
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spontaneous generation of singlet oxygen on microemulsion-derived manganese oxides with rich oxygen vacancies for efficient aerobic oxidation†
Jun
Tang
 ab,
Junbao
Chen
a,
Zhanyu
Zhang
a,
Qincheng
Ma
a,
Xiaolong
Hu
a,
Peng
Li
a,
Zhiqiang
Liu
a,
Peixin
Cui
c,
Chao
Wan
*de,
Qingping
Ke
*a,
Lei
Fu
e,
Jeonghun
Kim
f,
Takashi
Hamada
g,
Yunqing
Kang
*e and
Yusuke
Yamauchi
*fgh
ab,
Junbao
Chen
a,
Zhanyu
Zhang
a,
Qincheng
Ma
a,
Xiaolong
Hu
a,
Peng
Li
a,
Zhiqiang
Liu
a,
Peixin
Cui
c,
Chao
Wan
*de,
Qingping
Ke
*a,
Lei
Fu
e,
Jeonghun
Kim
f,
Takashi
Hamada
g,
Yunqing
Kang
*e and
Yusuke
Yamauchi
*fgh
aCollege of Chemistry and Chemical Engineering, Anhui University of Technology, Maanshan 243002, Anhui, P. R. China. E-mail: Qingke@ahut.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, P. R. China
cKey Laboratory of Soil Environment and Pollution Remediation, Institute of Soil Science, The Chinese Academy of Sciences, Nanjing 210008, P. R. China
dCollege of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: wanchao@zju.edu.cn
eResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: yqkang@toki.waseda.jp
fDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, South Korea
gDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan
hAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: y.yamauchi@uq.edu.au
First published on 12th October 2023
Abstract
Developing innovative catalysts for efficiently activating O2 into singlet oxygen (1O2) is a cutting-edge field with the potential to revolutionize green chemical synthesis. Despite its potential, practical implementation remains a significant challenge. In this study, we design a series of nitrogen (N)-doped manganese oxides (Ny-MnO2, where y represents the molar amount of the N precursor used) nanocatalysts using compartmentalized-microemulsion crystallization followed by post-calcination. These nanocatalysts demonstrate the remarkable ability to directly produce 1O2 at room temperature without the external fields. By strategically incorporating defect engineering and interstitial N, the concentration of surface oxygen atoms (Os) in the vicinity of oxygen vacancy (Ov) reaches 51.1% for the N55-MnO2 nanocatalyst. This feature allows the nanocatalyst to expose a substantial number of Ov and interstitial N sites on the surface of N55-MnO2, facilitating effective chemisorption and activation of O2. Verified through electron paramagnetic resonance spectroscopy and reactive oxygen species trapping experiments, the spontaneous generation of 1O2, even in the absence of light, underscores its crucial role in aerobic oxidation. Density functional theory calculations reveal that an increased Ov content and N doping significantly reduce the adsorption energy, thereby promoting chemisorption and excitation of O2. Consequently, the optimized N55-MnO2 nanocatalyst enables room-temperature aerobic oxidation of alcohols with a yield surpassing 99%, representing a 6.7-fold activity enhancement compared to ε-MnO2 without N-doping. Furthermore, N55-MnO2 demonstrates exceptional recyclability for the aerobic oxidative conversion of benzyl alcohol over ten cycles. This study introduces an approach to spontaneously activate O2 for the green synthesis of fine chemicals.
Introduction
Singlet oxygen (1O2), recognized as a highly active and environmentally friendly reactive oxygen species (ROS), has garnered substantial attention from researchers in recent years.1–4 Benefiting from its remarkable oxidative capabilities and eco-friendliness, 1O2 has found widespread applications in green catalysis,5–7 photocatalytic degradation,8 tumor diagnosis and treatment,9 and fluorescence probes.10 Nevertheless, the practical generation of 1O2 typically necessitates intense photoexcitation due to the spin transition between ground and excited state molecular oxygen.11 Throughout the last few decades, a range of photosensitizers, including photosensitive organic dyes,12 organometallic complexes,13 and noble metals,14,15 have been devised to produce 1O2 by harnessing photogenerated excitons and energy transfer pathways. However, the scarcity of noble metals and the susceptibility of organic photosensitizers to degradation render them inadequate to fulfill the escalating needs of green chemistry and sustainable development.11,16 Therefore, the construction of cost-effective and stable triggers for self-activating O2 in a mild environment through catalyst innovation holds great significance.Transition metal oxides are being considered as suitable candidates to replace noble metal catalysts due to their significant advantages, including low cost, excellent stability, and abundant reserves.17–20 Among these, manganese dioxides (MnO2) stand out due to the multiple valence states and intricate electronic structures, which can be readily modified through structural-tailoring strategies to enhance their redox capabilities. Our previous studies have demonstrated the effectiveness of in situ heteroatom insertion techniques in producing highly active catalysts with ample oxygen vacancies (Ov).17,19 These Ov play a pivotal role in promoting the mobility of lattice oxygen (Ol), consequently enhancing oxidative capability through the Mars–Van Krevelen (MVK) mechanism, rather than inducing spontaneous O2 transformation into 1O2.21 While theoretical studies have suggested that O2 adsorbed on the surface of metal oxides can lead to the direct generation of 1O2, experimental breakthroughs in this area have been limited.22–24 Fortunately, the defect-engineering approach, particularly by modulating rich Ov, promotes the chemisorption of O2, offering a promising avenue for activating O2 to form ROS.25–31 For instance, the {111} facet of MgO, with its abundance of Ov, enables chemisorbed O2 to undergo molecule transitions and electron rearrangements, resulting in the production of 1O2 even in the absence of light. However, the application in catalysis remains rarely explored.24 It can be inferred that an increase in Ov content facilitates O2 chemisorption on the surface of the catalyst, thereby promoting the formation of 1O2.24,25 These inspire us to devise catalysts with enhanced activity by creating ample surface chemisorption sites through innovative defect-engineering strategies.
The microemulsion composed of immiscible oleic and aqueous phases serves as a versatile platform for fabricating various nanocatalysts, distinguishing it significantly from conventional methods.32,33 Additionally, the utilization of microemulsions as templates for MnO2 preparation offers unique advantages: (1) confined aqueous microdroplets create a conducive environment for effective heteroatom doping and the formation of abundant Ov; (2) uniformly sized MnO2 nanocatalysts formed within these microdroplets tend to expose a higher proportion of surface Ov.32,33 However, the strategy of constructing high-concentration Ov based on a microemulsion microreactor is rarely reported.25
In this study, we synthesized N-doped MnO2 (Ny-MnO2, where y represents the molar amount of urea used) nanocatalysts with adjustable Ov through crystallization within compartmentalized droplets of a microemulsion, followed by calcination. The resulting Ny-MnO2 nanocatalysts are employed to activate O2 and generate 1O2 for green oxidation processes. As demonstrated in the aerobic oxidation of benzyl alcohol, an important reaction pathway for the synthesis of fine chemicals, the catalytic activity of N55-MnO2 at room temperature significantly outperforms that of pristine ε-MnO2 and commercially activated MnO2 (C-MnO2). We further elucidate how Ov promote O2 chemisorption and spontaneous activation through structural characterizations and density functional theory (DFT) calculations. Moreover, the pivotal roles of ROS in the oxidation process through controlled quenching experiments are also verified. This work offers a practical strategy and theoretical insights for constructing environmentally friendly Mn-based catalysts and producing highly desirable 1O2 for efficient aerobic oxidation.
Results and discussion
Synthesis and characterization
Compartmentalized microemulsions, stabilized by an excess of surfactants, serve as versatile miniature reactors for the synthesis of Ny-MnO2 to spontaneously produce 1O2 (Scheme 1). Within the confined aqueous microdroplets, isonitrile acid produced by urea decomposition reacts with Mn2+ to form catalyst precursor, manganese carbonate (MnCO3), as verified by X-ray diffraction (XRD) patterns in Fig. S1a.† Fourier transform infrared (FTIR) spectra in Fig. S1b† display the presence of new peaks at 1443 cm−1, 866 cm−1, and 720 cm−1, further confirming the successful formation of MnCO3.33,34 Following calcination in air, MnCO3 decomposes into MnO2. The XRD patterns of the Ny-MnO2 nanocatalysts, as shown in Fig. 1a, are identified as ε-MnO2. The disappearance of the broad band at 1443 cm−1 and the emergence of peak at 1385 cm−1 in FTIR spectra indicates the incorporation of N atoms into ε-MnO2 (Fig. S1b†). Additionally, X-ray photoelectron spectroscopy (XPS) was employed to analyze the embedded N in N55-MnO2. The distinct peak at 399.6 eV in the N 1s spectra of Ny-MnO2 (Fig. 1b and S2†) is unequivocally attributed to N atoms inserted at interstitial sites.35 The introduction of interstitial N minimizes the formation energy of Ov and facilitates the generation of high-concentration Ov.35 As a result, the N55-MnO2 catalyst, with an optimal interstitial N content, achieves a maximum concentration of surface oxygen atoms (Os) at 51.1% in the vicinity of Ov (Fig. 1c, S3, and Table S1†).36,37 In addition, the Os content and the unsaturated Mn coordination (Mn3+/Mn4+ ratio of 0.90, as shown in Fig. S4†), in N55-MnO2, significantly exceed that of similar catalysts (Table S2†).37–41 Furthermore, the evolutionary trend facilitated by interstitial N-doping is reflected in the increased-intensity peak of Ov at g = 2.003 in the electron paramagnetic resonance (EPR) spectrum in Fig. 1d. However, excessive N-doping in N70-MnO2 tends to occupy the formed vacancies, negatively affecting Ov formation (Fig. S3e†). As observed by transmission electron microscopy (TEM) in Fig. 2a, the N55-MnO2 catalyst possesses a size of ∼115 nm, which is consistent with the results from dynamic light scattering (DLS, Fig. S1c†). This demonstrates the distinctive compartmentalization effect of microemulsions in synthesizing uniform-size nanocatalysts. Additionally, High-resolution TEM (HRTEM) image of N55-MnO2 reveals d-spacings of 0.24 nm and 0.16 nm (Fig. 2b), corresponding to the (100) and (102) facets, respectively, of ε-MnO2.42 Energy dispersive spectroscopy (EDS) element mapping images of N55-MnO2 demonstrate that interstitial N is uniformly embedded in the MnO2 lattice (Fig. 2c). Doping interstitial N into MnO2 leads to alterations in the original electronic structure and unsaturated coordination environment, thus enhancing the catalytic oxidation capacity.37 This is confirmed through hydrogen temperature-programmed reduction (H2-TPR), with N55-MnO2 displaying the most prominent redox capability (Fig. S1d†), consistent with previous reports.37,43–47 Additionally, Brunauer–Emmett–Teller (BET) analysis of N2 adsorption–desorption measurements was conducted to assess the BET parameters of Ny-MnO2. As shown in Fig. S5 and Table S3,† N55-MnO2 exhibits the highest BET surface area of 78.7 m2 g−1 among the Ny-MnO2 samples and an average mesopore size of 11.41 nm. This can be attributed to the presence of Ov in Ny-MnO2, which contribute to the formation of abundant mesopores on the surface of Ny-MnO2.48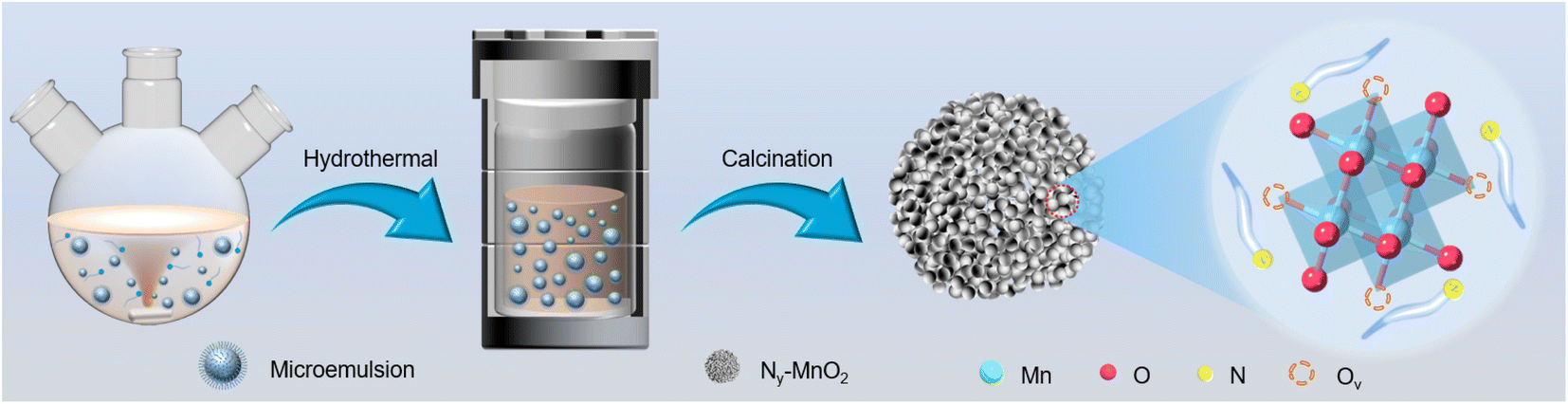 | ||
| Scheme 1 Schematic illustration of the preparation of Ny-MnO2 nanocatalysts using compartmentalized microdroplets. | ||
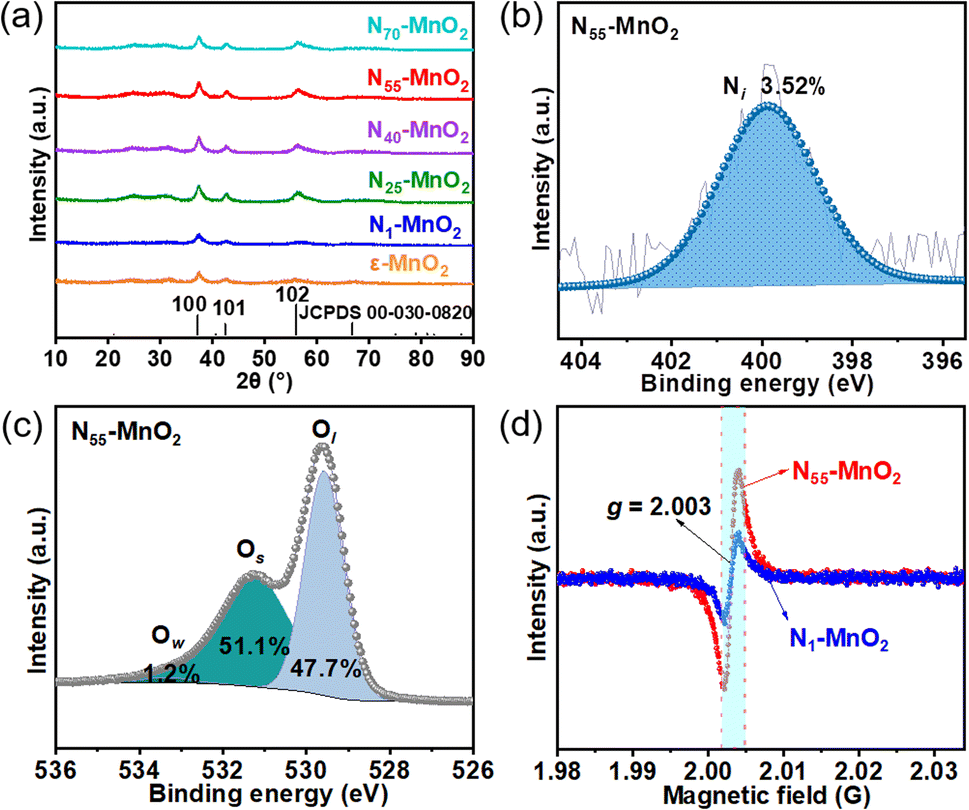 | ||
| Fig. 1 (a) XRD patterns of Ny-MnO2 (y = 1, 25, 40, 55, 70) and ε-MnO2; (b) N 1s and (c) O 1s XPS spectra of N55-MnO2 (Ow: oxygen from water adsorption); (d) EPR spectra of N1-MnO2 and N55-MnO2. | ||
 | ||
| Fig. 2 (a)TEM, (b) HRTEM, and (c) EDS elemental mapping images (scale bars, 50 nm) of N55-MnO2 nanocatalyst. | ||
Adsorption of O2 and production of 1O2
Previous studies have demonstrated that an increased concentration of Ov enhances the chemical adsorption of O2, leading to the elongation of the O–O bond and facilitating O2 activation.24,25,48 Our DFT calculations reveal that the adsorption energy of O2 on the surface of ε-MnO2 is −1.24 eV, whereas it decreases to −2.18 eV on N55-MnO2 (Fig. 3a). This indicates that the augmented Ov resulting from N-doping reduces the adsorption energy of O2, thereby promoting its chemisorption and excitation.25 To gain further insights, we employed in situ IR spectroscopy for real-time monitoring of O2 adsorption and activation on the surface of N55-MnO2. As shown in Fig. 3b, a chemisorption peak of O2 emerges at 1401 cm−1 in an O2 atmosphere, followed by the gradual formation of a robust band corresponding to surface-bonded superoxide anion species (O2−*) at 1052 cm−1.49,50 This illustrates the role of Ov in promoting O2 activation. Conversely, no discernible peaks are observed under an O2 + H2O vapor atmosphere, possibly due to H2O occupying the Ov and preventing O2 adsorption (Fig. S6a†).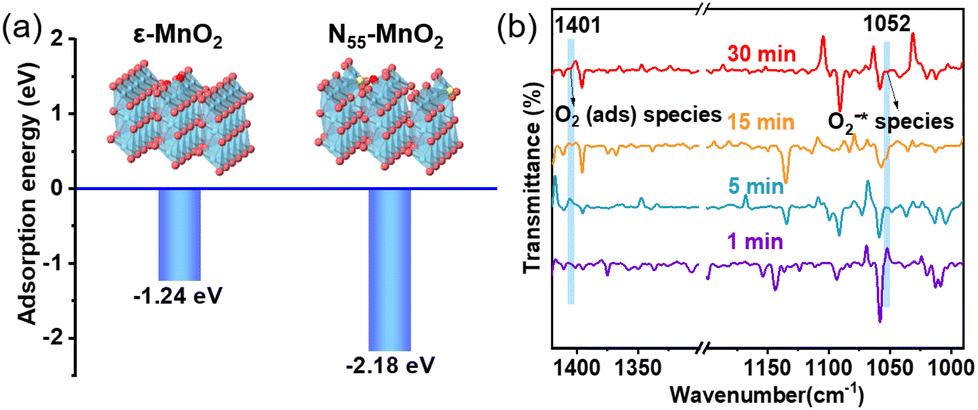 | ||
| Fig. 3 (a) Adsorption energy of O2 on the surface of pristine ε-MnO2, and N55-MnO2 obtained by DFT calculation; (b) in situ IR spectra of O2 adsorbed onto the surface of N55-MnO2. | ||
The direct interactions between O2 and Ny-MnO2 were investigated without light irradiation using EPR. 2,2,6,6-tetramethylpiperidine (TEMP) was employed as a quencher to capture 1O2 activated by Ny-MnO2 at room temperature. As depicted in Fig. 4a and b, N1-MnO2, which exhibits fewer Ov, generates only ˙O2− and not 1O2. With an increase in urea feeding, enhanced N-doping promotes Ov formation in Ny-MnO2. Notably, distinct triple peaks corresponding to TEMP-1O2 are evident for both N55-MnO2 and N25-MnO2, with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.25 The intensity of the 1O2 peak strengthens with higher Ov content, underscoring the significant role of Ov in activating O2. Quantitative EPR analysis indicates that N55-MnO2 generates 1O2 at an average rate of 5.94 mol gcat−1 L−1 min−1 (Fig. 4c). Importantly, Ny-MnO2 doesn't produce highly reactive hydroxyl radicals (˙OH, Fig. S6†).51,52 To investigate the contribution of Ov to the excitation of O2, various ε-MnO2 nanocatalysts were synthesized and characterized without N doping (Fig. S7–S9†). Interestingly, ε-MnO2-n-350 (where 350 indicates the thermal treatment temperature), with the highest Os concentration of 44.4%, demonstrates the capability to generate 1O2 under mild conditions (Fig. S10†). These findings underscore the pivotal role played by abundant surface-exposed Ov in nano-sized MnO2 for the self-activation of O2. However, the catalytic activity of ε-MnO2-n-350 is still much lower than that of N55-MnO2 can be attributed to the doping effect of interstitial N, which will be further discussed later.
:1:1.25 The intensity of the 1O2 peak strengthens with higher Ov content, underscoring the significant role of Ov in activating O2. Quantitative EPR analysis indicates that N55-MnO2 generates 1O2 at an average rate of 5.94 mol gcat−1 L−1 min−1 (Fig. 4c). Importantly, Ny-MnO2 doesn't produce highly reactive hydroxyl radicals (˙OH, Fig. S6†).51,52 To investigate the contribution of Ov to the excitation of O2, various ε-MnO2 nanocatalysts were synthesized and characterized without N doping (Fig. S7–S9†). Interestingly, ε-MnO2-n-350 (where 350 indicates the thermal treatment temperature), with the highest Os concentration of 44.4%, demonstrates the capability to generate 1O2 under mild conditions (Fig. S10†). These findings underscore the pivotal role played by abundant surface-exposed Ov in nano-sized MnO2 for the self-activation of O2. However, the catalytic activity of ε-MnO2-n-350 is still much lower than that of N55-MnO2 can be attributed to the doping effect of interstitial N, which will be further discussed later.
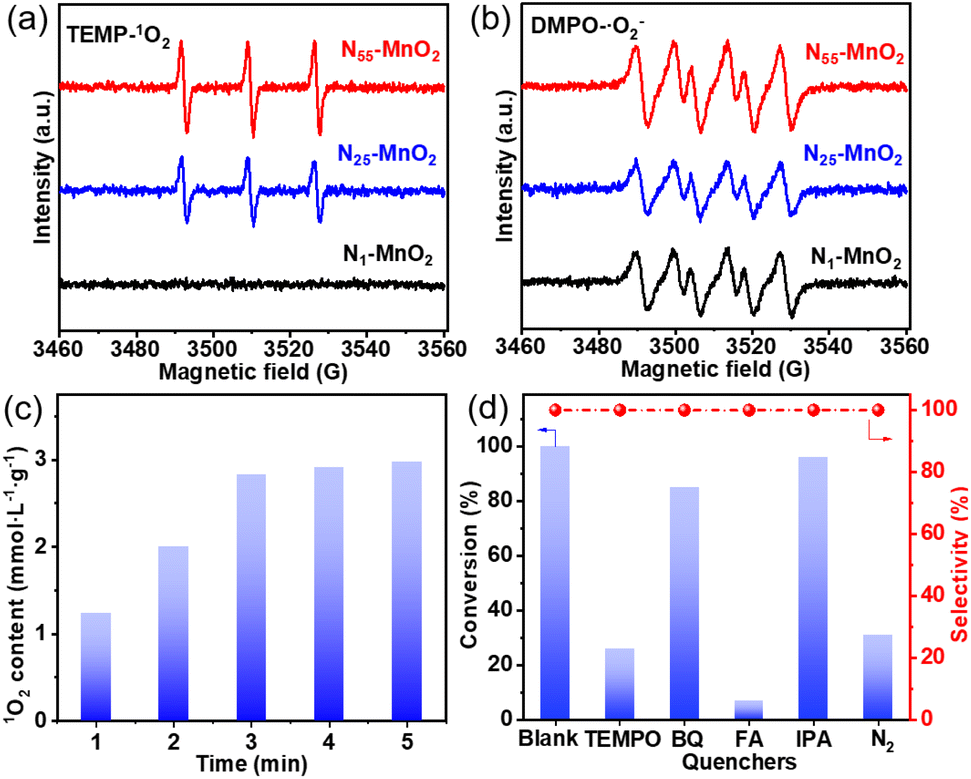 | ||
| Fig. 4 The characteristic peaks of (a) TEMP-1O2 and (b) DMPO-˙O2− captured by EPR; (c) the content of 1O2 produced by N55-MnO2; (d) the controlling experiment catalyzed by N55-MnO2 with different quenchers. | ||
To investigate the roles of ROS in the reaction, controlled quenching experiments were conducted. As shown in Fig. 4d, the conversion of benzyl alcohol sharply decreases from >99.9% to 26% after the addition of TEMPO as a ROS quencher, indicating that ROS mediate the oxidative reaction. Upon separately introducing 1,4-benzoquinone (BQ) and isopropyl alcohol (IPA), the conversion of benzyl alcohol decreases slightly, illustrating that ˙O2− and ˙OH play subsidiary roles in the selective oxidation process. Surprisingly, the oxidation reaction is nearly halted when furfuryl alcohol (FA) is used to capture 1O2. This undeniable evidence confirms 1O2 as the primary ROS in this O2-mediated oxidative reaction. In summary, 1O2 is spontaneously generated through the direct activation of O2, rather than the oxidation of ˙O2− observed in photocatalytic processes.52 Despite O2 being the source of ROS, the conversion of benzyl alcohol can be achieved at 31% and 25.5% under N2 and Ar atmospheres, respectively (Fig. 4d, S11, and Table S4†). This is attributed to the involvement of Ol in the aerobic oxidation process, following the MVK mechanism, which is consistent with previous reports.17,44
Selective oxidation of alcohols
Aerobic oxidation of alcohols serves as a widely important model reaction for assessing the oxidative activity of the catalyst. However, effectively exciting O2 with catalysts remains a challenge.15,20 If a catalyst can spontaneously generate 1O2 from the ground-state oxygen, it will significantly advance the development of environmentally friendly catalysis. The plausible reaction process for efficient oxidation by Ny-MnO2 is presented in Fig. 5. Ov and interstitial N sites promote O2 adsorption on the surface of Ny-MnO2, while Ov elongate the O–O bonds, facilitating direct excitation of O2 to 1O2 for efficient alcohol oxidation. We evaluate the catalytic performance of Ny-MnO2 for the aerobic oxidation of benzyl alcohol under O2 bubbling conditions. We observe that micrometer-scale ε-MnO2 displays low activity (conversion rate of 18.2% in 2.5 hours) due to its inability to activate O2 to form 1O2 (Fig. 6a and S10†). The C-MnO2 exhibits negligible activity (Fig. S12 and S13†). In contrast, N55-MnO2 achieves 99.9% conversion and 99.9% selectivity in 2.5 hours, with a turnover frequency (TOF) 7.4 and 6.7 times higher than that of ε-MnO2 and ε-MnO2-n-350, respectively (Fig. 6b and Table S5†).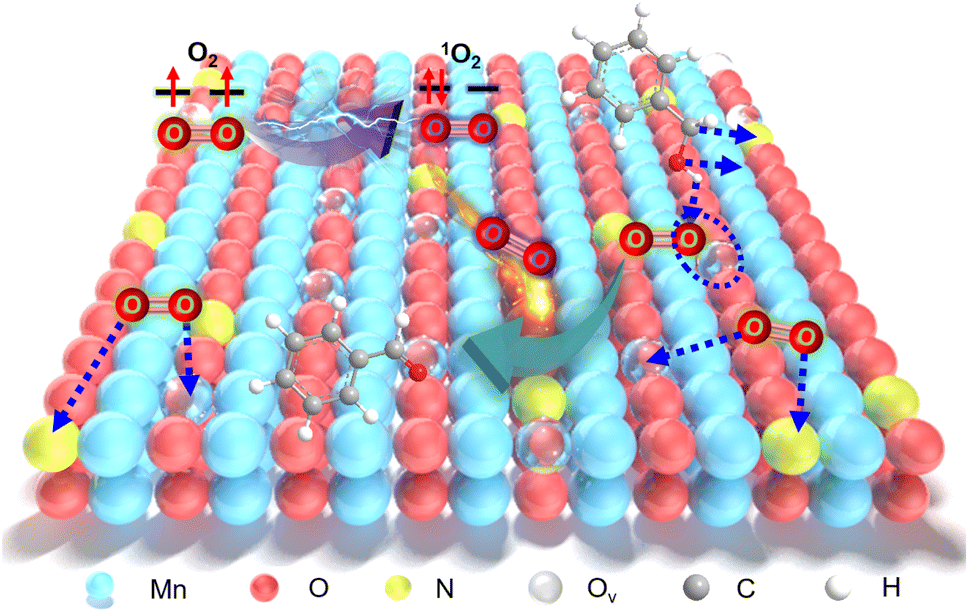 | ||
| Fig. 5 Plausible reaction process for efficient oxidation by Ny-MnO2 activating O2 to form 1O2. | ||
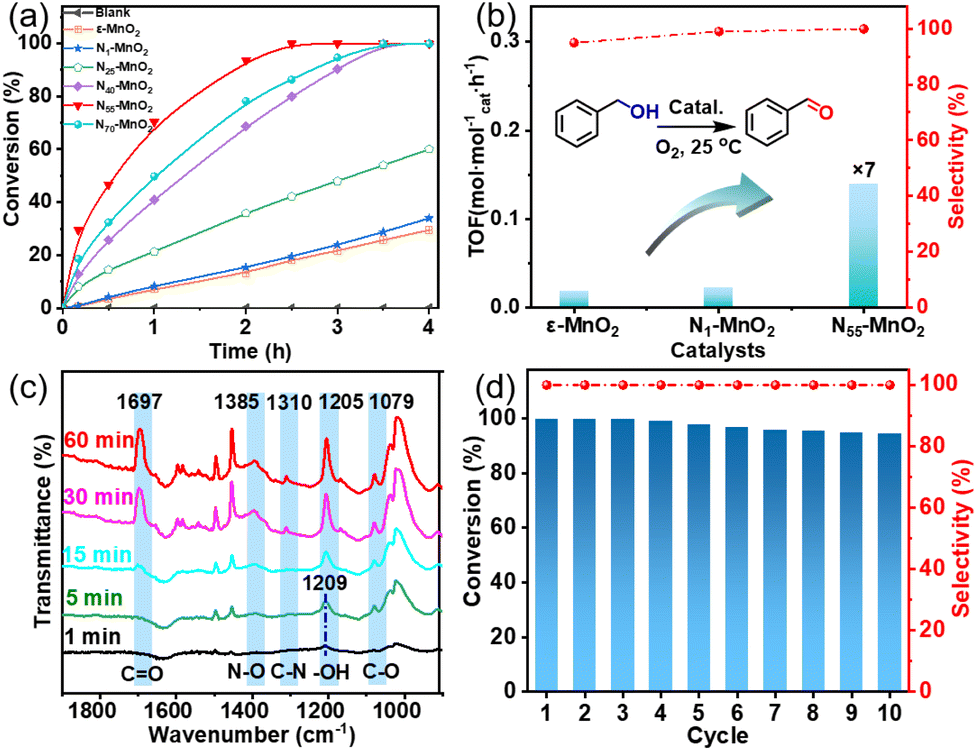 | ||
| Fig. 6 (a) Aerobic oxidation of benzyl alcohol using activated ε-MnO2 and Ny-MnO2 (y = 1, 25, 40, 55, 70), (b) the comparison of TOF of ε-MnO2, N1-MnO2, and N55-MnO2 (TOF = moles of benzyl alcohol converted per mole of catalyst/reaction time); (c) in situ IR spectra of oxidation of benzyl alcohol on the surface of N55-MnO2; (d) the recyclability of N55-MnO2 for aerobic oxidation of benzyl alcohol. | ||
To unravel the catalytic reaction mechanism, we conducted kinetics studies using various catalysts. It can be observed that the initial reaction rate remains constant under different O2 pressures, indicating independence from O2 pressure variations (Fig. S14 and Tables S6–S8†). Through calculation and fitting, the reaction is found to be correlated with the concentration of benzyl alcohol, further demonstrating a first-order reaction (Tables S9 and S10†). Furthermore, we calculate the activation energies (Ea) of the N1-MnO2, N25-MnO2, and N55-MnO2 catalysts using the Arrhenius equation, resulting in values of 55.04 kJ mol−1, 45.87 kJ mol−1, and 43.17 kJ mol−1, respectively (Fig. S15 and Tables S11–S13†). Evidently, the catalytic performance of Ny-MnO2 is positively correlated with its Ov content, providing further evidence of the role of Ov in the oxidation process. In situ IR spectra were utilized to analyze the dynamic process of benzyl alcohol oxidation on the surface of N55-MnO2, revealing enhanced catalytic performance. Under an O2 atmosphere, N55-MnO2 serves as the background for data acquisition. In the first five minutes, four characteristic peaks emerge in Fig. 6c. Compared with C-MnO2 (Fig. S16†), the newly appeared N–O peak at 1385 cm−1 on N55-MnO2 is attributed to O2 adsorption by interstitial N sites. At 1310 cm−1, the band corresponds to the stretching vibration of C–N bonds. The formation of C–N bonds is facilitated by the chemisorption of –OH groups from alcohol onto the interstitial N sites. After exposure to an O2 flow for 15 minutes, the O–H band shifts from 1209 cm−1 to 1205 cm−1, indicating an interaction between the adsorbed O2 species and -C-OH groups on benzyl alcohol. Continuing the exposure for 30 minutes, the broad C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1697 cm−1 signifies the formation of benzyl aldehyde catalyzed by N55-MnO2.
O band at 1697 cm−1 signifies the formation of benzyl aldehyde catalyzed by N55-MnO2.
Semiconductor catalysts have recently demonstrated an enhanced capacity for generating 1O2 when exposed to light, resulting in improved oxidation performance.24,25 In contrast, N55-MnO2 exhibits virtually unchanged activity under both light and dark conditions (Table S4†). This observation suggests that the inherent capability of N55-MnO2 to spontaneously generate 1O2 operates independently of external factors, marking a significant difference from previously reported studies.25,50,53,54 Compared to recently reported heterogeneous catalysts, the well-designed Ny-MnO2 nanocatalysts display a unique ability to spontaneously generate sufficient 1O2 under mild reaction conditions. This enables the green and efficient conversion of alcohols without the need for light or additives, showcasing their superior catalytic activity (Table S14†).
Furthermore, we investigated the applicable substrate scope of aerobic oxidation. As shown in Table 1, aromatic primary alcohols, including benzyl alcohol and its derivatives, exhibit yields of over 99.9% for the corresponding aldehydes. Even for aromatic secondary alcohols, yields exceeding 99.9% are obtained when the reaction time extends to 6.0 hours. In comparison, the catalytic performance of N55-MnO2 in the oxidation of aliphatic alcohols (e.g., 1-hexanol) is hampered by significant steric hindrance effects. Using benzyl alcohol as the model substrate, we evaluate the recyclability of N55-MnO2 in aerobic oxidation. As depicted in Fig. 6d, N55-MnO2 maintains >94% conversion of benzyl alcohol and >99.9% selectivity toward aldehydes over ten cycles. Additionally, XRD and XPS characterizations confirm the stability of the recycled N55-MnO2 structure (Fig. S17†). To the best of our knowledge, results demonstrating both sufficient formation of 1O2 and the green oxidation of alcohols under mild conditions have been scarcely reported.
|
|
||||
|---|---|---|---|---|
| Entry | Product | Conv. (%) | Sel. (%) | Yield (%) |
| a Reaction condition: 5.0 mL of toluene, 0.5 mmol of alcohols, 150.0 mg of N55-MnO2, 1200 rpm, 25.0 ± 1.0 °C, O2 flow 16.0 mL min−1 1.0 bar, reaction time 2.5 h, n. d. not detect. b Reaction time 6.0 h. | ||||
| 1 |
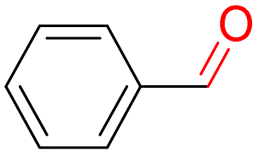
|
99.9 | >99.9 | >99.9 |
| 2 |
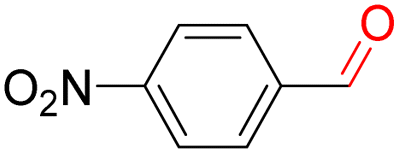
|
99.9 | >99.9 | >99.9 |
| 3 |
|
99.9 | >99.9 | >99.9 |
| 4 |
|
82.6 | >99.9 | >82.6 |
| 5 |
|
99.9 | >99.9 | >99.9 |
| 6 |
|
99.9 | >99.9 | >99.9 |
| 7 |
|
99.9 | >99.9 | >99.9 |
| 8 |
|
99.9 | >99.9 | >99.9 |
| 9 |
|
99.9 | >99.9 | >99.9 |
| 10b |
|
99.9 | >99.9 | 30.5 |
| 11b |
|
99.9 | >99.9 | 47.3 |
| 12 |
|
n.d. | — | n.d |
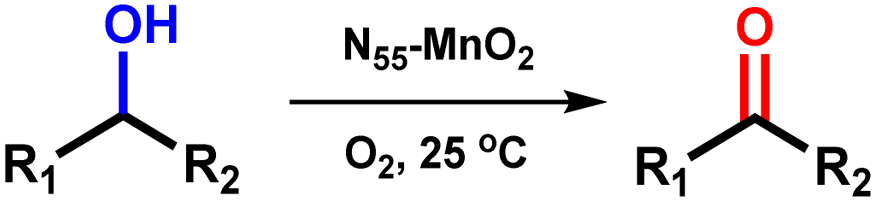
Conclusions
In conclusion, we have successfully developed a range of Ny-MnO2 nanocatalysts capable of directly generating 1O2 at room temperature for the aerobic oxidation of alcohols. The in situ insertion of interstitial N into MnO2 within encapsulated aqueous microreactors can lead to Os concentrations of up to 51.1% for N55-MnO2 and enhance chemisorption of reactants. Consequently, these well-synthesized Ny-MnO2 nanocatalysts have demonstrated enhanced redox performance due to their higher surface Ov exposure and N-doping. DFT calculations have revealed that the interstitial N and increased Ov content facilitates the chemisorption of O2, resulting in the generation of sufficient 1O2. Under aerobic oxidation conditions, the N55-MnO2 catalyst has achieved alcohol conversion rates exceeding 99.9% and aldehyde selectivity of over 99.9%, without the use of additives. Additionally, the N55-MnO2 catalyst has displayed a TOF value of 0.14 mol molcat−1 h−1, 6.7 times higher than that of the N-undoped ε-MnO2 catalyst. Furthermore, N55-MnO2 has shown excellent catalytic stability after ten cycles, remaining robust even after recycling and calcination under an air atmosphere. This study introduces a strategy for fabricating efficient ROS triggers within compartmentalized microdroplets, resulting in the generation of favorable 1O2 to facilitate the aerobic oxidation of alcohols.Experimental
Synthesis of Ny-MnO2 catalyst
A homogeneous oil phase was achieved through vigorous mechanical stirring of sodium dodecyl benzene sulfonate (10.5 g) and xylene (90 mL). Subsequently, a mixture containing manganese(II) nitrate tetrahydrate (10 mmol), water (5.4 mL), and urea (1, 25, 40, 55, 70 mmol) was gently introduced into the aforementioned oil phase, resulting in the formation of a water-in-oil (W/O) microemulsion. After stirring for 30 min, the resulting microemulsion was transferred to a 100 mL Teflon autoclave and hydrothermally treated at 160 °C for 8 h. Finally, the received gray precursor was dried overnight under vacuum, and heated at 350 °C in a muffle furnace to obtain Ny-MnO2 catalyst, where y represents the molar amount of urea used.Catalytic performance of aerobic oxidation
Ny-MnO2 (150 mg), toluene (5 mL), and benzyl alcohol (0.5 mmol) were charged into a Schleck flask, and the temperature was maintained at 25.0 ± 1.0 °C. The reaction mixture was stirred magnetically at 1200 rpm, while O2 was continuously bubbled at a rate of 16 mL min−1. After several hours of reaction, the mixture was analyzed by gas chromatography (PANNA A91Plus). The used Ny-MnO2 was recycled by high-speed centrifugation and subsequently calcined at 350 °C for 4 h.ROS trapping experiment
The ROS control experiments followed procedures similar to the aerobic oxidation process. In these experiments, benzyl alcohol and FA were employed as the model substrate and 1O2 scavenger, respectively. Ny-MnO2 (150.0 mg), toluene (5.0 mL), benzyl alcohol (0.5 mmol), and FA (1.0 mmol) were sequentially introduced into a reaction tube, maintaining the temperature at 25.0 ± 1.0 °C. The mixture underwent magnetic stirring (1200 rpm) with a continuous O2 flow (16.0 mL min−1). Upon completion of the reaction, the conversion of benzyl alcohol was assessed using gas chromatography. Additionally, experiments involving TEMPO, BQ and IPA quenchers were conducted following the aforementioned procedures. The roles of ROS (1O2, ˙O2− and ˙OH) in oxidative reaction were analyzed based on the observed oxidation of benzyl alcohol.Author contributions
J. T. and J. C.: conceptualization, methodology, writing – original draft. Y. Z. Q. M. and P. C.: chemical experiments and analysed the data. Y. K., L. F., J. K., and T. H. participated in various aspects of the experiments and discussions. X. H., P. L. and Z. L.: recycling experiments. C. W., Q. K., Y. K., and Y. Y. review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Y. K. thanks the support from JSPS Postdoctoral Fellowships for Research in Japan. This work was financially supported by the National Natural Science Foundation of China (22302001, 22108238), Key Projects of the Department of Education of Anhui Province of China (RZ2000003450, 2022AH050314), Anhui Provincial Natural Science Foundation of China (2008085MB47), the China Postdoctoral Science Foundation (2019M662060, 2020T130580, PC2022046), and Scientific Research Training Program for College Students of Anhui University of Technology (202110360040, S202110360213, S202310360207, 202310360037). This work was supported by the JST-ERATO Yamauchi Materials Space-Tectonics Project (JPMJER2003) and the UQ-Yonsei International Research Project. The authors would like to thank Zhengjie Chen and Zhangli Pei from SCI-GO (https://www.sci-go.com) for the EPR analysis. The authors also thank Shiyanjia Lab (https://www.shiyanjia.com) for the support of the XRD, XPS, and BET tests. This work used the Queensland node of the NCRIS-enabled Australian National Fabrication Facility (ANFF).References
- C. Lu, C. Zhang, P. Wang, Y. Zhao, Y. Yang, Y. Wang, H. Yuan, S. Qu, X. Zhang, G. Song and K. Pu, Chem, 2020, 6, 2314–2334 CAS.
- C. Bloyet, F. Sciortino, Y. Matsushita, P. A. Karr, A. Liyanage, W. Jevasuwan, N. Fukata, S. Maji, J. Hynek, F. D'Souza, L. K. Shrestha, K. Ariga, T. Yamazaki, N. Shirahata, J. P. Hill and D. T. Payne, J. Am. Chem. Soc., 2022, 144, 10830–10843 CrossRef CAS PubMed.
- X. Bao, H. Li, Z. Wang, F. Tong, M. Liu, Z. Zheng, P. Wang, H. Cheng, Y. Liu, Y. Dai, Y. Fan, Z. Li and B. Huang, Appl. Catal., B, 2021, 286, 119885 CrossRef CAS.
- X. Liu, H. Yu, J. Huang, J. Su, C. Xue, X. Zhou, Y. He, Q. He, D. Xu, C. Xiong and H. Ji, Chem. Sci., 2022, 13, 9560–9568 RSC.
- D. Kalaitzakis, A. Bosveli, K. Sfakianaki, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2021, 133, 4381–4387 CrossRef.
- W. Wu, C. Han, Q. Zhang, Q. Zhang, Z. Li, D. J. Gosztola, G. P. Wiederrecht and M. Wu, J. Catal., 2018, 361, 222–229 CrossRef CAS.
- Q. Ke, S. Fang, J. Tang, F. Li, C. Ning, Z. Tang, Q. Ling, X. Liu and P. Cui, ChemPhotoChem, 2022, 6, e202200075 CrossRef CAS.
- L. Kong, G. Fang, X. Xi, Y. Wen, Y. Chen, M. Xie, F. Zhu, D. Zhou and J. Zhan, Chem. Eng. J., 2021, 403, 126445 CrossRef CAS.
- H. Ma, S. Long, J. Cao, F. Xu, P. Zhou, G. Zeng, X. Zhou, C. Shi, W. Sun, J. Du, K. Han, J. Fan and X. Peng, Chem. Sci., 2021, 12, 13809–13816 RSC.
- C. Lin, S. M. Bachilo and R. B. Weisman, J. Am. Chem. Soc., 2020, 142, 21189–21196 CrossRef CAS PubMed.
- R. Gao, X. Mei, D. Yan, R. Liang and M. Wei, Nat. Commun., 2018, 9, 2798 CrossRef PubMed.
- N. Singh, P. Kumar, R. Kumar and U. Riaz, Ind. Eng. Chem. Res., 2019, 58, 14044–14057 CrossRef CAS.
- Y. Chen, Z. Wang, H. Wang, J. Lu, S. Yu and H. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS PubMed.
- J. Huang, S. He, J. L. Goodsell, J. R. Mulcahy, W. Guo, A. Angerhofer and W. D. Wei, J. Am. Chem. Soc., 2020, 142, 6456–6460 CrossRef CAS PubMed.
- W. Zhang, W. Huang, J. Jin, Y. Gan and S. Zhang, Appl. Catal., B, 2021, 292, 120197 CrossRef CAS.
- J. Tang, Y. Cao, F. Ruan, F. Li, Y. Jin, M. N. Ha, X. Han and Q. Ke, Ind. Eng. Chem. Res., 2020, 59, 9408–9413 CrossRef CAS.
- J. Chen, H. Tang, M. Huang, Y. Yan, J. Zhang, H. Liu, J. Zhang, G. Wang and R. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 26960–26970 CrossRef CAS PubMed.
- F. Ruan, F. Li, Z. Dong, Q. Ke, Y. Jin, W. Zhan, M. N. Ha and J. Tang, Green Synth. Catal., 2021, 2, 38–44 CrossRef.
- M. Koutani, E. Hayashi, K. Kamata and M. Hara, J. Am. Chem. Soc., 2022, 144, 14090–14100 CrossRef CAS PubMed.
- Q. Ke, Y. Jin, F. Ruan, M. N. Ha, D. Li, P. Cui, Y. Cao, H. Wang, T. Wang, V. N. Nguyen, X. Han, X. Wang and P. Cui, Green Chem., 2019, 21, 4313–4318 RSC.
- Y. Pérez-Badell, X. Solans-Monfort, M. Sodupe and L. A. Montero, Phys. Chem. Chem. Phys., 2010, 12, 442–452 RSC.
- J. Oviedo and M. J. Gillan, Surf. Sci., 2001, 490, 221–236 CrossRef CAS.
- Y. Hao, B. Liu, L. Tian, F. Li, J. Ren, S. Liu, Y. Liu, J. Zhao and X. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 12687–12693 CrossRef CAS.
- J. Wang, X. Xu, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, ChemSusChem, 2020, 13, 3488–3494 CrossRef CAS PubMed.
- S. Tan, Y. Ji, Y. Zhao, A. Zhao, B. Wang, J. Yang and J. G. Hou, J. Am. Chem. Soc., 2011, 133, 2002–2009 CrossRef CAS PubMed.
- M. Setvín, U. Aschauer, P. Scheiber, Y. Li, W. Hou, M. Schmid, A. Selloni and U. Diebold, Science, 2013, 341, 988–991 CrossRef.
- M. Li, S. You, X. Duan and Y. Liu, Appl. Catal., B, 2022, 312, 121419 CrossRef CAS.
- X. Kang, G. Dong and T. Dong, ACS Appl. Energy Mater., 2023, 6, 1025–1036 CrossRef CAS.
- Y. Zhuo, X. Guo, W. Cai, T. Shao, D. Xia, C. Li and S. Liu, Appl. Catal., B, 2023, 333, 122789 CrossRef CAS.
- M. Yang, K. Wu, S. Sun, J. Duan, X. Liu, J. Cui, S. Liang and Y. Ren, ACS Catal., 2023, 13, 681–691 CrossRef CAS.
- J. Tang, Q. Zhang, K. Hu, P. Zhang and J. Wang, J. Catal., 2017, 353, 192–198 CrossRef CAS.
- M. Liu, Q. Wang, Z. Liu, Y. Zhao, X. Lai, J. Bi and D. Gao, Chem. Eng. J., 2020, 383, 123161 CrossRef CAS.
- P. V. Vardhan, M. B. Idris, H. Y. Liu, S. R. Sivakkumar, P. Balaya and S. Devaraj, J. Electrochem. Soc., 2018, 165, A1865 CrossRef CAS.
- T. He, X. Zeng and S. Rong, J. Mater. Chem. A, 2020, 8, 8383–8396 RSC.
- T. J. Frankcombe and Y. Liu, Chem. Mater., 2023, 35, 5468–5474 CrossRef CAS.
- G. Qi, X. Liu, C. Li, C. Wang and Z. Yuan, Angew. Chem., Int. Ed., 2019, 58, 17406–17411 CrossRef CAS.
- L. Liu, R. Liu, T. Xu, Q. Zhang, Y. Tan, Q. Zhang, J. Ding and Y. Tang, Inorg. Chem., 2020, 59, 14407–14414 CrossRef CAS.
- S. Rong, K. Li, P. Zhang, F. Liu and J. Zhang, Catal. Sci. Technol., 2018, 8, 1799–1812 RSC.
- Y. Huang, Y. Liu, W. Wang, M. Chen, H. Li, S. Lee, W. Ho, T. Huang and J. Cao, Appl. Catal., B, 2020, 278, 119294 CrossRef CAS.
- F. Morales, D. Grandjean, A. Mens, F. M. F. Groot and B. M. Weckhuysen, J. Phys. Chem. B, 2006, 110, 8626–8639 CrossRef CAS PubMed.
- Y. Xu, J. Dhainaut, G. Rochard, J. Dacquin, A. Mamede, J. Giraudon, J. Lamonier, H. Zhang and S. Royer, Chem. Eng. J., 2020, 388, 124146 CrossRef CAS.
- J. Tang, B. Jiao, W. Chen, F. Ruan, F. Li, P. Cui, C. Wan, M. N. Ha, V. N. Nguyen and Q. Ke, Nano Res., 2022, 15, 6076–6083 CrossRef CAS.
- Y. Jin, F. Li, P. Cui, Y. Yang, Q. Ke, M. N. Ha, W. Zhan, F. Ruan, C. Wan, Z. Lei, V. N. Nguyen, W. Chen and J. Tang, Nano Res., 2021, 14, 2637–2643 CrossRef CAS.
- Q. Ke, D. Yi, Y. Jin, F. Lu, B. Zhou, F. Zhan, Y. Yang, D. Gao, P. Yan, C. Wan, P. Cui, D. Golberg, J. Yao and X. Wang, ACS Sustain. Chem. Eng., 2020, 8, 5734–5741 CrossRef CAS.
- H. Li, Y. Zhang, J. Tang, G. Huang, P. Cui and Q. Ke, Green Synth. Catal., 2022 DOI:10.1016/j.gresc.2022.09.005.
- J. Yu, T. Zeng, H. Wang, H. Zhang, Y. Sun, L. Chen, S. Song, L. Li and H. Shi, Chem. Eng. J., 2020, 394, 124458 CrossRef CAS.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- Y. Lin, Z. Liu, L. Yu, G. Zhang, H. Tan, K. Wu, F. Song, A. K. Mechler, P. P. M. Schleker, Q. Lu, B. Zhang and S. Heumann, Angew. Chem., Int. Ed., 2021, 60, 3299–3306 CrossRef CAS PubMed.
- F. Li, J. Tang, Q. Ke, Y. Guo, M. N. Ha, C. Wan, Z. Lei, J. Gu, Q. Ling, V. N. Nguyen and W. Zhan, ACS Catal., 2021, 11, 11855–11866 CrossRef CAS.
- J. Li, X. Wang, J. Tian, X. Zhang and F. Shi, Rare Met., 2023, 42, 1877–1887 CrossRef CAS.
- B. He, H. Jin, Y. Wang, C. Fan, Y. Wang, X. Zhang, J. Liu, R. Li and J. Liu, Rare Met., 2022, 41, 132–143 CrossRef.
- N. Siemer, A. Lüken, M. Zalibera, J. Frenzel, D. Muñoz-Santiburcio, A. Savitsky, W. Lubitz, M. Muhler, D. Marx and J. Strunk, J. Am. Chem. Soc., 2018, 140, 18082–18092 CrossRef CAS PubMed.
- T. Xia, W. Gong, Y. Chen, M. Duan, J. Ma, X. Cui, Y. Dai, C. Gao and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202204225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04418a |
| This journal is © The Royal Society of Chemistry 2023 |