–1248 RSC
- Y. Zhang, Y. Zhang, C. Ye, X. Qi, L.-Z. Wu and X. Shen, Nat. Commun., 2022, 13, 6111–6121 CrossRef CAS PubMed
- M. Bera, D. S. Lee and E. J. Cho, Trends Chem., 2021, 3, 877–891 CrossRef CAS
- T. Rigotti and J. Alemán, Chem. Commun., 2020, 56, 11169–11190 RSC
- P. Franceschi, S. Cuadros, G. Goti and L. Dell'Amico, Angew. Chem., Int. Ed., 2023, 62, e202217210 CrossRef CAS PubMed
- J. J. McCullough, W. K. MacInnis, C. J. L. Lock and R. Faggiani, J. Am. Chem. Soc., 1980, 102, 7780–7782 CrossRef CAS
- Y. Hanzawa and L. Paquette, Synth., 1982, 661–662 CrossRef CAS
- P. J. Wagner and K. Nahm, J. Am. Chem. Soc., 1987, 109, 4404–4405 CrossRef CAS
- N. Zupančič and B. Šket, J. Chem. Soc., Perkin Trans. 1, 1992, 179–180 RSC
- N. Hoffmann and J.-P. Pete, Tetrahedron Lett., 1996, 37, 2027–2030 CrossRef CAS
- J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef CAS PubMed
-
N.-Y. Fu, S.-H. Chan and H. N. C. Wong, in The Chemistry of Cyclobutanes, John Wiley & Sons, Ltd, 2005, pp. 357–440 Search PubMed
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624–8628 CrossRef CAS PubMed
- M. Zhu, C. Zheng, X. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2636–2644 CrossRef CAS PubMed
- E. A. Martynova, V. A. Voloshkin, S. G. Guillet, F. Bru, M. Beliš, K. Van Hecke, C. S. J. Cazin and S. P. Nolan, Chem. Sci., 2022, 13, 6852–6857 RSC
- Z. Zhang, D. Yi, M. Zhang, J. Wei, J. Lu, L. Yang, J. Wang, N. Hao, X. Pan, S. Zhang, S. Wei and Q. Fu, ACS Catal., 2020, 10, 10149–10156 CrossRef CAS
- M. Zhu, X. Zhang, C. Zheng and S.-L. You, ACS Catal., 2020, 10, 12618–12626 CrossRef CAS
- M. Zhu, X.-L. Huang, H. Xu, X. Zhang, C. Zheng and S.-L. You, CCS Chem., 2021, 3, 652–664 CrossRef CAS
- T. Hudlicky and J. W. Reed, Angew. Chem., Int. Ed., 2010, 49, 4864–4876 CrossRef CAS PubMed
- P. Li, J. Zhao, L. Shi, J. Wang, X. Shi and F. Li, Nat. Commun., 2018, 9, 1972–1980 CrossRef PubMed
- M. Zhu, X.-L. Huang, S. Sun, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2021, 143, 13441–13449 CrossRef CAS PubMed
- M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. K. Gupta, N. A. Meanwell, A. Mathur and T. G. M. Dhar, J. Am. Chem. Soc., 2020, 142, 3094–3103 CrossRef CAS PubMed
- E. R. Sauvé, D. M. Mayder, S. Kamal, M. S. Oderinde and Z. M. Hudson, Chem. Sci., 2022, 13, 2296–2302 RSC
- A. B. Rolka and B. Koenig, Org. Lett., 2020, 22, 5035–5040 CrossRef CAS PubMed
- N. Arai and T. Ohkuma, Org. Lett., 2019, 21, 1506–1510 CrossRef CAS PubMed
- J. Proessdorf, C. Jandl, T. Pickl and T. Bach, Angew. Chem., Int. Ed., 2022, 61, e202208329 CrossRef CAS PubMed
- N. Hu, H. Jung, Y. Zheng, J. Lee, L. Zhang, Z. Ullah, X. Xie, K. Harms, M.-H. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242–6246 CrossRef CAS PubMed
- M. S. Oderinde, A. Ramirez, T. G. M. Dhar, L. A. M. Cornelius, C. Jorge, D. Aulakh, B. Sandhu, J. Pawluczyk, A. A. Sarjeant, N. A. Meanwell, A. Mathur and J. Kempson, J. Org. Chem., 2021, 86, 1730–1747 CrossRef CAS PubMed
- P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC
- M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046–14128 CrossRef CAS PubMed
- M. Golfmann and J. C. L. Walker, Commun. Chem., 2023, 6, 9–21 CrossRef CAS PubMed
- M. A. A. Walczak, T. Krainz and P. Wipf, Acc. Chem. Res., 2015, 48, 1149–1158 CrossRef CAS PubMed
- W. J. Leigh, Chem. Rev., 1993, 93, 487–505 CrossRef CAS
- R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477–482 CrossRef CAS PubMed
- S. Agasti, F. Beltran, E. Pye, N. Kaltsoyannis, G. E. M. Crisenza and D. J. Procter, Nat. Chem., 2023, 15, 535–541 CrossRef CAS PubMed
- R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed
- Y. Liu, S. Lin, Y. Li, J.-H. Xue, Q. Li and H. Wang, ACS Catal., 2023, 13, 5096–5103 CrossRef CAS
- R. Kleinmans, S. Dutta, K. Ozols, H. Shao, F. Schäfer, R. E. Thielemann, H. T. Chan, C. G. Daniliuc, K. N. Houk and F. Glorius, J. Am. Chem. Soc., 2023, 145, 12324–12332 CrossRef CAS PubMed
- H. Wang, H. Shao, A. Das, S. Dutta, H. T. Chan, C. Daniliuc, K. N. Houk and F. Glorius, Science, 2023, 381, 75–81 CrossRef CAS PubMed
- X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515–5546 CrossRef CAS PubMed
- B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC
- J. A. Norton, Chem. Rev., 1942, 31, 319–523 CrossRef CAS
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS
- M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438–453 CrossRef CAS PubMed
- K. N. Houk, Acc. Chem. Res., 1975, 8, 361–369 CrossRef CAS
- S. J. Hamrock and R. S. Sheridan, J. Am. Chem. Soc., 1989, 111, 9247–9249 CrossRef CAS
- A. Gilbert and G. Taylor, J. Chem. Soc., Chem. Commun., 1978, 129–130 RSC
- D. Döpp, H. R. Memarian, C. Krüger and E. Raabe, Chem. Ber., 1989, 122, 585–588 CrossRef
- F. Birbaum, A. Neels and C. G. Bochet, Org. Lett., 2008, 10, 3175–3178 CrossRef CAS
- T. W. Bingham, L. W. Hernandez, D. G. Olson, R. L. Svec, P. J. Hergenrother and D. Sarlah, J. Am. Chem. Soc., 2019, 141, 657–670 CrossRef CAS PubMed
- W. C. Wertjes, M. Okumura and D. Sarlah, J. Am. Chem. Soc., 2019, 141, 163–167 CrossRef CAS PubMed
- L. W. Hernandez, J. Pospech, U. Klöckner, T. W. Bingham and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 15656–15659 CrossRef CAS PubMed
- E. H. Southgate, J. Pospech, J. Fu, D. R. Holycross and D. Sarlah, Nat. Chem., 2016, 8, 922–928 CrossRef CAS PubMed
- K. Ikeda, R. Kojima, K. Kawai, T. Murakami, T. Kikuchi, M. Kojima, T. Yoshino and S. Matsunaga, J. Am. Chem. Soc., 2023, 145, 9326–9333 CrossRef CAS PubMed
- D. J. Bellville, D. W. Wirth and N. L. Bauld, J. Am. Chem. Soc., 1981, 103, 718–720 CrossRef CAS
- D. J. Bellville, N. L. Bauld, R. Pabon and S. A. Gardner, J. Am. Chem. Soc., 1983, 105, 3584–3588 CrossRef CAS
- M. Sicignano, R. I. Rodríguez and J. Alemán, Eur. J. Org Chem., 2021, 3303–3321 CrossRef CAS PubMed
- K. Kishikawa, S. Akimoto, S. Kohmoto, M. Yamamoto and K. Yamada, J. Chem. Soc., Perkin Trans. 1, 1997, 77–84 RSC
-
L. Meites, P. Zuman and E. B. Rupp, CRC handbook series in organic electrochemistry, CRC Press, 1977 Search PubMed
- J. Ma, F. Strieth-Kalthoff, T. Dalton, M. Freitag, J. L. Schwarz, K. Bergander, C. Daniliuc and F. Glorius, Chem, 2019, 5, 2854–2864 CAS
- G. Zhen, G. Zeng, F. Wang, X. Cao and B. Yin, Adv. Synth. Catal., 2023, 365, 43–52 CrossRef CAS
- M. Zhu, H. Xu, X. Zhang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2021, 60, 7036–7040 CrossRef CAS PubMed
- M. Chiminelli, A. Serafino, D. Ruggeri, L. Marchiò, F. Bigi, R. Maggi, M. Malacria and G. Maestri, Angew. Chem., Int. Ed., 2023, 62, e202216817 CrossRef CAS
- J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338–1345 CrossRef CAS
- T. Morofuji, S. Nagai, Y. Chitose, M. Abe and N. Kano, Org. Lett., 2021, 23, 6257–6261 CrossRef CAS PubMed
- R. Guo, S. Adak, P. Bellotti, X. Gao, W. W. Smith, S. N. Le, J. Ma, K. N. Houk, F. Glorius, S. Chen and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 17680–17691 CrossRef CAS PubMed
- M. Bhakat, B. Khatua, P. Biswas and J. Guin, Org. Lett., 2023, 25, 3089–3093 CrossRef CAS PubMed
- P. Rai, K. Maji, S. K. Jana and B. Maji, Chem. Sci., 2022, 13, 12503–12510 RSC
- W. Wang, Y. Cai, R. Guo and M. K. Brown, Chem. Sci., 2022, 13, 13582–13587 RSC
- W. Wang and M. K. Brown, Angew. Chem., Int. Ed., 2023, e202305622 CAS
Footnote |
| † These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |